
radicale
Raggruppamento di atomi presente nelle molecole di numerosi composti che si conserva inalterato in molte trasformazioni (di doppio scambio, acido-base ecc.) non distruttive.
Cenni storici
Il termine r. fu introdotto nel 1787 da A.L. Lavoisier, G. de Morveau, A.F. de Fourcroy e P.J. Macquer, nell’ambito della loro nuova nomenclatura, per indicare gruppi di atomi che stavano alla base della costituzione di acidi e basi e quindi dei sali da loro derivati; J.J. Berzelius l’utilizzò in chimica organica: basandosi sul fatto che nelle reazioni organiche di sostituzione gruppi di atomi erano capaci di trasferirsi inalterati da un composto all’altro, formulò la teoria dei r. secondo la quale raggruppamenti atomici indipendenti, formati da idrogeno e carbonio, erano in grado di legarsi a elementi di «diversa polarità» come l’ossigeno. La teoria, che si basava sull’ipotesi dualistica sostenuta da Berzelius, entrò in crisi quando si mostrò che l’idrogeno poteva essere sostituito, in un r., da elementi di polarità opposta come il cloro; tuttavia essa fu importante sia perché ebbe il merito di aprire la strada alla successiva teoria della struttura, sia perché permise l’introduzione del concetto di r. anche in chimica organica.
Tipi di radicali
Formalmente, un r., come sopra definito, si caratterizza per la presenza di una o più valenze libere; per es., il r. solfato, SO4, è presente in molti sali, nonché nell’acido solforico, ed è bivalente.
In particolare, in chimica organica, un r. è la parte idrocarburica, alifatica o aromatica, di una molecola organica che rimane inalterata nelle reazioni non distruttive e non demolitive (per es., nelle reazioni di sostituzione). Si possono avere r. monovalenti (r. alchilici) come il metile, −CH3, e l’etile, −C2H5, bivalenti (r. alchilenici), come l’etilene −CH2CH2−, trivalenti (r. alchenilici) come il metenile, ≡CH. I r. ottenuti dagli idrocarburi aromatici sono chiamati r. arilici. Solitamente i r. alchilici sono indicati con la lettera R, quelli arilici con Ar; così la formula di un alcol alifatico è ROH mentre un cloruro arilico è ArCl.
R. liberi
Sono così dette le specie chimiche che, avendo uno o più elettroni di valenza spaiati non impegnati in legami chimici, risultano estremamente reattive e con vita media molto breve. L’uso del termine si giustifica considerando il fatto che nei r. liberi vi è almeno un atomo che, a causa dell’elettrone spaiato, risulta avere una valenza libera; per tale motivo i r. liberi corrispondono formalmente a r. propriamente detti. Nelle formule, un r. libero viene rappresentato apponendo un puntino accanto al simbolo dell’atomo che ha l’elettrone spaiato.
A causa della loro labilità, i r. liberi non sono isolabili ma costituiscono un’importante classe di intermedi di molte reazioni chimiche (reazioni radicaliche). Solo in particolari circostanze è possibile preparare r. liberi dotati di una certa stabilità, come nel caso del r. trifenilmetilico, (C6H5)3C, preparato per azione dell’argento metallico sul cloruro di trifenilmetano in benzene; la struttura di questo r. è stabilizzata per delocalizzazione dell’elettrone spaiato sul sistema aromatico.
La prova dell’esistenza dei r. liberi è dovuta a F.A. Paneth e W. Hofeditz che nel 1929, in un esperimento divenuto classico, decomposero termicamente il piombo tetrametile, Pb(CH3)4, in piombo elementare e in un prodotto gassoso che convogliarono tramite gas inerte su un filo di piombo metallico, ottenendo nuovamente piombo tetrametile. Per interpretare l’esperimento è necessario supporre la formazione di radicali metile e la loro successiva combinazione, secondo le equazioni:
I r. liberi si formano per rottura omolitica di un legame covalente, generalmente a temperatura elevata; solo pochi legami, come quello perossidico, si rompono a temperature al di sotto dei 200 °C.
La scissione omolitica del legame si ottiene anche per azione della radiazione ultravioletta: l’alterazione di alimenti, il deterioramento delle proprietà degli elastomeri e il processo di indurimento delle vernici sono fenomeni in larga misura attribuibili alla formazione, nel corso del tempo, di r. liberi da parte delle radiazioni ultraviolette naturali. Particolare rilievo ha l’azione dei r. liberi nei confronti degli organismi. I r. liberi hanno una notevole tendenza a ricombinarsi tra loro per formare specie con elettroni accoppiati; per tale motivo, al contrario dei prodotti di scissione eterolitica, cioè gli ioni, i r. non possono essere conservati in soluzione se non a bassissima concentrazione e solo se generati di continuo.
I r. liberi possono essere rivelati sulla base del loro paramagnetismo, dovuto alla presenza dell’elettrone non accoppiato: questa caratteristica dà luogo a proprietà spettroscopiche che possono essere messe in rilievo tramite l’uso di particolari tecniche strumentali. Un r. libero, una volta formatosi, può dare luogo a vari processi: in alcuni si ha la formazione di nuovi r., come nel processo di estrazione, quando un r. estrae un atomo da una molecola, o in quello di addizione, quando si lega a un legame multiplo, o in quello di frammentazione (che dà origine a r. di minor peso molecolare); in altri si ha la scomparsa dei r. liberi, come nel processo di ricombinazione o in quello di disproporzionamento. La sequenza di formazione dei r., propagazione della reazione con formazione di nuovi r., e terminazione con la ricombinazione dei r. è caratteristica delle reazioni radicaliche a catena. (➔ radicale libero)
R. liberi dell’ossigeno
I r. liberi dell’ossigeno hanno acquistato una particolare importanza per la loro implicazione in numerosi processi fisiopatologici degli organismi viventi (invecchiamento, ischemie, infiammazioni acute e croniche ecc.).
Origine. La configurazione elettronica esterna dell’ossigeno è 1s22s2p4. La molecola di O2 presenta, perciò, due elettroni spaiati, ciascuno localizzato in un differente orbitale π* antilegante, e caratterizzati dallo stesso numero quantico di spin. Quando l’O2 si trova in questa forma, si parla di stato fondamentale della molecola o stato di minima energia. Sono possibili, però, due stati eccitati, ad alta energia, della molecola di O2, noti come ossigeno di singoletto, nei quali gli elettroni spaiati presentano spin opposti: lo stato eccitato 1ΔgO2, in cui i due elettroni occupano lo stesso orbitale e hanno lo stesso momento angolare, che presenta un’energia di 98 kJ/mol in più rispetto all’O2; lo stato eccitato 1Σ+O2, in cui i due elettroni occupano orbitali diversi con spin antiparalleli, che presenta un’energia di 157 kJ/mol in più rispetto all’O2. Lo stato eccitato 1Σ+O2 ha un tempo di emivita di circa 10–11 s e, prima di poter reagire, decade allo stato eccitato 1ΔgO2. Poiché la riduzione bivalente del diossigeno è cineticamente limitata dal processo, relativamente lento, di inversione di spin, la molecola di O2 può essere facilmente ridotta in passaggi successivi monoelettronici. È attraverso questi passaggi che si generano le specie radicaliche dell’ossigeno, di cui lo ione superossido O∙2− rappresenta il primo elemento che si ottiene dall’acquisto di un elettrone da parte dell’O2.
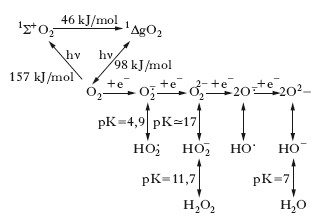
L’acquisto successivo di elettroni fino alla completa riduzione ad acqua può generare altri r., come indicato in fig. In condizioni fisiologiche, circa l’1% dell’O2 che viene ridotto a H2O dall’enzima citocromoossidasi, durante il metabolismo ossidativo mitocondriale, è trasformato in ione superossido O∙2− che può anche essere prodotto dall’attività di alcuni enzimi, per es., l’NADPH-citocromo-P450-reduttasi. L’O∙2− prodotto può andare incontro a una dismutazione spontanea secondo la reazione:
La stessa reazione, ovviamente con un’efficienza e una velocità diverse, è catalizzata dall’enzima detossificante superossidodismutasi. In presenza di tracce di metalli di transizione, come per es. il ferro, che è contenuto in quantità rilevanti nelle cellule, l’ O∙2− e il perossido di idrogeno (H2O2) vanno incontro alla cosiddetta reazione di Fenton (➔ Fenton, Henry John Horstman):
Uno dei prodotti finali è lo ione ossidrile, una delle specie radicaliche più reattive e dannose.
Reazioni. L’elevata reattività delle specie radicaliche dell’O2 (in particolare dell’ossigeno di singoletto, del r. peridrossile e del r. ossidrile) fa sì che esse possano reagire con una serie di macromolecole biologiche capaci di accettare elettroni che subiscono, perciò, modificazioni strutturali e, in seguito, funzionali irreversibili. Per es., i r. dell’ossigeno provocano l’ossidazione di proteine, acidi nucleici, e dei fosfolipidi di membrana, innescando in questi ultimi reazioni a catena di perossidazione lipidica (➔ malonico, acido; perossidi).
In alcune situazioni patologiche, quali le infiammazioni acute e croniche, il diabete, i fenomeni di ischemia tissutale, si verifica un aumento molto considerevole dei r. dell’ossigeno. In particolare, nei tessuti ischemici e successivamente riperfusi, i r. dell’O2 possono essere prodotti da 4 meccanismi differenti: a) l’attività della xantinaossidasi (➔ xantina), che nella reazione di ossidazione dell’ipoxantina e xantina a acido urico genera ioni O∙2−; b) la perdita della funzionalità mitocondriale, che provoca la riduzione incompleta dell’ossigeno molecolare con formazione di O∙2−; c) l’attivazione dei granulociti polimorfonucleati (neutrofili) che, richiamati per chemiotassi nell’area ischemica, generano durante la fagocitosi rilevanti quantità di O∙2−; d) l’ossidazione delle catecolammine che produce un flusso di ioni superossido O∙2−. Si può quindi dire che la produzione di O∙2− sia il punto di inizio per la comparsa di altre specie radicaliche più dannose, che si verifica attraverso la reazione di Fenton.
Meccanismi di difesa Tutti gli organismi viventi hanno sviluppato dei sistemi di difesa, enzimatici e non enzimatici, per limitare i danni derivanti dalla tossicità dei r. dell’O2. Così, l’enzima superossidodismutasi dismuta il superossido a H2O2 e O2, mentre la catalasi e la glutationeperossidasi trasformano l’H2O2 in H2O e O2. Inoltre, numerose molecole non enzimatiche, quali la vitamina E (α-tocoferolo), il glutatione, la vitamina C (acido ascorbico) e l’acido urico sono in grado di interagire aspecificamente con i r. dell’ossigeno, subendo un processo di ossidazione che riduce così la concentrazione dei r. presenti e impedisce, almeno parzialmente, l’ossidazione e il danneggiamento irreversibile di macromolecole biologiche fondamentali per la sopravvivenza cellulare (➔ antiossidanti).
Il trattamento e la prevenzione delle patologie da r. liberi dell’ossigeno (neoplasie, ischemie, processi aterosclerotici) si basa sulla somministrazione (alimentare o medicamentosa) degli antiossidanti.