
Eredità biologica
Eredita biologica
di Giuseppe Montalenti
Eredità biologica
sommario: 1. Concetti generali: a) introduzione: concetti tradizionali di eredità; b) il mendelismo; c) carattere e norma di reazione; d) il coefficiente di ereditarietà; e) pleiotropia, polimeria, penetranza, espressività; f) eredità generale ed eredità speciale: i geni letali. 2. L'eredità nell'uomo: a) introduzione; b) l'assetto cromosomico umano; c) mutazioni del genoma e mutazioni cromosomiche; d) mutazioni geniche; e) frequenza di mutazione nell'uomo; f) studi di genetica sulle cellule coltivate in vitro; g) polimorfismi genetici in popolazioni umane; h) consanguineità; i) conclusioni. □ Bibliografia.
1. Concetti generali
a) Introduzione: concetti tradizionali di eredità
Il problema dell'eredità biologica o ereditarietà, cioè come vengano trasmessi ai discendenti i caratteri morfologici, fisiologici, psichici, ed eventualmente patologici, ha sempre interessato, fin dai tempi antichi, i naturalisti, i medici, i filosofi. In passato, e fino a tutto il sec. XIX, s'intendeva per eredità la trasmissione dei caratteri dai genitori ai figli, cioè si fermava l'attenzione sul fatto che i figli rassomigliano ai propri genitori. Si sapeva anche che talvolta un carattere ‛salta una generazione' cioè si trasmette dai nonni ai nipoti e, per i casi in cui in una generazione compaiono caratteri che esistevano, o si presumeva esistessero, negli antenati più o meno remoti, si era creato il concetto e il termine di ‛atavismo' o ‛eredità atavica o ancestrale'. Nel caso in cui un carattere si manifesta in parecchi membri di uno stesso ceppo familiare, si parlava di ‛familiarità'.
Il ramo delle scienze biologiche che studia l'eredità biologica si è molto sviluppato nel sec. XX e costituisce una disciplina autonoma cui è stato dato il nome di genetica (W. Bateson, 1906). I dati fondamentali relativi ai problemi dell'ereditarietà sono trattati nell'articolo genetica, al quale rinviamo.
Molte teorie furono proposte, fin dai tempi dell'antica Grecia, per cercare di spiegare il fenomeno dell'ereditarietà. In un libro, pubblicato in seconda edizione nel 1903 col titolo: L'hérédité et les grands problèmes de la biologie générale, l'autore, Y. Delage, ignaro ancora delle leggi di Mendel, che erano state riscoperte tre anni prima, enumera una quarantina d'ipotesi che classifica in quattro categorie: ipotesi animiste, evoluzioniste (nel senso del preformismo settecentesco), micromeriste e organiciste. Le più numerose, soprattutto nei tempi moderni, sono le micromeriste, cioè quelle che fanno appello a particelle materiali piccolissime, che sarebbero depositarie di ciò che oggi si conviene di chiamare l'informazione genetica. Alle ipotetiche particelle furono dati vari nomi: bioblasti, plastidule, granuli, gemmule, micelle, idioblasti, determinanti, ecc.
Il fiorire di tante ipotesi dimostra il travaglio intellettuale che ha impegnato molti biologi nel tentativo di trovare una soluzione al problema dell'ereditarietà che le teorie dell'evoluzione, e in particolare il darwinismo, avevano portato in campo come uno dei problemi fondamentali per comprendere il meccanismo che sta alla base dell'evoluzione biologica (v. evoluzione). Queste teorie avevano anche spostato il punto focale dell'indagine dalla considerazione delle ‛somiglianze' a quella delle ‛differenze' fra genitori e figli, le quali sono più delle prime importanti, anzi essenziali per l'evoluzione.
Le ipotesi micromeriste sono le più numerose e le più accreditate, perché hanno una base meccanicistica che evita di fare appello a ‛forze' vitali, o ad altri principî metafisici, non dimostrabili scientificamente. Fra tutte, quella che più si accosta alle moderne vedute, anzi le precorre con sorprendente approssimazione, è la teoria di A. Weismann (1884), il quale ideò un'architettura del patrimonio ereditario, che sarebbe costituito da unità submicroscopiche, i ‛biofori'; parecchi biofori riuniti costituiscono i ‛determinanti', i quali sono raggruppati in strutture d'ordine superiore, denominate ‛idi' e ‛idanti'. Questi ultimi sarebbero individuabili nei cromosomi, che da poco erano stati scoperti, mentre i determinanti corrispondono press'a poco ai geni della moderna genetica.
Negli ultimi decenni del sec. XIX gli studi sull'ereditarietà condussero a tre fondamentali acquisizioni. La prima fu l'identificazione di una parte del protoplasma che è depositana dei caratteri ereditari, cioè dell'informazione genetica: fu chiamata ‛idioplasma' (K. Nägeli), e presto si riconobbe che l'idioplasma è localizzato nel nucleo della cellula, e si ipotizzò che fosse identificabile con i cromosomi, come appunto fece il Weismann nella teoria sopra ricordata. Questo fatto fu poi dimostrato da Th. H. Morgan e dai suoi collaboratori nel secondo decennio del sec. XX. Complemento della teoria dell'idioplasma è la teoria di Nussbaum-Weismann della separazione tra ‛soma' e ‛germe', cioè tra il corpo, perituro, e il ‛plasma germinale' che si trasmette di generazione in generazione ed è quindi potenzialmente immortale.
La seconda acquisizione è di metodo, più che non di fatti. Poiché è evidente che i figli non sono identici ai genitori, ma se ne discostano più o meno ampiamente, F. Galton si propose di determinare di quanto essi differiscano dai genitori e quindi, reciprocamente, quanto i figli ereditino dai genitori. La ricerca fu condotta su di un carattere quantitativo della specie umana, la statura, e i risultati furono interpretati dall'autore come la dimostrazione di due ‛leggi' dell'eredità: la ‛regressione filiale' e l'eredità ancestrale'. Le leggi di Galton non hanno più alcun valore, in quanto sono superate dalla concezione mendeliana; ma l'impianto quantitativo, su basi statistiche, dell'indagine galtoniana ha dato origine a importanti sviluppi metodologici (la scuola biometrica inglese) e di conoscenze di fatti (lo studio dei caratteri quantitativi; v. genetica).
Dalle ricerche del Galton prese lo spunto anche un'indagine che condusse a un terzo gruppo di risultati della massima importanza, in quanto diedero risposta negativa all'antica e diffusissima credenza dell'ereditarietà dei caratteri acquisiti. Questi sarebbero indotti nell'individuo dall'azione dei fattori esterni, ambientali, che agiscono sul ‛soma', cioè sul corpo, e che s'iscriverebbero poi nel plasma germinale trasmettendosi, per tale via, ai discendenti. Il botanico danese W. Johannsen, nell'intento di verificare i dati e la teoria di Galton, istituì gli esperimenti sulle ‛linee pure' dei fagioli, cioè su ceppi derivati da un unico individuo per successive autofecondazioni. In questi ceppi la variabilità genetica (derivata dalla anfimissi, cioè dalla miscela di patrimoni ereditari diversi, che si realizza con la riproduzione sessuale) è praticamente annullata. La variabilità residua che si riscontra negli individui della linea pura è dovuta all'azione dei fattori ambientali che agiscono sulla pianta durante il suo sviluppo. Johannsen dimostrò che i caratteri acquisiti dai singoli individui per questa via non sono trasmessi ai discendenti. Ulteriori prove della non ereditarietà dei caratteri acquisiti si ottennero in seguito su altri organismi, con esperimenti altrettanto rigorosi di quelli del Johannsen, e quindi la teoria che J.-B. Lamarck e i neolamarckiani avevano posto a base della loro interpretazione dell'evoluzione si è dimostrata fallace. Sono stati vani i molti tentativi, anche recenti, di farla rivivere (v. evoluzione: l'evoluzionismo nella cultura del XX secolo).
b) Il mendelismo
Nell'anno 1900 furono riscoperte le leggi che Gregorio Mendel aveva trovato e pubblicato fin dal 1866 e che era no rimaste completamente inosservate per più di trent'anni. Nei primi due decenni del sec. XX si dimostrò, dopo aspre polemiche, la validità universale delle leggi di Mendel in tutti i casi in cui la riproduzione avviene per via sessuale, e si ricondussero allo schema mendeliano anche i caratteri quantitativi per i quali sembrava doversi ammettere un altro modo di eredità, l'eredità mista o intermedia.
Le conseguenze più importanti del mendelismo (come fu chiamato l'insieme dei fenomeni interpretabili in base alle leggi di Mendel) sono due: in primo luogo, la dimostrazione che la struttura del ‛patrimonio ereditario', cioè della base fisica dell'eredità, è discreta, o micromeristica. Alle unità genetiche elementari, che devono essere particelle materiali capaci di riprodursi conservando le proprie caratteristiche, e di rimescolarsi ricombinandosi in vari modi, è stato dato il nome di ‛geni' (Johannsen, 1909). In secondo luogo, il fatto che i geni siano presenti a coppie (alleli) in ogni individuo che provenga dalla fecondazione di due gameti, e che uno dei due alleli possa dominare sull'altro, il quale rimane allo stato recessivo, spiega come alcuni caratteri possano rimanere latenti per più generazioni, e poi ricomparire in alcuni individui, quando si realizzi la condizione omozigote. Cade quindi la necessità di fare ricorso al concetto nebuloso dell'atavismo.
La concezione mendeliana ha dunque modificato profondamente il concetto tradizionale di eredità. Si è, fra l'altro, fatta una netta distinzione fra fenotipo e genotipo (termini introdotti da W. Johannsen nel 1909). Il primo è l'insieme dei caratteri di un organismo, quali appaiono ai nostri sensi; il secondo è la costituzione genetica, la quale, per effetto della recessività e di altri fenomeni che hanno effetto simile, non è necessariamente rappresentata fedelmente dal fenotipo. Un individuo fenotipicamente normale può portare nel proprio genotipo geni recessivi allo stato eterozigote, così che, dall'incrocio fra due eterozigoti possono nascere, in proporzioni prevedibili, individui omozigoti in cui si manifesta il carattere recessivo (v. genetica).
c) Carattere e norma di reazione
Nel periodo premendeliano era anche molto diffusa l'opinione che l'eredità fosse un fenomeno fatale e irrevocabile. Con il progresso delle indagini si dimostrò che ciò non è sempre vero. Il gene, per lo più, non determina necessariamente un dato carattere, o non ne determina la sua espressione più intensa. Esso stabilisce una ‛norma di reazione' ai fattori esterni, per cui l'espressione del carattere controllato da quel gene è il risultato di un'interazione del fattore genetico con i fattori ambientali che agiscono durante lo sviluppo individuale. Uno degli esempi più tipici per illustrare questo principio è dato da una pianta, Primula sinensis rubra, che, coltivata a temperatura ordinaria, forma fiori rossi, se invece è mantenuta in serra sopra i 30 °C dà fiori bianchi. La varietà affine Primula sinensis alba, invece, forma fiori bianchi a qualunque temperatura. Evidentemente il colore dei fiori nelle due varietà è controllato da geni diversi, che hanno una diversa norma di reazione. Analogamente, l'albinismo del coniglio di razza Imalaia è condizionato dalla temperatura: i peli che si formano in regioni del corpo più esposte al raffreddamento (estremità delle zampe, delle orecchie, del muso) sono di color bruno, anziché bianco. In base a questo principio si spiega come una costituzione genetica sfavorevole possa essere corretta con particolari interventi curativi (per es. somministrazione di ormoni e di vitamine, o di medicamenti). Inversamente una costituzione normale può essere profondamente modificata e resa anormale dall'azione di determinati fattori esterni: per esempio la somministrazione alla gestante di un farmaco, la talidomide, provoca una grave disfunzione dello sviluppo fetale che determina la formazione di un individuo focomelico; la rosolia contratta dalla madre durante la gestazione può indurre gravi malformazioni nel feto.
Queste alterazioni, sia che riconducano un individuo verso la norma, sia che consistano in anomalie più o meno gravi, non sono di per sé ereditarie. Il sistema genico rimane inalterato e come tale viene trasmesso ai discendenti: è soltanto la sua estrinsecazione fenotipica nei singoli individui che può venire modificata.
Comunque, la possibilità di modificare l'espressione dei geni consente molti interventi terapeutici atti a correggere condizioni ereditarie, o costituzionali, come si suol dire, il che ha una grande importanza nell'uomo e del resto era noto, più o meno vagamente, fin da tempi antichi.
L'eredità perde così il carattere di una fatalità ineluttabile; ma bisogna tener presente che non sempre è possibile l'intervento correttivo: vi sono molte condizioni genetiche di fronte alle quali la scienza è attualmente impotente, e che sono pertanto, almeno per ora, incurabili. Fra queste vanno ricordate numerose anomalie cromosomiche e in particolare il mongolismo.
d) Il coefficiente di ereditarietà
Molti caratteri sono controllati, anziché da una sola, da numerose coppie di geni, le cui azioni si sommano; ciò vale soprattutto per i caratteri quantitativi (per es. peso, statura, quantità della produzione di determinate sostanze, ecc.). Ciascun gene può avere una norma di reazione diversa, per cui in definitiva il carattere che c'interessa risulta determinato in parte geneticamente, in parte dalle condizioni ambientali. Si può enunciare schematicamente questo fatto, riferendosi a un insieme d'individui, indicando con G la variabilità genetica esistente in una popolazione, con E quella ambientale e con P la variabilità totale, rilevabile fenotipicamente, per cui si ha l'equazione:
G + E = P;
dividendo per P si ha:
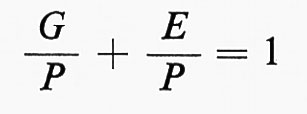
che si può esprimere con la formula
h2 + e2 = 1,
dove h2 è la variabilità genetica, cioè il coefficiente di ereditarietà (heritabllity), e2 la variabilità residua, cioè quella indotta dall'ambiente; questa si chiama anche il complemento dell'ereditarietà, ossia quanto di variabilità rimane quando si annulli la variabilità genetica.
Il calcolo del coefficiente di ereditarietà è importante nella genetica agraria e zootecnica, per stabilire quanto l'intensità di un carattere (per es. produzione del latte nei bovini o delle uova nei polli, resistenza all'allettamento nei cereali, ecc.) di una data popolazione d'individui dipenda dalla costituzione genetica e quanto dalle circostanze ambientali (qualità e quantità del cibo, tipo della concimazione, ecc.). Nella pratica del miglioramento delle piante e degli animali d'interesse economico è infatti assai utile fare una valutazione di questo tipo (ancorché non sia sempre facile ottenerla con precisione) per sapere quali risultati si possono ottenere con il miglioramento genetico (selezione) e quali col migliorare le condizioni di allevamento. La condizione ideale per determinare il valore del coefficiente di ereditarietà è l'annullamento della variabilità genetica, ottenendo una ‛linea pura' secondo la tecnica di W. Johannsen, a cui si è fatto cenno sopra. Negli animali si può procedere con una serie d'incroci consanguinei. Risultati soddisfacenti si possono ottenere anche riducendo al minimo la variabilità ambientale e operando la selezione.
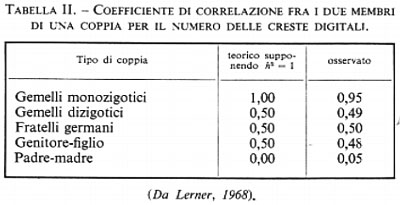
Anche nell'uomo sarebbe molto interessante poter conoscere il coefficiente di ereditarietà dei caratteri. In particolare, per i caratteri intellettuali e psicologici, il problema sconfina in quello del libero arbitrio e della determinazione: le circostanze ambientali, in questi casi, si possono riassumere con la parola educazione. La valutazione precisa è assai difficile. Uno dei mezzi più adeguati per raggiungere lo scopo è la comparazione delle coppie di gemelli monozigotici (MZ), che sono genotipicamente identici, con le coppie dizigotiche (DZ) in cui i due individui sono genotipicamente diversi (v. tabb. I e II).
È stata proposta da K. J. Holzinger la formula seguente:

dove r DZ è l'indice di correlazione, per un determinato carattere, fra le coppie dizigotiche e r MZ quello fra le coppie monozigotiche.
Il valore di h2 può variare fra 1 e 0. Nel primo caso il carattere è determinato interamente dalla costituzione genetica e l'ambiente non ha alcuna influenza; nel secondo caso è vero il contrario; nei casi intermedi, la funzione 1 − h2 dà una misura dell'importanza dell'azione ambientale.
e) Pleiotropia, polimeria, penetranza, espressività
Non esiste un rapporto univoco fra gene e carattere: un singolo gene non controlla, di solito, un solo carattere. E, viceversa, un carattere non è, generalmente, sotto il controllo di un solo gene. Si chiama ‛pleiotropia' il fatto che un gene faccia sentire la sua influenza su diverse strutture o funzioni. Per esempio il gene per l'albinismo nei topi non determina soltanto l'assenza di pigmento, e quindi la colorazione bianca, ma anche maggior mansuetudine, minor resistenza ad alcune malattie infettive, ecc.
Alcuni caratteri sono controllati principalmente da una coppia genica (major gene), ma oltre a questa possono entrare in gioco altri geni, che funzionano come modificatori o, nel caso estremo, come soppressori dell'azione del gene principale. In altri casi, come nell'esempio tipico del colore della pelle nell'uomo, sono implicate parecchie coppie di geni, che hanno azione additiva: quanti più alleli per la pigmentazione sono presenti nel genotipo di un individuo, tanto più intensa è la colorazione. Il riconoscimento di questo fenomeno, chiamato ‛polimeria' (o eredità da geni multipli o poligeni) è stato decisivo per dirimere la controversia fra i sostenitori dell'ipotesi dell'eredità mista o intermedia (blending inheritance) e i sostenitori dell'eredità alternativa, secondo lo schema mendeliano. Verso il 1920 la disputa si chiuse, perché si riconobbe che la cosiddetta eredità intermedia rientra nell'ambito dell'eredità mendeliana, come un caso di poliibridismo: numerose coppie di geni, con effetto simile e azione additiva, danno segregazioni molto più complesse di quella del monoibridismo (1:2:1) o del diibridismo (1:4:6:4:1), con frequenze relativamente molto alte dei genotipi aventi composizione intermedia fra quelle dei due omozigoti.
I geni manifestano una certa variabilità nel modo e nell'intensità della propria espressione, non soltanto in di- pendenza dalle condizioni esterne in cui avviene lo sviluppo di ciascun individuo, ma anche in funzione della presenza di altri geni che, come si è detto, possono funzionare da modificatori o inibitori dell'azione del gene principale. La ‛costellazione genica' in cui opera un singolo gene ha grande importanza per la sua manifestazione. La dominanza di un allele sul suo recessivo può essere più o meno completa, e la sua intensità modificabile da condizioni esterne o da costellazioni geniche diverse. Così, per esempio, il gene della calvizie precoce è dominante nei maschi (i quali sono calvi anche se sono eterozigoti, Aa), recessivo nelle femmine, le quali se eterozigoti (Aa) sono capellute, e soltanto quando siano omozigoti (aa) sono calve. Il colore bruno dei capelli è recessivo nell'età infantile e diviene dominante nell'adulto: qui, evidentemente, sono le condizioni fisiologiche varianti con l'età che determinano la dominanza di un gene sull'altro. L'età ha molta importanza nell'espressione dei geni: alcuni entrano in azione precocemente, altri, come quello della calvizie, più tardivamente, altri, come per esempio quello che determina la corea di Huntington, in età relativamente avanzata, tra i quaranta e i cinquant'anni. Nelle piante e in molti animali la temperatura a cui sono sottoposti durante lo sviluppo può far variare la dominanza di un allele rispetto a un altro.
Un carattere può quindi manifestarsi fenotipicamente in tutti gli individui che posseggono il relativo gene, oppure soltanto in una percentuale di essi. Questa diversa probabilità di manifestazione è stata chiamata ‛penetranza'. Se, invece, il carattere si manifesta in modo più o meno intenso nei vari individui, si parla di ‛espressività'. Sia l'una che l'altra proprietà si possono esprimere numericamente con una certa approssimazione. Ambedue hanno notevole importanza in genetica medica.
Un chiaro esempio di queste proprietà è dato dalla osteogenesi imperfetta nell'uomo. Si tratta di un gene dominante, pleiotropo, che determina tre caratteri principali: colore azzurro della sclera, fragilità delle ossa e sordità. La penetrazione è prossima al 100%, cioè tutti i portatori dell'allele dominante manifestano almeno uno di questi caratteri, ma l'espressività è molto variabile, nel senso che, nei vari individui, i caratteri possono manifestarsi con una certa indipendenza l'uno dall'altro e con intensità molto diversa.
Talvolta, soprattutto negli organismi più usati negli esperimenti di genetica, le condizioni ambientali possono produrre variazioni fenotipiche assai simili agli effetti di particolari geni. Tali variazioni, chiamate ‛fenocopie', evidentemente non sono ereditarie.
f) Eredità generale ed eredità speciale: i geni letali
Un pregiudizio un tempo assai diffuso riguarda il presunto differente apporto del padre e della madre alla costituzione ereditaria dei figli. Si riteneva che i caratteri ‛generali' dell'organizzazione (così, per es., per un cavallo l'essere vertebrato, quadrupede, mammifero, perissodattilo, equide) fossero trasmessi dalla madre, mentre il padre avrebbe contribuito con l'apporto di caratteri più ‛speciali' come, per esempio, la dimensione corporea, il colore e la pezzatura della pelliccia, l'attitudine alla corsa o al lavoro pesante, ecc. Anche in medicina era opinione diffusa che la madre avesse maggiore importanza del padre nel determinare la costituzione dei figli. Espressa in termini citologici, questa teoria equivale a dire che i caratteri cosiddetti ‛generali' sono trasmessi tramite il citoplasma, di cui la cellula uovo, a differenza dello spermatozoo, è ricca; mentre quelli ‛speciali' sarebbero trasmessi dal nucleo, che è presente in modo quantitativamente eguale sia nell'uovo, sia nello spermatozoo.
L'ipotesi di questo dualismo era basata sulla convinzione che i caratteri generali non obbedissero alle leggi di Mendel e perciò non fossero controllati da geni localizzati nei cromosomi. Ma la scoperta dei geni letali (L. Cuénot, 1905) ha dimostrato che non è così. Si tratta di geni recessivi che, quando siano in condizione omozigote, determinano la morte dell'individuo. Possono sussistere e venire trasmessi di generazione in generazione soltanto in condizione eterozigote, in quanto il loro effetto deleterio è annullato dalla dominanza dell'allele normale.
Si è constatato che i geni che così si comportano controllano strutture o processi fisiologici di fondamentale importanza per l'organismo: per esempio la formazione della clorofilla nelle piante, la formazione dello scheletro osseo o del sistema nervoso, o la ematopoiesi nei Vertebrati, ecc. Che obbediscano alle leggi di Mendel è provato dal fatto che, se si fa un incrocio fra due eterozigoti, gli individui malformati, che muoiono, sono nella proporzione di 1/4 del totale della figliolanza, come ci si attende in base alla legge della segregazione.
I geni letali (molti ne sono conosciuti nei microrganismi, nelle piante, negli animali, nell'uomo) forniscono quindi la prova che anche i caratteri ‛generali' dell'organizzazione sono sotto il controllo di geni localizzati nei cromosomi, così come i caratteri ‛speciali', e non è quindi necessario ricorrere all'ipotesi dell'eredità citoplasmatica. Inoltre essi danno informazioni sul processo embriologico o fisiologico che è controllato dall'allele normale. Se non si conoscesse la mutazione letale, e le malformazioni che essa determina in condizione omozigote, sarebbe impossibile, con il metodo genetico dell'incrocio, svelare la presenza del gene normale.
2. L'eredità nell'uomo
a) Introduzione
Le conoscenze relative all'eredità nell'uomo rimasero molto scarse e frammentarie fino alla seconda metà del sec. XIX. F. Galton si propose di studiare l'argomento con metodo scientifico e con impostazione quantitativa, come si è detto precedentemente, al fine di migliorare la costituzione genetica della specie umana; diede vita così alla disciplina che chiamò eugenica (v. eugenica). Nel 1869 pubblicò un libro dal titolo Hereditary genius, in cui raccolse dati sulle attività e il successo di molti membri di eminenti famiglie inglesi. Nel 1876 propose lo studio dei gemelli identici (monozigotici) in paragone con i non identici (dizigotici) per decidere sull'importanza relativa della costituzione genetica e dell'ambiente per la realizzazione di un carattere e introdusse l'espressione, tratta da una frase di Shakespeare, nature and nurture per indicare l'uno e l'altro complesso di fattori, espressione che fu poi largamente usata nella letteratura scientifica inglese. Sempre al Galton si deve l'introduzione del metodo delle impronte digitali per il riconoscimento dell'identità personale, con il libro Finger prints (1892). Successore di Galton alla direzione del laboratorio da lui fondato nel 1907 allo University College di Londra, e che dopo la sua morte (1911) fu denominato The Galton Laboratory, fu K. Pearson, a cui si deve principalmente lo sviluppo della scuola biometrica, che ha avuto tanta importanza nel fornire gli strumenti matematici necessari allo studio della genetica, e in particolare della genetica umana.
Dopo la riscoperta delle leggi di Mendel nel 1900, Galton e i biometrici non accettarono il mendelismo come unica base per l'interpretazione dei fenomeni ereditari, e diedero avvio alla polemica di cui si è fatto cenno (v. sopra, cap. 1, § a), che si estinse soltanto alla fine del secondo decennio di questo secolo.
Fra i primi a riconoscere che le leggi di Mendel si applicano anche all'uomo fu un medico inglese, A. Garrod, che pubblicò nel 1909 un libro oggi considerato classico: Inborn errors of metabolism. Egli osservò che alcune rare malattie come l'alcaptonuria, la cistinuria, la pentosuria e una anomalia della pigmentazione, l'albinismo, si comportano come caratteri recessivi: si manifestano in individui nati da genitori sani e, nonostante la loro rarità nella popolazione generale, compaiono spesso in parecchi membri di uno stesso ceppo familiare. Per l'alcaptonuria il Garrod enunciò (1902) l'ipotesi, per quel tempo molto ardita e innovatrice, che l'anomalia derivasse dalla mancanza di un enzima.
Gli sviluppi delle ricerche sull'eredità dei caratteri normali o patologici nell'uomo richiesero l'elaborazione di particolari metodologie per la raccolta dei dati e per la loro elaborazione statistica. L'uomo, infatti, mal si presta al tipo d'indagine che si può eseguire nelle piante e negli animali: l'impossibilità di fare incroci sperimentali, lo scarso numero della figliolanza di ciascuna coppia, la durata della vita, che è eguale nell'oggetto di studio e nell'osservatore, e la rarità di molti caratteri che si vogliono studiare, costituiscono altrettanti impedimenti, che si devono aggirare escogitando opportuni accorgimenti. In linea di massima si può dire che la genetica umana e la genetica medica (la quale si occupa più specificatamente dei caratteri patologici) si sono sviluppate e hanno raggiunto un alto grado di perfezionamento, soprattutto paragonando i risultati ottenuti per mezzo dell'indagine genealogica, o statistica, con i paradigmi ottenuti dalla sperimentazione sugli animali e sulle piante, e collaudandone la validità sui dati umani.
b) L'assetto cromosomico umano
Nel decennio 1910-1920 Th. H. Morgan e i suoi collaboratori dimostrarono che i geni sono localizzati nei cromosomi. La dimostrazione ebbe inizio dalla constatazione che il carattere ‛occhi bianchi' del moscerino Drosophila melanogaster si comporta in modo particolare perché il relativo gene è localizzato nel cromosoma X. Lo schema cosiddetto dell'eredità ‛legata al sesso' si dimostrò subito adatto a rendere ragione del particolare comportamento ereditario di alcune anomalie dell'uomo, quali l'emofilia e il daltonismo, che colpiscono i maschi, e sono trasmesse dalle femmine, nelle quali il carattere generalmente non si manifesta. Ciò costituiva una prova indiretta che, anche nell'uomo, i geni sono localizzati nei cromosomi. Tuttavia il numero dei cromosomi umani non era conosciuto con sicurezza. In base a ricerche eseguite con la tecnica usuale delle sezioni dei tessuti al microtomo, si erano raggiunti risultati discordanti. Per lo più era accolto nei trattati il numero 48 cromosomi, compresa la coppia dei cromosomi sessuali, XY nel maschio e XX nella femmina. Soltanto nel 1956 J. H. Tjio e A. Levan (v., 1956) dimostrarono che tale numero è sbagliato: i cromosomi sono 46, cioè 22 coppie di autosomi (eguali nei due sessi) più la coppia XX nella femmina e XY nel maschio. Tale risultato fu confermato da tutte le ricerche successive ed è ormai definitivamente acquisito. Gli accorgimenti tecnici che hanno permesso l'identificazione e, negli anni più recenti, lo studio della struttura dei singoli cromosomi dell'uomo (e di altri organismi con numero di cromosomi piuttosto elevato) sono i seguenti: preparati microscopici per schiacciamento anziché per sezioni microtomiche; uso della colchicina per bloccare le mitosi in metafase e far contrarre di molto i cromosomi; uso delle fitoemoagglutinine per agglutinare i globuli rossi (che essendo privi di nucleo, nei Mammiferi, non presentano interesse per questo tipo di osservazione) e per stimolare le mitosi; e, infine, l'introduzione delle tecniche per il ‛bandeggio' (banding), che consentono di riconoscere una struttura zonale caratteristica dei singoli cromosomi. Inoltre, l'uso dell'isotopo radioattivo tritio (3H) con cui si può marcare la timina, base pirimidinica che entra nella costituzione del DNA, consente di studiare il processo di replicazione dei cromosomi.
Nel 1960 si tenne a Denver, nel Colorado, una conferenza allo scopo di uniformare la numerazione e la nomenclatura dei cromosomi umani. Si stabilì di classificarli secondo la dimensione e la posizione del centromero (o punto di attacco al fuso della mitosi) che può essere mediana (cromosomi mediocentrici) o terminale (acrocentrici) o subtermibale o submediana. Si convenne di numerare gli autosomi da 1 a 22, cominciando dai più lunghi e procedendo verso i più corti e riservando i simboli tradizionali X e Y ai cromosomi sessuali. Si convenne inoltre di raggrupparli in sette gruppi sempre secondo le dimensioni: il gruppo A comprende le coppie 1, 2, 3; B, le coppie 4 e 5; C, le coppie 6-12 più il cromosoma X; D, 13, 14, 15; E, 16-18; F, 19 e 20; G, 21, 22 più l'Y.
Per la tecnica del ‛bandeggio', cioè per riconoscere le strutture zonali caratteristiche dei singoli cromosomi, T. O. Caspersson e collaboratori (1971) usarono alcuni derivati della quinacrina, che si legano specificamente ad alcune zone dei cromosomi e ne determinano la fluorescenza (bande Q). Altri mezzi escogitati in seguito (denaturazione con calore, acidi, alcali; colorante di Giemsa) consentono altri tipi di bandeggio. Anche la nomenclatura delle bande ha richiesto un lavoro di unificazione, che è stato concordato dalla Conferenza Internazionale di Parigi del 1971, supplemento 1975.
Si è quindi attualmente in grado d'identificare con sufficiente approssimazione i singoli cromosomi delle cellule umane, il che ha una grande importanza teorica e pratica, per i motivi che saranno chiari nei paragrafi seguenti (v. anche genetica: citogenetica).
c) Mutazioni del genoma e mutazioni cromosomiche
Poco dopo la determinazione esatta del cariotipo umano fu pubblicata un'importante scoperta dovuta a J. Lejeune, M. Gautier e R. Turpin (v., 1959). Era nota da molto tempo un'anomalia abbastanza frequente (circa 1,5 su mille nati) conosciuta con i nomi di sindrome di Langdon Down, o idiozia mongoloide, o mongolismo, della quale non si conosceva la causa né si sapeva con quale regola si trasmettesse ai discendenti. Lejeune e collaboratori dimostrarono che gli individui affetti da mongolismo hanno un corredo cromosomico anormale: 47 cromosomi, perché il cromosoma 21, anziché essere rappresentato da due elementi, come di norma, è presente tre volte; questa condizione è perciò chiamata trisomia 21. La presenza di un piccolo cromosoma soprannumerario determina numerose anomalie somatiche, caratteristiche del mongolismo, e deficienza mentale. L'anomala condizione cromosomica è dovuta al fenomeno noto col nome di ‛mancata disgiunzione' (non-disjunction) di quei cromosomi alla meiosi, da cui risultano gameti che invece di un solo 21 ne hanno due. Dall'unione di un gamete con due 21 e uno normale con un 21, risulta uno zigote con tre cromosomi 21. Le cause della mancata disgiunzione non sono conosciute. Si sa che la frequenza di nati mongoloidi cresce con l'aumentare dell'età della madre.
Dopo la scoperta di Lejeune furono trovate altre mutazioni del genoma, del tipo aneuploide (cioè variazioni numeriche di una coppia cromosomica). Per gli autosomi sono piuttosto limitate e rare: la trisomia 13 (sindrome di Pätau) produce gravi anomalie della regione cefalica, che portano a morte nei primi mesi dopo la nascita. La trisomia 18 (sindrome di Edwards) provoca anch'essa gravi malformazioni e morte entro il primo anno. Probabilmente altre mutazioni aneuploidi sono letali a stadi più precoci, e provocano l'aborto.
Molto importanti e relativamente frequenti (da 0,5 a 2 per mille) sono le anomalie della distribuzione dei cromosomi sessuali. Le più conosciute sono la presenza di due cromosomi X e un Y supplementare (XXY), che determina la sindrome di Klinefelter: fenotipo maschile, con eunucoidismo, spesso associato con debolezza mentale; e la presenza di un solo X (XO), che determina la sindrome di Turner: fenotipo femminile, con ovari poco sviluppati, amenorrea, statura bassa, intelligenza normale (v. sesso).
Si conoscono anche molti altri casi d'irregolarità dei cromosomi sessuali. Grande scalpore ha fatto l'osservazione di P. Jacobs e collaboratori (v., 1965) che la frequenza degli individui con genotipo XYY è più elevata nei manicomi criminali che non nella popolazione generale. Il cromosoma Y supplementare sarebbe responsabile di una statura più elevata e di una forte tendenza all'aggressività. Tale affermazione, secondo la quale gli individui con due Y avrebbero maggior disposizione alla violenza e alla criminalità, fu contestata in base ad altri dati raccolti dalla stessa Jacobs e da altri autori. Tuttavia la questione non è risolta e dati più recenti sembrano concordare con l'ipotesi primitiva (v. Hook, 1973).
Un altro tipo di mutazioni si riferisce, anziché al numero, alla struttura di singoli cromosomi: sono le mutazioni cromosomiche, di cui la più conosciuta nell'uomo è quella che determina la sindrome del cri-du-ehat (v. Lejeune e altri, 1963). Si tratta della mancanza (delezione) del braccio corto del cromosoma 5°, che ha come conseguenza grave deficienza mentale e varie malformazioni, che portano precocemente a morte il bambino.
La traslocazione consiste nel trasferimento di una parte di un cromosoma su di un altro appartenente a un'altra coppia. In alcuni mongoloidi che presentano apparentemente 46 cromosomi, anziché 47, si è trovato che il cromosoma 21 supplementare è traslocato, e si trova attaccato al cromosoma 14 o al 15. Quindi questi casi, anziché un'eccezione, risultano a conferma della trisomia 21 come causa del mongolismo. Oltre alle delezioni e alle traslocazioni, altri tipi di mutazioni cromosomiche (cromosomi ad anello, inversioni) sono stati riscontrati, sebbene raramente; ma è probabile che, con l'estendersi e il perfezionarsi delle ricerche, si scopriranno molti altri casi e sarà probabilmente possibile metterli in relazione con determinate sindromi patologiche.
Si è anche pensato alla possibilità che i tumori maligni fossero dovuti ad aberrazioni cromosomiche. Il solo caso finora chiaramente individuato è il cosiddetto cromosoma Philadelphia, indicato con il simbolo Ph1, che si trova con alta frequenza nelle cellule del midollo osseo di persone che soffrono di una rara forma di leucemia mieloide cronica. Si tratta della delezione del braccio lungo di uno dei due cromosomi della coppia 22.
d) Mutazioni geniche
La maggior parte delle differenze dei caratteri morfologici, fisiologici e psichici fra gli uomini è dovuta alla presenza di diversi alleli dei singoli geni e alle loro combinazioni, che avvengono secondo le regole scoperte con la sperimentazione su piante e animali. I diversi alleli si sono originati per mutazione. Si conoscono oggi numerosi caratteri che ‛mendelizzano' e perciò sono controllati da singoli geni: V. A. McKusick (1971) ne ha compilato un elenco di circa 1.800.
Ci limitiamo qui a ricordare i geni meglio conosciuti fino al livello della loro struttura molecolare, cioè i geni che controllano le varianti dell'emoglobina (v. sangue: emoglobina) e i geni che controllano alcuni processi metabolici. La linea d'indagine aperta dal Garrod nei primi anni del secolo fu perseguita sia dal punto di vista biochimico, sia dal punto di vista genetico. Furono riconosciuti i passi successivi di diverse ‛vie metaboliche', che conducono alla formazione di un prodotto terminale, e si riconobbe che le trasformazioni che si susseguono a partire da una sostanza iniziale (precursore) sono catalizzate da enzimi specifici. L'indagine genetica, d'altra parte, riconobbe che l'azione primaria del gene consiste nella produzione di una proteina che può avere funzione enzimatica. Una mutazione del gene, che ha come conseguenza la produzione di una proteina inattiva, determina un blocco metabolico, che non consente il progresso delle reazioni biochimiche che conducono al prodotto terminale. Il blocco può avvenire a vari livelli, a seconda del gene interessato, e possono così verificarsi stati patologici diversi. Una delle catene di reazioni meglio conosciute è quella relativa al metabolismo della fenilalanina, che può essere bloccata in diversi punti dalla mancanza degli enzimi specifici, determinando o fenilchetonuria, o tirosinosi, o alcaptonuria, o albinismo.
La base ereditaria dei gruppi sanguigni, cioè la presenza di particolari antigeni situati sulla membrana dei globuli rossi del sangue, che reagiscono con anticorpi presenti nel siero, ha avuto grande importanza nello sviluppo delle conoscenze di genetica umana (v. Race e Sanger, 1975a). Basterà ricordare la dimostrazione (F. Bernstein, 1923) che il sistema ABO è controllato da una serie di alleli multipli; la scoperta del sistema Rh (K. Landsteiner e A. S. Wiener, 1940) e l'elaborazione della sua base genetica a opera di R. Fisher, che riconobbe la presenza di tre geni distinti, strettamente associati; l'identificazione di numerosi altri sistemi di gruppi sanguigni, la cui base genetica è attualmente abbastanza ben conosciuta (v. sangue: genetica del sangue).
Di particolare interesse pratico è l'individuazione di antigeni che si trovano in cellule diverse dai globuli rossi. Quando si operi il trapianto di tessuti, o di organi, da una persona a un'altra, tali antigeni possono determinare la formazione di anticorpi da parte dell'ospite e dar luogo quindi al rigetto del trapianto. L'istocompatibilità e il suo contrario, l'istoincompatibilità, sono controllate da una serie di geni indicati con il simbolo HL-A. È evidente l'importanza di conoscere se il donatore e l'ospite posseggono geni che consentono la compatibilità del trapianto; in caso contrario esso viene rigettato (v. trapianti; v. immunologia e immunopatologia: malattie immunoproliferative).
Oltre ad avere i sistemi di antigeni e di anticorpi presenti nell'organismo e governati dai corrispondenti sistemi genetici, i Vertebrati, com'è noto, hanno la capacità di produrre anticorpi contro antigeni che provengono dall'esterno (Batteri o altri microrganismi, sostanze varie; v. immunologia e immunopatologia: immunologia generale). Le ricerche genetiche in questo campo dell'immunologia hanno portato all'identificazione di una serie di alleli Gm, che controllano la formazione di alcune immunoglobuline che hanno funzione di anticorpi.
Le ricerche in questi campi sono in fase di vivace sviluppo, dato il loro alto interesse sia teorico sia pratico, tanto da giustificare il nome d'immunogenetica che si è dato a questo capitolo della scienza dell'eredità.
e) Frequenza di mutazione nell'uomo
Le mutazioni geniche sono eventi che avvengono spontaneamente, cioè per cause ignote, con frequenza per lo più molto bassa. Con metodi di valutazione indiretti si è riusciti a stabilire la frequenza di mutazione di alcuni geni in alcune popolazioni umane. Dalle tabb. III e IV, in cui sono riportate alcune stime considerate come le più attendibili, si vede come la frequenza di mutazione varia da 2 a 100 per milione di gameti: la frequenza media è dell'ordine di 2 × 10-5
Tabella III
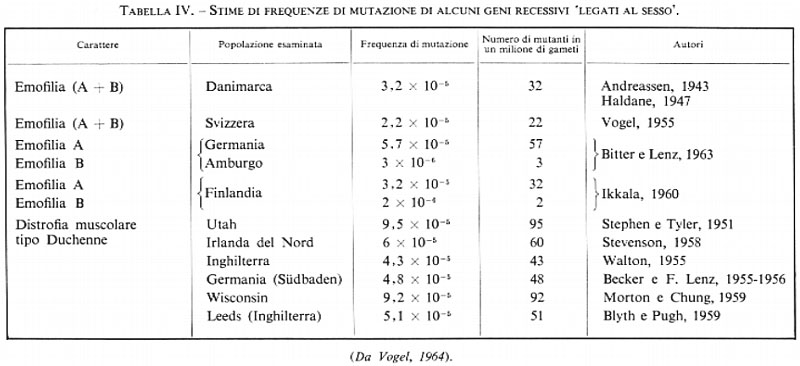
Come è noto dalle ricerche sugli animali, sulle piante, sui microrganismi, la frequenza di mutazione è accresciuta da parecchi agenti fisici e chimici (agenti mutageni). Fra questi i più efficaci e conosciuti da più lungo tempo (H. J. Muller, 1927) sono le radiazioni ionizzanti.
Ciò pone dei problemi per la protezione dell'uomo contro dosi di radioattività che possono produrre un eccessivo aumento delle mutazioni. Condizione questa che conviene evitare, perché la maggior parte delle mutazioni che compaiono sono sfavorevoli. Poiché molte persone sono soggette a un aumento della radioattività ambientale (lavoratori degli istituti di ricerca e delle aziende di produzione in cui si usano sostanze radioattive, pazienti che si espongono a radiazioni per scopo diagnostico o terapeutico) si è posto il problema della protezione dal danno genetico.
Commissioni internazionali hanno studiato questo argomento e hanno consigliato di non superare la dose totale che determina il raddoppiamento della frequenza di mutazione spontanea. Si è raccomandato, quindi, che non si superi, nei primi trent'anni di vita, la dose di 25-30 r a livello delle gonadi. Ma i calcoli precisi sono estremamente ardui e non è facile ottenere cifre attendibili; quindi conviene porre dei limiti cautelativi molto bassi (v. radioprotezione).
Nel 1940 C. Auerbach dimostrò che anche alcune sostanze chimiche hanno forte azione mutagena. La lista delle sostanze attive è andata aumentando enormemente negli anni successivi. La determinazione dell'attività mutagena dei composti chimici è anche più difficile che non per le radiazioni. Perciò anche in questo caso non è possibile indicare misure precauzionali precise; si sono costituiti gruppi internazionali per studiare i problemi della mutagenesi ambientale e stabilire le misure atte a proteggere i lavoratori delle industrie chimiche e le popolazioni in genere contro i danni genetici dovuti agli inquinamenti chimici.
f) Studi di genetica sulle cellule coltivate in vitro
La coltura dei tessuti in vitro, che fu introdotta nei primi anni di questo secolo, è stata largamente applicata per l'indagine di vari problemi biologici, quali fisiologia cellulare, differenziamento, ecc. Recentemente la coltivazione di cellule ha recato importanti contributi alla genetica. Molti difetti metabolici si manifestano anche nelle singole cellule in coltura, dovè si possono studiare molto più accuratamente che non nel paziente da cui le cellule provengono.
Questo metodo si è rivelato particolarmente utile nella diagnosi prenatale. Questa si può effettuare prelevando alcuni centimetri cubici di liquido amniotico dalla gestante per puntura trans-addominale (amniocentesi). Nel liquido sono sospese cellule di desquamazione degli epiteli del feto; da queste si possono ottenere, per coltura, numerose cellule di provenienza fetale sulle quali è possibile effettuare alcune ricerche diagnostiche. Oltre alla possibilità di mettere in luce anomalie cromosomiche, si possono effettuare indagini su determinati enzimi, la cui assenza indica che il nascituro è affetto da un disordine metabolico che può dar luogo a gravi, incurabili malattie, spesso letali, che si manifestano dopo la nascita. Attualmente si è in grado di diagnosticare prima della nascita circa una quarantina di malattie, tutte fortunatamente rare, ma per lo più gravi.
Con l'ausilio di particolari tecniche d'ibridazione interspecifica di cellule somatiche (per es., uomo-topo o uomo- ratto) a cui consegue, nel susseguirsi delle generazioni cellulari, l'eliminazione dei cromosomi di una delle due specie, e applicando metodi selettivi piuttosto complicati che non è possibile descrivere qui, si può arrivare a stabilire la localizzazione di alcuni geni in determinati cromosomi. Quindi le ‛mappe' genetiche, che negli organismi in cui è possibile la sperimentazione (per es. Drosophila, granoturco, topo) si ottengono in base a incroci da cui risulti il grado di associazione (linkage), si possono costruire, nell'uomo, utilizzando le colture di cellule somatiche. È prevedibile che, nel corso di alcuni anni, si possa disporre di una mappa abbastanza completa della distribuzione dei geni sulle 23 coppie dei cromosomi umani sulle quali, a tutt'oggi, sono stati localizzati poco più di un centinaio di geni.
g) Polimorfismi genetici in popolazioni umane
La specie umana, poco adatta alle ricerche di genetica formale per le ragioni esposte nell'introduzione, si è rivelata invece particolarmente favorevole per le indagini di genetica di popolazioni. Per quei caratteri di cui si conosce il determinismo genetico, è relativamente facile stabilire le frequenze dei vari alleli nelle diverse popolazioni. Per esempio, le frequenze dei fenotipi del sistema dei gruppi sanguigni ABO nelle varie popolazioni sono ben note, e da queste si può risalire alle frequenze dei tre alleli principali IA, IB, i. Questo è un caso a cui si applica la definizione di polimorfismo genetico, che consiste nella presenza di due o più alleli in una popolazione, in proporzioni tali che non si possa ammettere che la frequenza dell'allele più raro sia mantenuta da mutazione ricorrente.
Molti sono oggi i polimorfismi studiati nelle popolazioni umane: si riferiscono sia a geni i cui effetti sono riconoscibili con i metodi tradizionali, sia a geni la cui attività primaria (produzione di un enzima) può essere svelata per mezzo dell'analisi elettroforetica, con la quale si mettono in evidenza alcune differenze delle molecole proteiche.
Diverse cause mantengono in essere i polimorlismi: la più efficace è la selezione che, operando a vantaggio di uno dei tre genotipi possibili con una coppia di alleli (AA, Aa, aa) ne mantiene alta la frequenza. Un'ipotesi alternativa è quella dei ‛geni neutrali', cioè geni che determinano caratteri che non hanno alcun valore selettivo. In tal caso la diversa frequenza degli alleli sarebbe dovuta a variazioni casuali (deriva genetica).
Di molti polimorfismi riscontrabili nelle popolazioni umane non si è riusciti a riconoscere la causa delle diverse frequenze geniche. In particolare, per quanto riguarda il sistema ABO, non è stato possibile trovare una chiara dimostrazione di un diverso valore selettivo dei vari alleli. Ciò non significa che si debba escludere senz'altro che questo esista: può essere difficile scoprirlo.
In altri casi, invece, la situazione è molto più chiara. Sono conosciuti alcuni geni di cui un allele è molto frequente nelle popolazioni che sono state per lungo tempo soggette all'infezione malarica. Tale è il caso della talassemia (gene Th) in Italia, e della falcemia (gene S, Sickle cell) nelle popolazioni africane. In ambedue i casi un allele è molto più frequente nelle popolazioni ove la malaria era o è tuttora endemica, che non in quelle in cui la malattia è assente. L'allele in questione, in condizione omozigote, è letale: il gene th nella combinazione thth determina il morbo di Cooley, che porta a morte i bambini nei primi anni di vita; il gene s, subletale, in condizione omozigote determina un'alta mortalità infantile e giovanile. Gli eterozigoti, Thth e Ss rispettivamente, sono individui pressoché normali; ma i loro globuli rossi presentano condizioni poco favorevoli allo sviluppo del Plasmodium falciparum, agente patogeno della terzana maligna. Questi individui sono quindi protetti rispetto all'infezione malarica, la quale miete molte più vittime, nell'età infantile, fra gli individui normali (ThTh o SS). In questo caso è evidente che un agente esterno, l'infezione malarica, opera una selezione a favore degli eterozigoti, e perciò la frequenza dell'allele th o s è mantenuta alta, a dispetto del fatto che esso viene costantemente eliminato dalla popolazione per la nascita degli omozigoti letali, o subletali, che costituiscono 1/4 della prole che discende dal matrimonio di due eterozigoti. Il polimorfismo, e in particolare l'alta frequenza di un gene sfavorevole, in questo caso è mantenuto dal fatto che gli eterozigoti sono avvantaggiati rispetto ad ambedue gli omozigoti. Anche nelle piante e negli animali il vantaggio dell'ibrido, o eterosi, è una delle cause più efficaci del mantenimento dei polimorfismi (v. sangue: anemie emolitiche).
h) Consanguineità
I matrimoni fra persone che abbiano prossimi ascendenti in comune sono per lo più sconsigliati o, nel caso di consanguineità stretta, sono considerati illegali o peccaminosi da quasi tutti i popoli, anche primitivi (tabù dell'incesto). Dal punto di vista biologico la giustificazione delle norme che si trovano nelle legislazioni civili o religiose sta nel fatto che i matrimoni fra consanguinei rendono più probabile la formazione di discendenti omozigoti. Poiché molti alleli che si trovano come eterozigoti recessivi hanno, in condizione omozigote, effetti deleteri o letali, l'unione fra individui che appartengono allo stesso ceppo familiare, e che quindi abbiano alta probabilità di essere eterozigoti per le stesse coppie geniche, può dar luogo a figli che presentino caratteri sfavorevoli con probabilità maggiore che non l'unione fra individui non imparentati.
Un certo grado di consanguineità esiste in tutte le popolazioni umane: basti pensare che il numero teorico di antenati che ciascuno di noi dovrebbe avere avuto nell'anno 1000, dal quale ci separano circa 32 o 33 generazioni, è 232, cioè una cifra che supera i quattro miliardi, mentre tutta la popolazione della terra a quel tempo era ben lontana dal raggiungere tale numero.
Gli effetti deleteri della consanguineità si fanno sentire soprattutto quando avvengono matrimoni fra parenti stretti o nei piccoli centri isolati (valli alpine, piccole isole, ecc.) in cui i matrimoni fra parenti più o meno stretti si sono susseguiti per numerose generazioni, con nullo o scarso apporto di geni provenienti da altre popolazioni.
Si deve tuttavia tener presente che i matrimoni consanguinei non comportano necessariamente una figliolanza difettosa: la prole di molti matrimoni fra parenti può essere normale, o anche particolarmente ben dotata. In alcune dinastie dei faraoni dell'antico Egitto si praticava addirittura il matrimonio fra fratello e sorella, per mantenere la purezza della stirpe reale; ma non si hanno dati sulla frequenza di anomalie fra i figli. Comunque, la nascita di figli difettosi è valutabile in termini di probabilità, di rischio.
In base alle formule mendeliane si può calcolare la probabilità che, in un dato tipo d'incrocio, i due alleli di un locus abbiano origine comune e siano perciò identici. Nei figli di cugini in primo grado tale probabilità è 1/16.
Il problema degli effetti della consanguineità si è posto anche per l'allevamento degli animali e la coltivazione delle piante. La pratica dello inbreeding (inincrocio) per ottenere razze pure si è dimostrata pericolosa, se continuata per parecchie generazioni, perché dà origine a molte nascite d'individui anormali, oppure sterili. S. Wright ha sviluppato la teoria matematica per il calcolo del ‛coefficiente di consanguineità' nei vari tipi di incrocio.
Varie ricerche sono state eseguite, sia sulla frequenza dei matrimoni fra consanguinei nelle diverse popolazioni (per l'Italia, v. Moroni, 1964 e 1967), sia sulla quantità di effetti deleteri realmente constatabili. L'indagine più completa, in questo campo, è quella di W. J. Shull e J. V. Neel (1965) sui bambini giapponesi di Hiroshima.
i) Conclusioni
La conoscenza dei fenomeni ereditari nell'uomo è lungi dall'essere completa; tuttavia i progressi compiuti nel corso del sec. XX hanno consentito di raggiungere alcune mete molto importanti; ma hanno anche aperto problemi pratici di grande momento e non tutti di facile soluzione.
Un primo punto che occorre chiarire è il seguente: la dimostrazione che gli uomini non sono biologicamente tutti eguali, anzi sono tutti diversi l'uno dall'altro (a eccezione dei gemelli monozigotici), non deve trarre in inganno e indurre a negare il principio di eguaglianza degli uomini. Come ha bene esposto Th. Dobzhansky in diverse pubblicazioni, il concetto di eguaglianza è un concetto morale, sul quale sarebbe inutile insistere se tutti gli uomini fossero identici. Il fatto che non lo siano impone che ciascun membro della comunità sia posto egualmente in condizione di sviluppare la propria personalità, per contribuire alla vita della società.
Si è detto (v. sopra, cap. 1, § c) che l'eredità, nella maggior parte dei casi, non è una fatalità ineluttabile che incombe sull'individuo. In taluni casi però la struttura ereditaria determina anomalie fisiche e psichiche, che la scienza non e attualmente in grado di modificare. Donde la necessità di far sì che agli individui anormali siano assicurate buone condizioni di vita, evitando che essi possano recar danno agli altri. Ma s'impone anche il dovere di trovare il modo di curare le malattie ereditarie: la medicina moderna è impegnata in questo tipo d'indagine e molte sono le conquiste già realizzate in questo campo. E prevedibile che la lista delle possibili cure delle malattie ereditarie si accresca negli anni futuri. Ma è certo che non tutte le anomalie si potranno curare con i metodi della medicina tradizionale.
Questa considerazione apre due specie di problemi: il problema eugenico e il problema delle possibilità della cosiddetta ingegneria genetica. Del primo si tratta nell'articolo eugenica. Esso comporta delicati quesiti di ordine morale e giuridico: se sia lecito imporre di non generare a individui, a coppie che abbiano un'alta probabilità di mettere al mondo figli anormali. In generale prevale il criterio di fornire la necessaria consulenza per mezzo dei consultori genetici, tanto più che il rischio di avere discendenti difettosi si può soltanto esprimere in termini di probabilità, quasi mai come certezza. Questa si può invece raggiungere in molti casi con la diagnosi prenatale per mezzo dell'amniocentesi. E allora sorge il problema della liceità del procurato aborto quando il nascituro sia sicuramente deficiente o gravemente tarato. Questo non è che un aspetto del più generale problema dell'aborto, che si dibatte attualmente in molti paesi ed è diversamente risolto nelle varie legislazioni e nelle diverse dottrine religiose.
Comunque si vogliano giudicare queste situazioni, è certo che s'impongono alcuni compiti urgenti nelle ricerche sull'eredità dell'uomo: innanzitutto giungere a identificare, per un gran numero di geni, i portatori eterozigoti, in modo da accrescere il numero dei casi in cui si può dare una buona consulenza genetica; accrescere il novero delle anomalie diagnosticabili in fase prenatale, sia per poter consigliare l'aborto, ove questo sia consentito, sia per poter eventualmente intervenire tempestivamente con terapie adeguate, se possibile. È necessario intensificare le ricerche in questo senso, per trovare cioè metodi curativi atti a correggere l'azione di geni deleteri.
L'altro argomento che è stato dibattuto è se sia possibile sostituire in un organismo un gene ‛malato' con un gene ‛sano' e, qualora la sostituzione dei geni risultasse possibile, se questa manipolazione genetica, che può alterare profondamente la composizione della personalità umana, sia o non sia lecita. Allo stato attuale, si può soltanto affermare che un'operazione d'‛ingegneria genetica' di questo tipo appare come una possibilità molto vaga e remota. È vero che simili operazioni sono riuscite nei Batteri o in cellule in coltura, ma per ora non si vede come si possa, in pratica, arrivare a manipolare il patrimonio genetico dei gameti prima della fecondazione, o dell'uovo appena fecondato, o di un'intera linea cellulare in cui agisca il gene difettoso. Non si può certamente escludere che il progresso della scienza raggiunga questo scopo, che per ora appare piuttosto lontano; ma soltanto quando si saprà fino a che punto e in che condizioni una siffatta operazione è realizzabile, avrà un senso discutere se essa sia lecita e a quali mani debba essere affidata (v. genetica: applicazioni della genetica).
Bibliografia
Becker, P. E. (a cura di), Humangenetik: ein kurzes Handbuch, 5 voll., Stuttgart 1964-1972.
Bresch, C., Klassische und molekulare Genetik, Berlin 1964 (tr. it.: Genetica classica e molecolare, Firenze 1968).
Cavalli Sforza, L. L., Bodmer, W. F., The genetics of human populations, San Francisco 1974.
D'Amato, F., Genetica vegetale, Torino 1971.
Delage, Y., L'hérédité et les grands problèmes de la biologie générale, Paris 19032.
Gedda, L. (a cura di), De genetica medica, 7 voll., Roma 1961 ss.
Hadorn, E., Developmental genetics and letal factors, London 1961.
Hamerton, J. L., Human cytogenetics, vol. I, General cytogenetics, vol. II, Clinical cytogenetics, New York 1971.
Hogben, L., Nature and nurture, London 1945.
Hook, E. B., Behavioral implications of the human XYY genotype, in ‟Science", 1973, CLXXIX, pp. 139-150.
Jacobs, P. A., Brunton, M., Melville, M. M., Brittain, R. P., McClemont, W. F., Aggressive behaviour, mental subnormality and the XYY male, in ‟Nature", 1965, CCVIII, pp. 1351-1352.
Johannsen, W., Über Erblichkeit in Populationen und reinen Linien, Jena 1903.
Johannsen, W., Elemente der exakten Erblichkeitslehre, Jena 1909.
Lejeune, J., Gautier, M., Turpin, R., Étude des chromosomes somatiques de neuf enfants mongoliens, in ‟Comptes rendus hebdomadaires des séances de l'Académie des Sciences", 1959, CCXLVIII, p. 1721.
Lejeune, J., Lafourcade, J., Berger, R., Vialatte, J., Boesweillwald, M., Seringe, P., Turpin, R., Trois cas de délétion partielle du bras court du chromosome 5, in ‟Comptes rendus hebdomadaires des séances de l'Académie des Sciences", 1963, CCLVII, pp. 3098-3102.
Lerner, J. M., Heredity evolution and society, San Francisco 1968 (tr. it.: Eredità, evoluzione, società, Milano 1972).
Lush, J. L., Animal breeding plans, Ames, Iowa, 1960.
McKusick, V. A., Mendelian inheritance in man: catalogue of autosomal dominant, autosomal recessive and X-linked recessive, Baltimore 19713.
Montalenti, G., Introduzione alla genetica, Torino 1971.
Moroni, A., Evoluzione della frequenza dei matrimoni consanguinei in Italia negli ultimi cinquant'anni, in ‟Atti. Associazione Genetica Italiana", 1964, IX, pp. 207-223.
Moroni, A., Andamento della consanguineità nell'Italia settentrionale negli ultimi quattro secoli, in ‟Atti. Associazione Genetica Italiana", 1967, XII, pp. 202-222.
Pollack, R. (a cura di), Readings in mammalian cell culture, Cold Spring Harbor 1973.
Priest, J. H., Human cell culture in diagnosis of disease, Springfield, Ill., 1971.
Race, R. R., Sanger, R., Blood groups in man, Oxford 19756.
Sermonti, G., Genetica generale, Torino 1971.
Stern, K., Principles of human genetics, San Francisco 19733.
Strickberger, M. W., Genetics, New York 1968 (tr. it.: Trattato di genetica, Padova, 1974).
Sutton, H. E., An introduction to human genetics, New York 19752.
The National Foundation, Standardization in human cytogenetics. Birth defects, New York 1975.
Tjio, J. H., Levan, A., The chromosome number of man, in ‟Hereditas", 1956, XLII, pp. 1-6.
Turpin, R., Lejeune, J., Les chromosomes humains, Paris 1965.
Vogel, F., Lehrbuch der allgemeine Humangenetik, Berlin 1961.
Vogel, F., Mutations in man, in Genetics today. Proceedings of the XI international congress of genetics, 1963, New York 1964, pp. 833-850.
Vogel, F., Rohrborn, G., Chemical mutagenesis in Mammals and man, Berlino 1970.
Weismann, A., Das Keimplasma, eine Theorie der Vererbung, Jena 1892.