
reattività
reattività In chimica, la tendenza di una sostanza a reagire facilmente al contatto con altre.
R. e struttura
Lo studio delle relazioni esistenti fra la struttura delle sostanze chimiche e la loro r. costituisce un aspetto importante della chimica moderna i cui obiettivi sono perseguibili al meglio solo se sono note le condizioni necessarie per trasformare una sostanza chimica in un’altra, con le massime rese, con il minor dispendio energetico, nel minor tempo. I gruppi funzionali presenti in una molecola ne determinano la r.: gli acidi carbossilici si ionizzano, le ammine sono basiche ecc.
Le r. dei gruppi funzionali sono state l’oggetto della chimica organica del 19° sec. e di parte del 20°. Ci si accorse subito, tuttavia, che non esiste una relazione biunivoca fra la struttura di una sostanza e la sua r.; quest’ultima, infatti, è correlata alla particolare reazione adoperata e le r. relative di due sostanze simili possono subire profonde variazioni al variare della reazione cui sono sottoposte. I primi passi nello studio delle relazioni tra r. e struttura sono stati quelli relativi agli effetti dei sostituenti nelle reazioni di sostituzione elettrofila aromatica. Si trovò che substrati contenenti gruppi elettron donatori (per es., −OCH3) reagivano più velocemente del benzene e che il gruppo entrante era indirizzato nelle posizioni orto e para e che, viceversa, substrati contenenti gruppi elettron attrattori (per es., −NO2) davano reazioni di sostituzione elettrofila più lentamente del benzene e che il nuovo gruppo entrava nell’anello aromatico in posizione meta.
Lo studio del rapporto tra r. e struttura si identifica anche con lo studio dei meccanismi di reazione, il cui approccio principale consiste nella determinazione quantitativa della velocità con cui una sostanza, A, è trasformata nei prodotti di reazione (B, C, …). Le leggi cinetiche che descrivono questi fenomeni possono essere anche molto complesse. Sostanze simili A1, A2, ..., An vengono consumate in una reazione con costanti cinetiche diverse k1, k2, ..., kn (➔ cinetica) legate fra loro da relazioni del tipo
[1] formula
Storicamente sono state le ricerche di L.P. Hammett ad aprire l’epoca degli studi del rapporto tra r. e struttura. Egli trovò (1937) che le costanti di acidità, K, degli acidi aromatici meta e para sostituiti e le costanti cinetiche, k, dell’idrolisi basica dei loro esteri etilici sono legate da una semplice legge di proporzionalità
[2] formula
dove K0 e k0 sono rispettivamente la costante di acidità dell’acido benzoico e la costante cinetica di idrolisi del suo estere. Hammett ne dedusse che i cambiamenti delle energie di ionizzazione, provocati dall’introduzione di una serie di sostituenti nell’anello aromatico, sono direttamente proporzionali ai cambiamenti delle energie di attivazione per l’idrolisi dei benzoati causati dalla stessa serie di sostituenti. Egli propose allora di scrivere il termine di destra della precedente equazione come il prodotto di due costanti empiriche, ρ e σ, rappresentanti rispettivamente la sensibilità della reazione agli effetti dei sostituenti e gli effetti elettronici dei sostituenti stessi. Pervenne quindi alle due seguenti correlazioni lineari di energia libera valide rispettivamente per le velocità e per gli equilibri:
Hammett propose anche di assegnare alla reazione di ionizzazione degli acidi benzoici un valore convenzionale unitario per la costante ρ. Le costanti σ, per una serie di sostituenti, furono quindi da lui determinate misurando le costanti di dissociazione di una serie di acidi benzoici sostituiti. I valori così trovati furono usati per altre serie di reazioni di cui determinò i valori di ρ. È chiaro che è sufficiente determinare due valori di ln (kx/ky) per sostituenti di cui siano note le costanti σ, per poter determinare il ρ di una reazione.
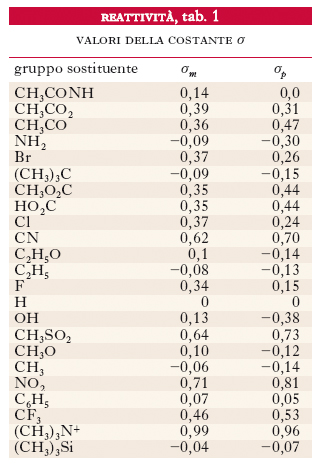

La tab. 1 dà i valori di σ di Hammett per una serie di sostituenti in posizione meta (σm) e para (σp): essi indicano la capacità complessiva di un sostituente di attrarre o di respingere elettroni, indipendentemente dal meccanismo (mesomerico, di campo o induttivo) con cui l’effetto si esplica. I gruppi con valori di σ positivi attirano elettroni; quelli con valori negativi li respingono. La tab. 2 contiene una serie di valori di ρ per reazioni di substrati aromatici. Le reazioni con valori positivi di ρ sono facilitate da sostituenti che attirano elettroni; viceversa quelle con valori negativi. In totale le due tabelle contengono 58 valori con i quali è possibile calcolare le costanti di dissociazione o le r. di 552 composti diversi. La tab. 1 indica già un limite del metodo nel fatto di contenere soltanto i valori di σm e σp dei sostituenti: gli effetti causati da sostituenti in posizione orto rispetto alla posizione reattiva non obbediscono infatti a leggi di correlazione lineare e ciò a causa del sovrapporsi di interazioni steriche, variabili da una reazione all’altra, fra gruppi reattivi e sostituenti. D’altra parte, se esiste coniugazione diretta fra centro reattivo e sostituente, è necessario usare valori di σm e σp diversi da quelli contenuti nella tab. 1.
Comportamento delle molecole
Spesso si verifica che una sostanza A sia consumata da più reazioni concorrenti in processo chimico così schematizzabile: A→B+C+D. In questi casi alla fine della reazione si ha una miscela di sostanze diverse. Ciò dipende da molti fattori, il più importante dei quali è il fatto che quello da noi chiamato sistema iniziale A è costituito da una moltitudine di singole molecole. Raramente nelle sue provette un chimico manipola meno di 1019 molecole, ciascuna delle quali può subire un processo chimico diverso dalle altre. A livello microscopico la spiegazione del diverso comportamento di molecole, peraltro tutte uguali fra loro, risiede nel fatto che l’atto reattivo è una collisione fra molecole: le energie e le geometrie delle collisioni fra corpi, che, come le molecole, in nessun caso possono essere assimilati a una sfera, determinano il destino di ciascuna molecola. La riproducibilità di un sistema chimico è soltanto statistica nel senso che, dato il sistema iniziale e le condizioni di reazione, il sistema finale è costituito sempre dalla stessa miscela di molecole diverse. Lo scopo pratico degli studi del rapporto tra r. e struttura è appunto quello di introdurre nel substrato, nei reattivi, nelle condizioni di reazione, quelle variazioni che rendano più probabile una delle possibili geometrie di collisione, o più vantaggioso in termini energetici uno dei possibili stati di transizione, o più stabile uno dei possibili stati finali.
Se tutte le reazioni concorrenti sono irreversibili, la composizione della miscela finale è determinata dalle velocità relative delle reazioni concorrenti. Si dice allora che la reazione è sotto controllo cinetico. Un aumento di temperatura può invece rendere reversibili le reazioni concorrenti. La composizione della miscela finale è determinata allora dalle differenze di energia libera fra tutti i prodotti del sistema chimico. In tal caso la reazione è sotto controllo termodinamico. Spesso un’accurata scelta delle condizioni di reazione può privilegiare il controllo cinetico o quello termodinamico.
La reazione di enolizzazione dei chetoni

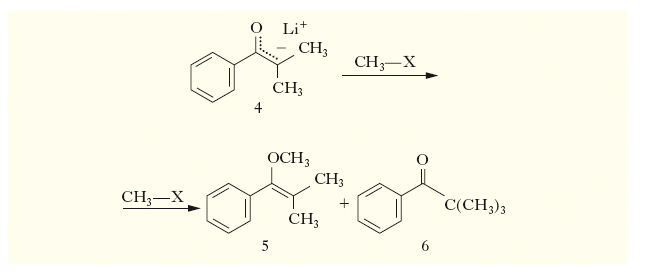
(una reazione molto importante per la chimica di sintesi) può illustrare bene questo punto: il metilisopropilchetone 1 può essere enolizzato per azione di una base in due diversi modi fornendo gli enolati 2 e 3. L’enolato 2, avendo il doppio legame più sostituito, è quello termodinamicamente più stabile e si forma in prevalenza se la base è debole (di basicità comparabile a quella dell’enolato stesso) e se il solvente di reazione è protico. Se il solvente è aprotico e la base è forte e stericamente impedita, la reazione di enolizzazione diviene sostanzialmente irreversibile ed essendo più veloce il processo che sottrae un protone dal più accessibile gruppo α-CH3, si forma l’enolato ‘cinetico’ 3. Gli enolati sono dei nucleofili bidentati e come tali possono essere alchilati (questo è il principale scopo della preparazione degli enolati) sia all’ossigeno sia al carbonio. Pur essendo la carica negativa degli enolati prevalentemente concentrata sull’atomo di ossigeno, è possibile trovare condizioni di reazione che favoriscano l’alchilazione dell’atomo di carbonio (v. fig.).
L’enolato 4 è alchilato all’ossigeno, per dare 5, se il gruppo uscente dell’agente alchilante è un nucleofilo piccolo e molto elettronegativo (è il caso dei solfati e dei solfonati alchilici) e il solvente è polare aprotico (per es., il dimetilsolfossido), e soprattutto in presenza di eteri corona che possano catturare il catione Li+. L’enolato 4 è invece alchilato al carbonio, per dare 6, se il gruppo uscente dell’agente alchilante è poco elettronegativo e molto polarizzabile (è il caso degli ioduri alchilici) e il solvente è meno polare (per es., tetraidrofurano).