
GENETICA MEDICA
Genetica medica
(App. V, ii, p. 377)
Il progresso della g. m. è stato fortemente influenzato in questi anni dallo sviluppo delle tecniche di biologia molecolare (v. App. V e in questa Appendice). L'analisi sistematica delle malattie umane ha dimostrato che la maggior parte di esse ha una base o una larga componente genetica (malattie poligeniche e multifattoriali; Collins 1995). L'identificazione dei geni responsabili delle malattie ha un impatto importante sui programmi dedicati alla loro assistenza e al loro controllo: in primo luogo, sulla diagnosi dei pazienti e delle persone potenzialmente a rischio; in secondo luogo, sulla qualità del trattamento, che viene razionalizzato dalla conoscenza della funzione del prodotto genico mutato nella malattia; infine, e in una dimensione a più lungo termine, sulla terapia genica, che fonda i suoi presupposti sulla conoscenza dell'espressione del gene-malattia.
Il trattamento delle malattie genetiche e in particolare la terapia genica non sono per il momento ancora soddisfacenti, quanto meno per la maggior parte delle affezioni. Le strategie generali della terapia genica (v. App. V) fino a oggi hanno perseguito quattro principali obiettivi: aumentare la dose genica, attraverso l'introduzione di coppie aggiuntive del gene normale nelle cellule dei pazienti affetti da malattie causate dalla perdita della funzione di un gene, e nelle quali la patogenesi è reversibile; uccidere in maniera mirata specifiche popolazioni cellulari, in particolare nei processi neoplastici; correggere una mutazione, soprattutto quando produce un effetto dominante negativo; inibire in maniera mirata l'espressione genica (Verma, Somia 1997).
Consulenza genetica
Il controllo più efficace delle malattie ereditarie resta perciò ancora affidato alla prevenzione, che ha i suoi fondamenti nella diagnosi. Questo obiettivo viene idealmente raggiunto attraverso la consulenza genetica, un complesso servizio medico rivolto ai pazienti e alle famiglie nelle quali è presente una patologia che può essere o non essere ereditaria, per la quale si tenta di definire l'origine e l'eventuale ricorrenza familiare e nei confronti della quale vengono avviati interventi utili a prevenirla e a migliorare la qualità di vita dei pazienti (Harper 1993). In questo senso, la consulenza genetica affronta i temi della diagnosi e dell'origine di una malattia, compresa la definizione del suo rischio di ricorrenza (v. oltre), del trattamento e della prevenzione (v. genetica medica: Diagnosi prenatale, App. V).
Genetica molecolare
Il Progetto genoma umano (v. progetto genoma, App. V e in questa Appendice) e più in generale la ricerca genetica e molecolare applicata alle malattie hanno sviluppato efficaci strumenti diagnostici. A livello citogenetico, all'era del bandeggiamento, all'inizio degli anni Settanta, che ha reso possibile la classificazione di tutti i cromosomi e ha definito una nuova categoria di sindromi cromosomiche, è subentrata l'alta risoluzione cromosomica e la citogenetica molecolare. Le tecniche ad alta risoluzione hanno individuato nuove 'sindromi da geni contigui', che originano dallo sbilanciamento di piccole regioni di cromosoma (Dallapiccola et al. 1995). Le tecniche di citogenetica molecolare, basate sulla ibridazione di sonde fluorescenti su interi cromosomi o su specifiche regioni, rendono possibili diagnosi molto precise. Utilizzando sonde specifiche e software sofisticati è possibile diversificare le singole coppie cromosomiche, nel cosiddetto cariotipo spettrale (Speicher et al. 1996); possono essere identificati riarrangiamenti complessi non diagnosticati con gli altri protocolli (Delaroche et al. 1995); è possibile analizzare sequenze genomiche sui nuclei in interfase ed effettuare diagnosi retrospettive su materiale biologico archiviato per molti anni (Calabrese et al. 1996); è possibile analizzare e mappare fisicamente i geni (Pizzuti et al. 1996). La citogenetica molecolare avvicina perciò la risoluzione del cariotipo dei mammiferi a quella della Drosophila, dove la particolare struttura dei cromosomi politenici rende possibile la visualizzazione dei geni, direttamente al microscopio ottico.
La comprensione delle basi biologiche delle malattie ha ricevuto uno straordinario impulso dallo sviluppo di strategie che consentono con relativa facilità di localizzare sul cromosoma (mappare) e di isolare (clonare) i geni di interesse. In particolare, la tecnica del clonaggio posizionale ha accelerato questo processo; esso si basa sull'analisi delle famiglie in cui segrega una malattia ereditaria, che vengono analizzate per intere serie di marcatori di DNA polimorfi, cioè individualmente variabili, omogeneamente distribuiti lungo le braccia cromosomiche. La localizzazione sul cromosoma del gene-malattia viene suggerita e validata da appropriate analisi matematiche (lod scores), che dimostrano la cosegregazione di alcuni marcatori, a posizione di mappa nota, in tutti i pazienti. La regione critica viene successivamente ristretta, analizzando altri marcatori che mappano nella stessa regione. Quando l'area è sufficientemente ridotta, può essere utilizzato l'approccio dei 'geni candidati', che mappano nella stessa regione e che potenzialmente sono correlati con la malattia. La prova formale che un gene è il gene-malattia viene fornita dalla dimostrazione che è mutato nei pazienti.
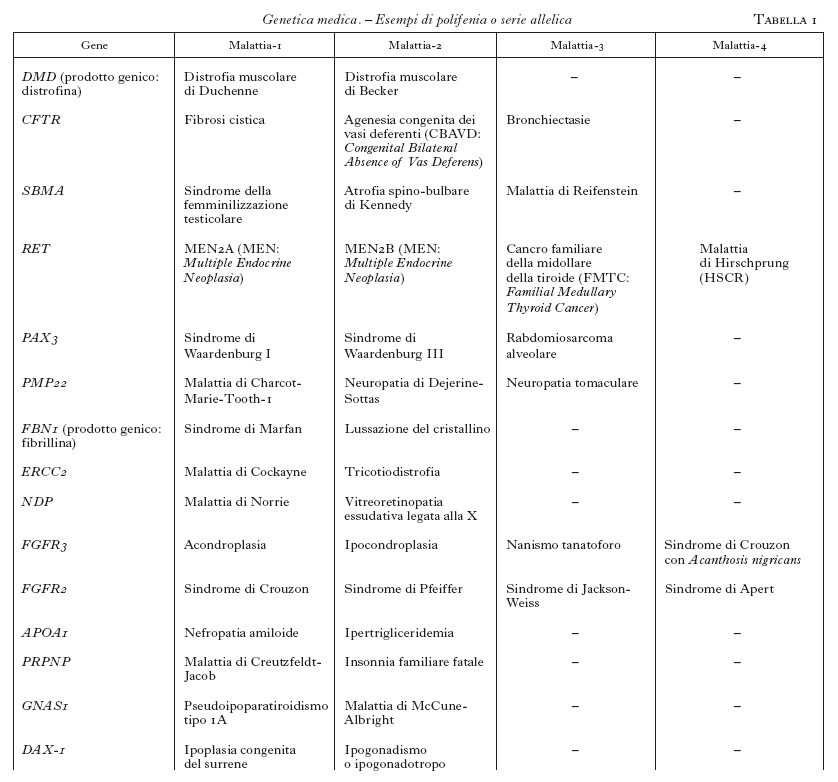
Questa tecnica, insieme ad altre, ha contribuito a mappare e a clonare svariate centinaia di geni correlati a malattie umane. L'analisi genetica ha rivelato una complessità biologica ben più sofisticata, rispetto a quanto era immaginabile. In questo contesto devono essere distinte le malattie mendeliane da quelle poligeniche e multifattoriali. Tuttavia, anche le malattie monogeniche presentano spesso aspetti di elevata complessità. Questo è, per es., il caso della cosiddetta polifenia o serie allelica. È esemplificativo il caso dell'acondroplasia (v. App. V), una osteocondrodisplasia con arti corti e macrocefalia, che è dovuta alla mutazione 1138 G→A nel gene che codifica per un recettore di un fattore di crescita dei fibroblasti (FGFR3; Shiang et al. 1994). Tenuto conto delle dimensioni relativamente grandi del gene, è senza dubbio sorprendente che tutti i pazienti presentino la stessa mutazione e che tutti i casi de novo abbiano un'origine paterna. Di notevole interesse è poi l'osservazione dell'elevata variabilità clinica associata alle altre mutazioni di FGFR3 che, oltre all'acondroplasia, causano l'ipocondroplasia, il nanismo tanatoforo, la malattia di Muenke, un tipo di sindrome di Pfeiffer, la sindrome di Crouzon con Acanthosis nigricans, alcuni tipi di craniostenosi isolata (Wilkie 1997). La polifenia indica dunque che lo stesso gene, mutato in domini funzionali diversi, può causare quadri clinici distinti. Questo fenomeno si applica attualmente a circa 200 geni-malattia (tab. 1). La serie allelica riconduce pertanto a uno stesso gene la patogenesi di malattie nosologicamente distinte, e in questo processo di accorpamento migliora la comprensione delle correlazioni tra il genotipo e il fenotipo.
Un fenomeno opposto e relativamente comune è quello dell'eterogeneità genetica, che definisce la presenza di mutazioni in loci diversi, associate a fenotipi simili. È illustrativo l'esempio della retinite pigmentosa (RP), una malattia degenerativa della retina che porta progressivamente alla cecità. Sono oggi noti oltre 15 geni-malattia, che causano forme isolate della malattia e altrettanti geni che causano forme sindromiche (Sullivan, Daiger 1996). L'eterogeneità genetica è espressione della ridondanza biologica. Proteine come la rodopsina, la periferina, la trasducina, la fosfodiesterasi sono espresse sulla membrana cellulare dei coni della retina e partecipano alla visione. Le modificazioni di questi prodotti, a seguito di mutazione nei geni che li codificano, causano un'alterazione in questo processo e la precipitazione di pigmento sul fondo della retina. Se a questa eterogeneità di locus si aggiunge l'eterogeneità allelica, cioè la variabilità delle mutazioni nei singoli geni-malattia, si comprende la difficoltà nella definizione del difetto molecolare della malattia, soprattutto quando è sporadica o è presente in una famiglia di ridotte dimensioni.
L'eterogeneità genetica suggerisce l'esistenza di rapporti funzionali indiretti tra i geni che entrano in una specifica via metabolica. In altri casi l'interazione tra gli alleli e tra i geni è maggiormente diretta. Questo è il caso dell'effetto di un determinato polimorfismo su una mutazione allelica. Per es., la mutazione ASN178 nel gene dei prioni (v. prione, App. V) in combinazione con il polimorfismo MET129, si associa all'insonnia fatale familiare, mentre in associazione al polimorfismo VAL129 causa la malattia di Creutzfeldt-Jakob (Goldfarb et al. 1992). Questa e altre simili osservazioni rivalutano l'importanza di alcuni polimorfismi sull'espressione fenotipica. In altri casi un fenotipo ritenuto mendeliano ha di fatto un'eredità digenica. Per es., la sindrome di Waardenburg (WS) di tipo 2, associata all'albinismo oculo-cutaneo ii, origina da una doppia mutazione, nel gene MIFT, che causa la WS, e in un gene che codifica per una tirosinasi, della quale il MIFT è un fattore di trascrizione (Morell et al. 1997). Ancora più complesse sono le interazioni nelle 'sindromi da geni contigui' e nelle malattie multifattoriali. La delezione della regione cromosomica 22q11.2 esemplifica un modello di sindrome da geni contigui. L'emizigosi di una regione di 300-2000 kb si associa a quadri clinici variabili, che comprendono tra l'altro la sindrome di DiGeorge, la sindrome velo-cardio-facciale, la sindrome di Cayler, la malattia di Opitz autosomica dominante, cardiopatie troncoconali isolate e un complesso di oltre 180 dismorfismi e malformazioni. Benché non sia ancora del tutto chiaro il meccanismo di origine di questi fenotipi e neppure l'elevata variabilità intrafamiliare, è comunque evidente che essi sono correlati all'emizigosi o a un'alterazione nella regolazione di geni fisicamente vicini nella regione critica (Dallapiccola et al. 1996): nel caso delle malattie multifattoriali il fenotipo è causato dall'interazione tra geni localizzati su cromosomi diversi, che interagiscono con un ambiente 'sfavorevole'. Sono esemplificativi gli oltre 20 geni della suscettibilità al diabete mellito di tipo 1, o i numerosi polimorfismi nei geni che conferiscono circa il 50% della suscettibilità ad ammalare di ictus o di infarto giovanile: PAI (Plasminogen Activator Inhibitor), ACE (Angiotensin Converting Enzyme), fibrinogeno, omocistinuria, fattore VII della coagulazione ecc. (Todd, Farrall 1996; D'Addedda et al. 1997).
Sebbene l'analisi molecolare renda possibile decifrare molte malattie a livello delle loro basi biologiche, il rapporto che intercorre tra il progresso scientifico e il suo impatto a livello clinico non è spesso diretto e immediato. La neurofibromatosi di tipo 1 (NF1) esemplifica una malattia comune, a elevata variabilità fenotipica, della quale è stato clonato da tempo il gene (nf1). Si tratta di un gene a organizzazione complessa, in quanto contiene al suo interno altri geni, e che si estende per una regione genomica di circa 350 kb. Mentre nei casi familiari (almeno due soggetti affetti) è possibile seguire la segregazione del cromosoma mutato, utilizzando polimorfismi intragenici, nei casi sporadici le grandi dimensioni del gene e la variabilità delle mutazioni rendono poco probabile la possibilità di caratterizzare la mutazione (Sheng et al. 1996). Questo si riflette, in pratica, nell'impossibilità di monitorizzare con la diagnosi prenatale una parte delle gravidanze a rischio.
In altri casi la mappatura e il clonaggio di un gene-malattia può incidere significativamente sulla malattia. Per es., i pazienti affetti da rene policistico tipo adulto sviluppano cisti renali in media nel 22% dei casi già all'età di 10 anni, nel 68% dei casi a 20 anni, nell'86% a 30 anni; a 40 anni presentano i primi sintomi di insufficienza renale, a 50 anni necessitano di trattamento dialitico e attorno ai 55 anni muoiono, a meno che non vengano sottoposti al trapianto renale. Le analisi molecolari consentono in tutte le famiglie di identificare le persone a rischio in epoca presintomatica e addirittura nella vita fetale. La diagnosi precoce non è ancora in grado di cambiare radicalmente la vita dei pazienti, ma può incidere sulla storia naturale della malattia, per es. ricorrendo a restrizioni dietetiche, e, in maniera più sostanziale, sulla pianificazione familiare.
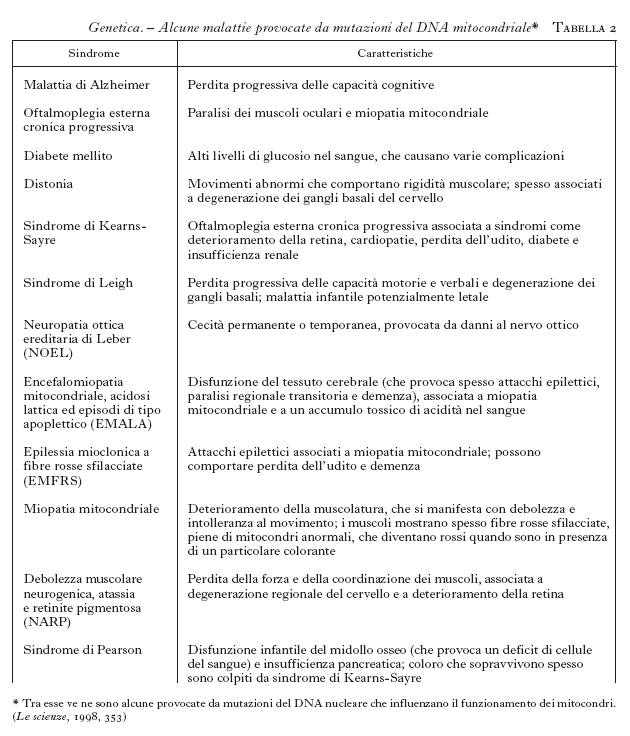
La possibilità di intervenire predittivamente sulle malattie costituisce uno degli aspetti più rivoluzionari della moderna g. m., che ha la sua espressione più conclamata nelle cosiddette malattie da mutazioni dinamiche (tab. 2). Si tratta di affezioni, prevalentemente di interesse neurologico, nelle quali la malattia origina dall'espansione di una sequenza di tre lettere (triplette) normalmente presenti in un basso numero di ripetizioni nel gene selvatico. È al riguardo esemplificativo il caso della distrofia miotonica (DM) nella quale la tripletta CTG, normalmente presente in un numero discreto di copie (〈50), si localizza al 3´ del gene della miotonina proteinchinasi (MT-PK), che è correlato alla malattia. La notevole instabilità nella trasmissione nelle famiglie a rischio comporta la progressiva espansione della sequenza dai genitori ai figli. È dimostrata una correlazione tra le dimensioni dell'espansione, l'età di esordio e l'espressività della malattia (Novelli et al. 1993; Ashizawa et al. 1994; Mastrogiacomo et al. 1994; Melacini et al. 1995), che spiegano la sua anticipazione, cioè la comparsa in età sempre più precoce con il passare delle generazioni. In conseguenza di questo meccanismo, una malattia tipicamente dell'adulto può presentarsi anche in forma congenita nella stessa famiglia. Queste evidenze hanno reso possibile lo sviluppo di un test predittivo per la DM, che, applicato in epoca prenatale o in un paziente non ancora sintomatico, consente di stabilire con buona approssimazione, in base all'espansione della sequenza instabile, l'età di esordio della malattia e la gravità dei sintomi clinici (Gennarelli et al. 1996).
Le mutazioni dinamiche nelle quali la tripletta espansa riguarda sequenze di glutammina (CAG) hanno nella corea o malattia di Huntington la condizione più rappresentativa. Questa malattia illustra almeno due problemi principali relativi all'impatto della biologia molecolare sulla diagnosi. Il primo riguarda il test predittivo e in particolare l'analisi del DNA che viene effettuata sui familiari a rischio, quando sono asintomatici. È stato dimostrato che i livelli di ansia e di depressione nei consanguinei dei pazienti si riducono significativamente già a un anno di distanza dalla comunicazione di un risultato negativo del test (assenza del rischio). D'altra parte, il risultato positivo non modifica significativamente i preesistenti livelli di ansia e di depressione. Inoltre, la comunicazione del risultato, in entrambi i casi, sembra avere un forte impatto sulle decisioni riproduttive (Decruyenaere et al. 1996). I test psicometrici che si effettuano prima dell'indagine possono perciò predire le reazioni che si verificheranno successivamente alla comunicazione dei risultati dell'analisi molecolare. Questa osservazione raccomanda quindi una stretta integrazione tra il genetista e lo psicologo medico, soprattutto quando il test viene applicato alle patologie a esordio tardivo, che incidono profondamente sulla qualità e sulle attese di vita. Una seconda considerazione, suggerita dall'analisi del gene della corea di Huntington, è che la differenziazione tra la 'normalità' e la 'malattia' non è sempre facile a livello molecolare. Uno studio collaborativo, che ha riguardato 178 campioni di persone con espansioni comprese tra 30 e 40 triplette CAG, ha dimostrato che l'amplificazione tra le 36 e le 39 triplette si può associare sia alla malattia, sia a un fenotipo non-affetto (Rubinsztein et al. 1996). Questa osservazione indica che la mutazione non è sempre penetrante e suggerisce cautela nell'interpretazione dei risultati delle indagini molecolari.
Test e screening genetici
Complessivamente i dati sopra riportati, se da un lato documentano l'importante impatto della biologia molecolare sulla diagnosi delle malattie umane, dall'altro raccomandano prudenza e umiltà nell'interpretazione dei risultati. Questo concetto è stato esemplarmente sintetizzato da R. Hubbard e R.C. Lewontin (1996) i quali hanno scritto che "l'aumento delle conoscenze sulle sequenze di DNA nei geni sta trasformando i concetti di gene selvatico o normale e di gene mutato. I rapporti tra queste sequenze nucleotidiche e le rispettive espressioni cliniche possono essere complessi e non prevedibili, neppure in caso di eredità mendeliana". Proprio in considerazione di questi limiti è necessario che le tecniche molecolari entrino nel bagaglio della medicina moderna, ma non prendano il sopravvento sulla clinica. Questo rischio è potenziale, in considerazione della tipologia dei test genetici, che li classifica come:
diagnostici, che pongono o confermano la diagnosi di una malattia genetica (per es., distrofia muscolare di Duchenne);
predittivi, che identificano un fattore di rischio per lo sviluppo di una malattia, quando non venga controllata la predisposizione (per es., fenilchetonuria ed effetto della dieta a basso contenuto di fenilalanina);
prognostici, che prevedono l'insorgenza futura di una malattia genetica (per es., corea o malattia di Huntington);
che conferiscono suscettibilità, ovvero stabiliscono la vulnerabilità a un fattore ambientale (per es., deficit di α-1- antitripsina ed enfisema polmonare);
che conferiscono resistenza, che cioè proteggono dallo sviluppo di certe malattie, in presenza di un gene mutato (per es., Par1, Par2, Par3 proteggono dagli effetti cancerogeni di Pst1 nel polmone);
probabilistici, che individuano un genotipo a rischio con una probabilità significativamente maggiore rispetto a quella della popolazione (per es., BRCA1 e carcinoma della mammella);
di profilo genetico, che identificano un'associazione empirica tra una mutazione e l'aumento dell'incidenza della malattia (per es., delezioni del gene ACE e rischio di infarto cardiaco).
Queste ampie possibilità diagnostiche e la documentata presenza di mutazioni nel genoma di ogni persona, nella quale queste ultime costituiscono potenziali fattori di malattia o di suscettibilità, propongono l'avvio di screening genetici finalizzati all'identificazione delle persone a rischio. I programmi di questo tipo, che si rivolgono a varie categorie di utenti, come i neonati, gli adulti o la popolazione che si riproduce, possono in teoria avere importanti ripercussioni sulla società, sulla frequenza delle malattie, sui singoli utenti e sulla consulenza genetica. È comunque implicito che il loro avvio è subordinato alla preventiva sensibilizzazione della popolazione oggetto dello screening. È illustrativa al riguardo l'esperienza effettuata sui consanguinei di primo grado dei pazienti affetti da tumori genetici, che hanno espresso un elevato interesse nei confronti degli screening molecolari di queste malattie (Lerman et al. 1996). Al contrario, a fronte di un potenziale interesse manifestato dai familiari dei pazienti con corea di Huntington prima che i test molecolari divenissero disponibili, si è verificato un significativo calo di attenzione quando l'analisi ha potuto essere attuata (Craufurd et al. 1989; Bloch, Hayden 1990). Questo contrastante atteggiamento è probabilmente dovuto alla diversa efficacia degli screening sulla prevenzione dei tumori, rispetto a una malattia degenerativa irreversibile del sistema nervoso, e sottolinea comunque che la messa a punto di questi programmi non deve ignorare la sensibilità e le reazioni future della popolazione che sarà sottoposta alle indagini.
D'altra parte gli screening sono soggetti anche a condizionamenti di tipo operativo e tecnico. Negli USA è stato calcolato che il 15% delle coppie in età fertile è interessato allo screening della mutazione associata al ritardo mentale legato all'X, che interessa circa una ogni 350 coppie. Questo significa una domanda teorica di 600.000 test di questo tipo ogni anno. Ammettendo che anche solo la metà del campione si sottoponga di fatto allo screening, è calcolato un impegno di circa 170 ore per anno per ognuno dei 600 genetisti medici in attività, da dedicare all'informazione delle coppie sul significato dell'analisi e alla comunicazione dei risultati del test. Questo impegno, considerato nel contesto dell'altra attività istituzionalmente affidata ai genetisti medici, rende questo screening impraticabile (Laxova 1995).
I test genetici che si rivolgono ad ampie fasce di popolazione devono anche tenere conto degli aspetti tecnici, dell'efficienza, dell'accuratezza, dei costi e delle priorità. L'aspetto tecnico appare quello più facilmente risolvibile, come sembra suggerire l'immissione sul mercato dei cosiddetti MASDA (Multiple Allele-Specific Diagnostic Assay), che consentono di analizzare contemporaneamente i campioni di 500 persone per 106 mutazioni in 7 geni-malattia. La tecnica dei DNA-chip è in grado di fissare 16.000 nucleotidi su un'area di 1,5 cm². Questo numero è all'incirca 25 volte inferiore rispetto alle potenzialità del sistema previste nell'immediato futuro (Abbott 1996). Appositi lettori ottici interpretano i risultati dell'analisi, che può riguardare interi tratti di genoma ed essere applicata a bassi costi ad ampie fasce di popolazione.
È perciò necessario prepararsi a questa fase di transizione che precederà l'impatto della biologia molecolare sulla medicina di massa attorno all'anno 2000. Secondo Jonsen et al. (1996), è prevedibile che i canonici principi della medicina ippocratica saranno investiti da una rivoluzione prognostica, nella quale entreranno pesantemente i geni della suscettibilità. La prognosi, fondata su criteri probabilistici, farà sempre maggiore riferimento al rischio congenito, e la consulenza genetica, piuttosto che utilizzare i rischi teorici ed empirici, formulerà rischi integrati con le informazioni fornite dai test genetici. In questo scenario, il medico sposterà il suo tradizionale interesse, dalla prevenzione e dalla rimozione della malattia nel singolo paziente, verso la medicina delle comunità, all'interno della quale la ricerca dei geni della suscettibilità finirà per creare una nuova classe di invalidi, quella dei non-pazienti. I rischi individuali evidenziati dai test genetici finiranno per creare problemi psicologici e malattie psicosomatiche, in antitesi con l'ippocratico primum non nocere.
Sul piano sociale, i test genetici troveranno, oltre al paziente, altri fruitori, per es. le compagnie di assicurazione, le compagnie industriali, i governi. Questo creerà problemi alla riservatezza dei risultati delle analisi, ma accenderà anche il dibattito sui doveri della persona identificata a rischio, per es. nei confronti dei suoi familiari. Un rischio potenziale è quello di creare tribù genetiche, formate da persone che condividono gli stessi marcatori, richiedono particolari programmi di sorveglianza, e che potrebbero venire diversamente discriminate. Si creerà il bisogno di una nuova legislazione per le popolazioni divise e ridistribuite in base alle caratteristiche genetiche, e dovranno essere emanate leggi che regolamentino l'uso dei test genetici da parte di terzi (Masood 1996).
In tale contesto la figura del medico, quanto meno di quello di famiglia, rischia di essere ridimensionata e forse destinata progressivamente a scomparire. Alla sanità pubblica spetta il compito di predisporre questa transizione che sarà forse traumatica, perché la sua rapidità non potrà essere controllata, in quanto strettamente scandita dal metronomo del progresso scientifico. L'evoluzione del modo di effettuare la diagnosi e la prevenzione delle malattie, che forse la medicina subirà con un certo fatalismo, dovrà comunque salvaguardare il principio dell'autonomia, quella che i genetisti identificano nella non-direttività della consulenza genetica, cioè la garanzia del rispetto, nel progresso, delle libertà individuali.
bibliografia
D. Craufurd, A. Dodge, L. Kerzin-Storrar et al., Uptake of presymptomatic predictive testing for Huntington's disease, in The Lancet, 1989, 2, pp. 603-05.
M. Bloch, M.R. Hayden, Opinion: Predictive testing for Huntington disease in childhood: challenges and implications, in American journal of human genetics, 1990, 46, pp. 1-4.
Y.H. Fu, P.D. Kuhl, A. Pizzuti et al., Variation of CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox, in Cell, 1991, 67, pp. 1047-58.
L.G. Goldfarb, R.B. Petersen, M. Tabaton et al., Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism, in Science, 1992, 258, pp. 806-08.
P.S. Harper, Practical genetic counselling, Oxford 1993.
G. Novelli, M. Gennarelli, E. Menegazzo et al., (CTG)in triplet mutation and phenotype manifestations in myotonic dystrophy patients, in Biochemical medicine and metabolic biology, 1993, 50, pp. 85-92.
T. Ashizawa, M. Anvret, M. Baiget et al., Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy, in American journal of human genetics, 1994, 54, pp. 414-23.
I. Mastrogiacomo, E. Pagani, G. Novelli et al., Male hypogonadism in myotonic dystrophy in relation to (CTG) in triplet mutation, in Journal of endocrinological investigation, 1994, pp. 381-83.
G. Romeo, V.A. McKusik, Phenotypic diversity, allelic series and modifier genes, in Nature genetics, 1994, 7, pp. 451-52.
R. Shiang, L.M. Thompson, Y.Z. Zhu et al., Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia, in Cell, 1994, 78, pp. 335-42.
F.S. Collins, Positional cloning moves from perditional to traditional, in Nature genetics, 1995, 9, pp. 347-50.
R. Dallapiccola, R. Mingarelli, G. Novelli, The link between cytogenetics and mendelism, in Biomedicine and pharmacotherapy, 1995, 49, pp. 83-93.
I. Delaroche, M. Sabani, G. Calabrese et al., Fetal translocation between chromosomes 2, 18 and 21 resolved by FISH, in Prenatal diagnosis, 1995, pp. 278-81.
R. Laxova, Fragile X screening: what is the real issue?, in American journal of medical genetics, 1995, 57, pp. 508-09.
P. Melacini, G. Villanova, E. Menegazzo et al., Correlation between cardiac involvement and CTG trinucleotide repeat lenght in myotonic dystrophy, in Journal of American college of cardiologists, 1995, pp. 239-45.
A. Abbott, DNA chips intensify the sequence search, in Nature, 1996, 379, p. 392.
G. Calabrese, R. Mingarelli, P. Francalanci et al., Diagnosis of DiGeorge syndrome in nuclei released from archival autoptic heart specimens using fluorescent in situ hybridization, in Human genetics, 1996, 97, pp. 414-17.
B. Dallapiccola, A. Pizzuti, G. Novelli, How many breaks do we need to CATCH on 22q11?, in American journal of human genetics, 1996, 59, pp. 7-11.
M. Decruyenaere, G. Evers-Kiebooms, A. Boogaerts et al., Prediction of psychological functioning one year after the predictive test for Huntington's disease and impact of the test results on reproductive decision making, in Journal of medical genetics, 1996, pp. 737-43.
M. Gennarelli, G. Novelli, F. Andreasi Bassi et al., Prediction of myotonic dystrophy clinical severity based on number of intragenic [CTG]n trinucleotide repeats, in American journal of medical genetics, 1996, 65, pp. 342-47.
R. Hubbard, R.C. Lewontin, Pitfalls of genetic testing, in New England journal of medicine, 1996, 334, pp. 1192-93.
A.R. Jonsen, S.J. Durfy, W. Burke et al., The advent of the "unpatiens", in Nature medicine, 1996, pp. 622-24.
C. Lerman, S. Narod, K. Schulman et al., BRCA1 testing in families with hereditary breast-ovarian cancer. A prospective study of patient decision making and outcomes, in Journal of American medical association, 1996, 275, pp. 1885-92.
E. Masood, Gene tests: who benefits from risk?, in Nature, 1996, 379, p. 389.
A. Pizzuti, F. Amati, G. Calabrese et al., cDNA characterization and chromosomal mapping of two human homologues of the Drosophila dishevelled polarity gene, in Human molecular genetics, 1996, pp. 953-58.
D.C. Rubinsztein, J. Leggo, R. Coles et al., Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington's disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats, in American journal of human genetics, 1996, 59, pp. 16-22.
M.H. Sheng, P.S. Harper, M. Upadhyaya, Molecular genetics of neurofibromatosis type 1 (NF1), in Journal of medical genetics, 1996, pp. 2-17.
M.R. Speicher, S. Gwyn Ballard, D.C. Ward, Karyotyping human chromosomes by combinatorial multi-fluor FISH, in Nature genetics, 1996, 12, pp. 368-75.
L.S. Sullivan, S.P. Daiger, Inherited retinal degeneration: exceptional genetic and clinical heterogeneity, in Molecular medicine today, 1996, pp. 380-86.
J.A. Todd, M. Farrall, Panning for gold: genome-wide scanning for linkage in type 1 diabetes, in Human molecular genetics, 1996, pp. 1443-48.
M. D'Addedda, G. Cappucci, D. Colaizzo et al., Interrelationship between ACE I/D and PAI-1 4G/5G polymorphisms on plasma PAI-1 antigen levels, in Thrombosis and haemostasis, 1997, 77 (suppl.), p. 391.
R. Morell, R.A. Spritz, L. Ho et al., Apparent digenic inheritance of Waardenburg syndrome type 2 (WS2) and autosomal recessive ocular albinism (AROA), in Human molecular genetics, 1997, pp. 659-64.
I.M. Verma, N. Somia, Gene therapy: promise, problems and prospects, in Nature, 1997, 389, pp. 239-42.
A.O. Wilkie, Craniosynostosis: genes and mechanisms, in Human molecular genetics, 1997, pp. 1647-56.