
GENETICA
Genetica
(XVI, p. 509; App. II, i, p. 1022; III, i, p. 716; IV, ii, p. 7; V, ii, p. 372)
Le tecniche veloci e sensibili sviluppatesi a partire dalla metà degli anni Settanta, che complessivamente prendono il nome di tecnologie del DNA ricombinante (v. ingegneria genetica e biologia molecolare, in questa Appendice), permettono di manipolare DNA, RNA e proteine; pertanto coinvolgono tutti gli aspetti della g., dalla ricerca di base a quella biotecnologica e clinica. Nella ricerca di base il campo della g. soggetto a più intense sperimentazioni è quello che riguarda l'organizzazione delle sequenze del DNA nei genomi complessi, per studiare in che misura le varie modificazioni del genoma possano regolare l'attività dei geni o fornire spiegazioni sulla loro evoluzione. Sostanzialmente queste ricerche hanno profondamente modificato le nostre conoscenze sul concetto di gene: i profili di eredità, i geni normali e anomali e le funzioni geniche devono essere interpretate non solo in termini di sequenze nucleotidiche, ma anche in termini di sequenze di DNA e proteine che regolano l'attivazione e la disattivazione della trascrizione (v. oltre). Linee di ricerca genetica di grande interesse sono inoltre quelle che riguardano lo studio delle mutazioni che insorgono nelle cellule somatiche. Esse comprendono lo studio delle mutazioni di geni coinvolti nel controllo della proliferazione cellulare, che costituiscono la base della g. dei tumori (v. oncogeni, App. V e in questa Appendice, e tumore, in questa Appendice); lo studio dei riarrangiamenti del DNA che accompagnano il differenziamento del sistema immunocompetente (v. immunologia, in questa Appendice), causa della suscettibilità individuale a differenti tipi di malattie; le mutazioni somatiche del DNA mitocondriale, causa di numerose malattie che si ereditano secondo un modello di eredità non mendeliano, detto eredità materna o citoplasmatica (v. oltre). Nel campo delle applicazioni cliniche le analisi basate sul DNA sono destinate a diventare sempre più numerose ed efficaci (v. genetica medica, in questa Appendice) e, dalla fine degli anni Settanta, quando è stato sequenziato il primo gene umano, il gene per la β-globina (Maniatis, Fritsch, Lauer et al. 1980), le conoscenze sulla base molecolare delle malattie ereditarie hanno subito una grande accelerazione: attualmente sono centinaia i geni umani clonati nell'ambito del Progetto genoma (v. App. V e in questa Appendice). In linea di principio tutte le malattie genetiche possono essere analizzate in termini molecolari, e la gran parte di esse potrebbe essere trattata o curata in seguito alla precisa descrizione molecolare dei difetti del DNA che ne stanno alla base e alle loro conseguenze sul metabolismo, anche se alcuni fra i più avanzati progetti della terapia genica (v. App. V) presumibilmente rimarranno nel campo della pura speculazione.
Regolazione dell'espressione dei genomi complessi
Lo studio della regolazione dell'espressione dei geni permette di comprendere importanti fenomeni biologici quali lo sviluppo, il differenziamento, la proliferazione cellulare e l'insorgenza di malattie. Le diverse cellule di un organismo multicellulare differiscono infatti enormemente per struttura e funzione. Talvolta le differenze sono così grandi che è difficile immaginare che tutte le cellule contengano la stessa informazione genetica: per lungo tempo i biologi sospettarono che i geni venissero persi in modo selettivo nel differenziamento cellulare.
La miglior prova della conservazione di tutto il genoma durante il differenziamento di qualsiasi tipo di cellula deriva dai classici esperimenti degli anni Sessanta effettuati da J. Gurdon sullo sviluppo delle uova di anfibi (v. clonazione, App. V e in questa Appendice). Una grande serie di dati sperimentali ha successivamente dimostrato che le proprietà specifiche di ogni cellula in un organismo pluricellulare dipendono dall'attivazione di geni diversi nell'ambito di un complesso programma genetico, che si attua nel corso dello sviluppo di ciascun organismo, a partire dalla cellula uovo fecondata. Le proteine presenti solo nelle cellule specializzate, nelle quali svolgono una funzione specifica, sono codificate da geni, detti di differenziamento, soggetti a forme complesse di regolazione; per es., le catene peptidiche dell'emoglobina sono esclusivamente presenti nei globuli rossi. Tuttavia, le cellule di un organismo hanno anche molte proteine in comune, codificate da geni chiamati complessivamente housekeeping: alcune di esse sono molto abbondanti e facili da analizzare, quali le proteine del citoscheletro, dei cromosomi, del reticolo endoplasmatico o dei ribosomi; altre sono meno abbondanti, per es. gli enzimi coinvolti nelle reazioni centrali del metabolismo; tutte comunque provvedono alle esigenze fondamentali delle singole cellule indipendentemente dalla loro specializzazione.
Nella via che porta il flusso delle informazioni genetiche dal DNA alle proteine vi sono molti passaggi e tutti possono essere regolati: il controllo trascrizionale determina quando e con che frequenza il gene viene trascritto; il controllo post-trascrizionale regola i processi di elaborazione del RNA trascritto e il suo trasporto dal nucleo al citoplasma; il controllo traduzionale seleziona i RNA messaggeri del citoplasma che devono essere tradotti sui ribosomi; il controllo post-traduzionale attiva, disattiva o compartimentalizza le proteine dopo che queste sono state sintetizzate. Tutti questi tipi di controllo, fondamentali per le corrette dinamiche cellulari, sono soggetti a intensi studi; tuttavia, per la maggior parte dei geni il controllo più importante, e conseguentemente il più studiato, è il controllo trascrizionale, che assicura che non vengano sintetizzate molecole intermedie superflue.
Nello studio della regolazione della trascrizione si prendono in esame i tre livelli di organizzazione del genoma: il primo riguarda i singoli geni e gli elementi regolativi a essi associati; il secondo è rappresentato dal numero e dalla posizione relativa dei geni e degli altri segmenti di DNA del genoma (a questo livello si ha una grande quantità di dati ma non è ancora emersa una regola di principio generale); il terzo livello riguarda l'organizzazione spaziale del DNA, cioè la struttura della cromatina e dei cromosomi.
Motivi strutturali delle proteine che legano il DNA
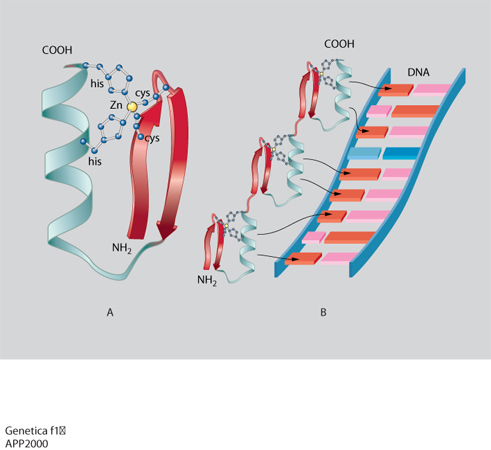
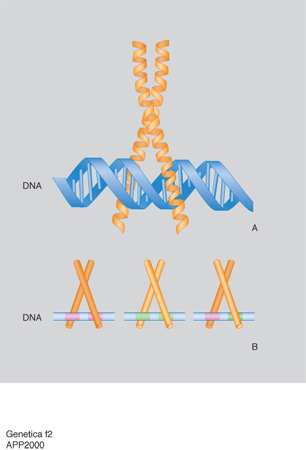
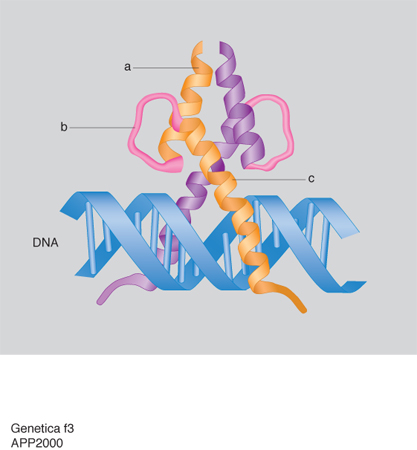
La trascrizione del DNA è controllata da alcune regioni regolatrici: alcune sono semplici e agiscono da interruttori che vengono accesi da un singolo segnale, altre sono più complesse e rispondono a una varietà di segnali. Questi interruttori sono costituiti da brevi tratti di DNA di sequenza ben definita e da proteine regolatrici che li riconoscono e li legano. La geometria dell'elica del DNA non è uniforme, ma dipende dalla sequenza nucleotidica e presenta irregolarità locali, quali appaiamenti di nucleotidi obliqui o angoli di giro dell'elica più piccoli o più grandi di 36°, che possono essere riconosciuti dalle proteine che legano il DNA. Inoltre il grado variabile di deformabilità della doppia elica consente una corrispondenza esatta fra le superfici di DNA e di proteine e quindi il loro riconoscimento molecolare basato su un gran numero di legami idrogeno, legami ionici o interazioni idrofobiche. La corrispondenza fra i motivi strutturali delle proteine e le sequenze specifiche del DNA è così ottimale che si può pensare che le unità strutturali di base degli acidi nucleici e delle proteine si siano evolute insieme per permettere una perfetta interazione. Sebbene ciascun esempio di interazione DNA-proteina sia unico nei suoi dettagli, studi di cristallografia ai raggi X hanno dimostrato che la maggior parte delle proteine regolatrici contengono pochi tipi di motivi strutturali, quali: il motivo a dita di zinco (zinc finger; fig. 1), il motivo a cerniera di leucine (leucin zipper; fig. 2) e il motivo elica-ansa-elica (HLH: Helic-Loop-Helic; fig. 3). Nel motivo a cerniera di leucine (fig. 2A) due proteine vengono tenute insieme dalle interazioni idrofobiche che si formano tra due α-eliche costituite da catene di leucine. Le due α-eliche si separano per formare una struttura a Y che abbraccia da ambo i lati il DNA come le branche di una pinza. Nella fig. 2B è anche illustrata la struttura di tre dimeri che partecipano a un processo nel quale combinazioni diverse di proteine controllano la stessa funzione. Questo controllo combinatorio costituisce uno dei meccanismi più importanti nel controllo dell'espressione dei geni nelle cellule. Nel motivo elica-ansa-elica (fig. 3) due monomeri sono costituiti da due α-eliche, una corta e una lunga, connesse da un'ansa e sono uniti in un fascio a quattro eliche. La sequenza specifica di DNA viene legata dalle due α-eliche più lunghe che sporgono dal fascio.
Elementi regolativi associati ai geni
La regione di controllo di un gene eucariotico è costituita dal promotore, sequenza di DNA a monte del gene, da una serie di proteine, chiamate fattori generali di trascrizione (TF, Transcription Factors), che devono essere assemblate al promotore, affinché la polimerasi possa iniziare la trascrizione, e da una serie di sequenze regolatrici, alle quali si legano le proteine regolatrici che controllano la frequenza dei processi di assemblaggio sul promotore. Le sequenze del promotore dei geni housekeeping e di quelli soggetti a regolazione presentano alcune differenze insieme ad alcune caratteristiche comuni.
Frequentemente il nucleotide +1 (il primo nucleotide trascritto) è un'adenina e si trova all'interno di una sequenza ricca di pirimidine detta iniziatore (inr) presente in quasi tutti i promotori. Una regione ricca di adenine timine ricorre fra i nucleotidi −26 e −34, a somiglianza della regione di sequenza timina-adenina-timina-adenina, detta TATA box, dei promotori batterici in posizione −10 (convenzionalmente i nucleotidi a monte del gene vengono preceduti dal segno −); questa regione è assente nei promotori dei geni housekeeping, che hanno però un elemento inr attivo. Sequenze citosina-citosina-adenina-adenina-timina (CCAAT box) e guanina-citosina (GC box) sono presenti in vario numero, orientamento e posizione nei promotori di molti geni, ma mancano nei geni housekeeping.
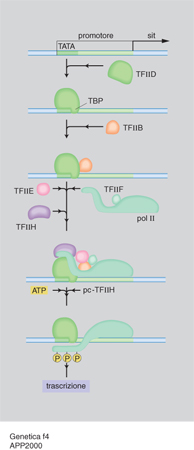
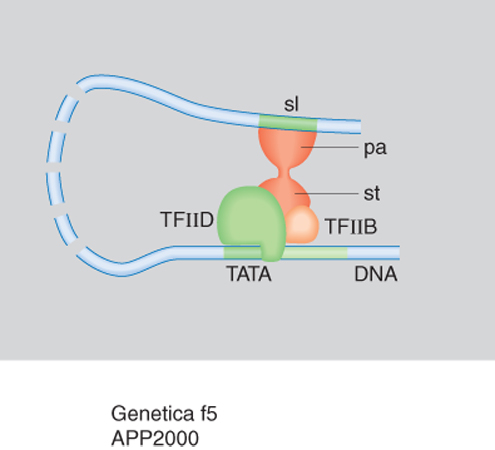
Tutti gli eucarioti possiedono tre diverse RNA polimerasi: la RNA polimerasi I che trascrive i geni per il RNA ribosomale a elevato peso molecolare; la RNA polimerasi II che trascrive il RNA messaggero necessario per la sintesi delle proteine; la RNA polimerasi III che trascrive il RNA transfer e altri RNA a basso peso molecolare. Con sofisticati esperimenti di biologia molecolare è stata identificata una serie di fattori necessari alle polimerasi per il corretto avvio della trascrizione, legati a specifici siti sul DNA. I fattori della trascrizione vengono anche detti fattori attivanti in trans in contrapposizione agli elementi che agiscono in cis, ossia ai tratti di DNA localizzati sullo stesso cromosoma dove è localizzato il gene che essi controllano (quali, per es., i promotori). La fig. 4 mostra un esempio di come alcuni TF si assemblano alla polimerasi II per iniziare la trascrizione del RNA messaggero. Inizialmente il fattore di trascrizione D riconosce la sequenza TATA del promotore di un gene mediante una sua subunità proteica (TPB); in successione si assemblano sul promotore, prima la polimerasi II insieme al fattore di trascizione F e poi i fattori di trascrizione E ed H. La polimerasi II viene fosforilata dalla subunità che funge da chinasi della TFIIH e la fosforilazione determina il suo distacco dal complesso dei fattori e l'inizio della trascrizione. A differenza del piccolo numero di TF, in grande quantità nelle cellule, che si assemblano su tutti i geni trascritti, esistono migliaia di proteine regolatrici diverse, prodotte in piccole quantità, che regolano l'espressione dei diversi geni. Esse si legano a tratti regolativi del DNA che possono trovarsi fra i CCAAT box e GC box del promotore e negli introni o a distanza di migliaia di basi a monte (estremità 5´) o a valle (estremità 3´) del gene che influenzano. Tali sequenze, scoperte per la prima volta nel 1979, vengono chiamate intensificatori (ingl. enhancer). Per spiegare come una proteina enhancer possa regolare un gene lontano, si deve ipotizzare che il DNA fra l'enhancer e il promotore si ripieghi, formando un'ansa per permettere alle proteine legate all'enhancer di interagire direttamente con uno dei TF o con il RNA polimerasi. La maggior parte delle proteine che attivano i geni presenta infatti, oltre a un motivo strutturale che lega il DNA (come precedentemente descritto), un dominio di transattivazione che, nei casi più semplici, prende contatto con il macchinario della trascrizione e ne accelera il ritmo di inizio (fig. 5). Dato che i geni devono rispondere nella maniera più flessibile ai segnali provenienti dall'esterno, essi devono essere attivati ma anche rapidamente disattivati. Questo può avvenire o attaccando o sottraendo gruppi fosforici ai TF, oppure mediante la funzione di appositi fattori a influenza negativa. Esperimenti di espressione transiente hanno infatti dimostrato non solo la presenza di enhancer, ma anche la presenza di tratti di DNA a influenza negativa, detti silenziatori (ingl. silencer). Grazie al rapido sviluppo degli studi si può ora osservare che il sistema di regolazione dei geni è particolarmente complesso: serie multiple di proteine regolatrici possono agire insieme su un promotore, ma non sono ancora chiari i dettagli del modo con il quale si ottiene l'integrazione di questi segnali multipli; il dato certo è che, qualunque sia l'esatto meccanismo di azione, una proteina regolatrice deve essere legata al DNA, sia direttamente sia indirettamente, per influenzare la trascrizione del suo gene bersaglio.
Il metodo di espressione transiente. - Per studiare la funzione delle regioni regolatrici di un gene (promotore enhancer o altro) il metodo di elezione è quello chiamato transient expression essay. Questo metodo si basa sul fatto che qualsiasi molecola di DNA estraneo può essere introdotta nelle cellule in coltura, in cui talvolta può esprimersi. Il fenomeno prende il nome di trasfezione e si può verificare in opportune condizioni sperimentali. Per es., se si mescolano molecole di DNA, cloruro di calcio e fosfato di sodio, si formano cristalli di fosfato di calcio insolubile che incorporano il DNA e possono essere introdotti nelle cellule per endocitosi. È possibile quindi studiare l'espressione di un gene, e quindi la funzione del suo promotore o delle sue sequenze regolatrici, introducendo nelle cellule in coltura il DNA interessato, clonato in un plasmide che funge da vettore. Dopo due o tre giorni dalla trasfezione, il gene viene trascritto se possiede un promotore capace di funzionare ordinatamente. Nel caso in cui il gene eucariotico sia troppo lungo per essere clonato nei plasmidi, la sequenza regolatrice della quale si vuole studiare la funzione viene legata a un gene procariotico di lunghezza adeguata, detto gene reporter (per es., il gene che codifica la β-galattosidasi o la cloramfenicolacetiltrasferasi CAT). Se la sequenza regolatrice è attiva, essa facilita l'espressione del gene reporter che viene misurata mediante la determinazione dell'attività dell'enzima nelle cellule trasfettate.
Struttura della cromatina e controllo epigenetico
La maggior parte dei dati finora riportati è il risultato di ricerche biochimiche e di esperienze di espressione genica transiente. È possibile pertanto che siano passati inosservati importanti elementi di controllo attivi in un genoma intatto. All'interno del nucleo cellulare il genoma è infatti costituito da molecole, organizzate in una gerarchia di strutture, che formano la cromatina. La prima struttura è costituita dai nucleosomi, cioè da particelle proteiche, ciascuna delle quali è formata da otto molecole di proteine basiche, uguali a due a due, chiamate istoni H2A, H2B, H3, H4. Il DNA si avvolge intorno ai nucleosomi formando due giri. In opportune condizioni sperimentali è possibile osservare al microscopio elettronico una fitta successione di nucleosomi legati al DNA che formano una fibra (a collana di perle) di 10 nm di diametro. Il secondo livello strutturale è costituito dall'avvolgimento su se stessa della collana di perle per formare una fibra più spessa (30 nm di diametro). Responsabile del compattamento di questa struttura è l'istone H1. La fibra di 30 nm forma infine delle anse in grado di legarsi a elementi strutturali del nucleo. Da lungo tempo è noto che i geni attivi si trovano a livello di una cromatina più lassa (eucromatina), mentre quelli silenti si trovano nella cromatina più condensata (eterocromatina). L'eterocromatina presenta una varietà di modificazioni - che la distinguono dallo stesso tratto di DNA nello stadio eucromatico - responsabili della modulazione dell'espressione dei geni. Dallo studio della struttura della cromatina e della sua influenza sull'espressione dei geni sono emersi alcuni principi generali. L'eterocromatina si replica più tardi dell'eucromatina, e questo suggerisce che la replicazione tardiva e l'inattivazione dei geni sono fenomeni strettamente correlati. I nucleosomi non costituiscono un ostacolo serio né per le proteine regolatrici dei geni né per le RNA polimerasi: è stato dimostrato che gli enhancer possono funzionare anche in loro presenza, che gli istoni possono essere spostati dal promotore per permettere l'assemblaggio dei fattori generali di trascrizione e che una volta iniziata la trascrizione la polimerasi può trascrivere attraversando i nucleosomi senza dissociarli; vi sono delle forme di compattamento della cromatina di ordine superiore che rendono il DNA inaccessibile sia alle proteine regolatrici sia ai fattori generali di trascrizione, mantenendo silenti grosse sezioni del genoma a volte irreversibilmente a volte no. Un ultimo principio generale deriva dall'osservazione che la cellula differenziata mantiene il suo stato nel corso di numerose generazioni; il fenomeno della memoria cellulare è un prerequisito per la creazione di tessuti organizzati e per il mantenimento di tipi cellulari differenziati in modo stabile. Il controllo superiore dei geni influenzato dalla struttura della cromatina e non da una differenza nelle sequenze del DNA prende il nome di controllo epigenetico. Il termine è stato introdotto inizialmente da C. Waddington per esprimere il cambiamento nell'espressione di alcuni geni durante lo sviluppo senza che si verificassero cambiamenti della sequenza nucleotidica. Esempi di regolazione epigenetica sono stati studiati in una grande varietà di organismi eucarioti; la Drosophila melanogaster, il nematode Caenorhabditis elegans e i lieviti, utilizzati come modelli, hanno fornito molto spesso la chiave di lettura per la comprensione dei processi molecolari coinvolti nel generare e mantenere lo stato di inattività dei geni (silencing) nelle cellule dei mammiferi. I temi comuni sembrano essere sia le modificazioni locali o estese della struttura della cromatina a opera di fattori che si legano al DNA, sia le modificazioni covalenti del DNA stesso.
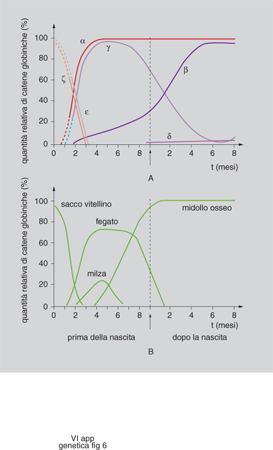
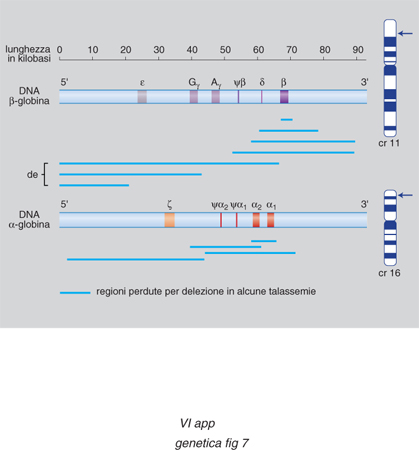
Controllo dell'espressione dei geni per le globine. - Un esempio del modo in cui la cromatina condensata può impedire l'espressione dei geni è fornito dallo studio delle famiglie geniche delle globine. È un sistema interessante perché i geni del gruppo vengono espressi con una stretta specificità cellulare solo nelle cellule eritroidi, e ciascun gene è acceso a uno stadio diverso dello sviluppo e in organi diversi secondo una precisa successione temporale. Per es. nell'embrione l'emoglobina è costituita da due catene α e da due catene π che, nel feto, sono sostituite da due γ (fig. 6). L'emoglobina è formata da quattro catene polipeptidiche, ciascuna legata a un gruppo eme contenente ferro. Il 97% dell'emoglobina totale nell'uomo adulto è costituito da due catene α e da due catene β (HbA), mentre la restante emoglobina (HbA2) contiene due catene α e due catene δ. Una volta sequenziati i geni e chiarita l'organizzazione delle due famiglie geniche (fig. 7), sono state individuate anche moltissime mutazioni che determinano anomalie nella corretta trascrizione dei geni per le globine α e β e che sono la causa di un importante gruppo di anemie ereditarie, chiamate rispettivamente talassemie α e β. Tutti i geni della stessa famiglia vengono trascritti nella stessa direzione a partire dall'estremità 5´ del DNA; la posizione del gene all'interno del gruppo rispecchia la successione della sua espressione. Nello studio dei processi di regolazione dei geni globinici si è scoperto che ciascun gene presenta elementi regolatori individuali, e che l'intero gruppo è soggetto a un controllo 'acceso-spento' che comporta cambiamenti globali nella struttura della cromatina. In alcuni individui affetti da una forma di talassemia nella quale i geni della famiglia β sono del tutto silenti è presente una delezione di una sequenza di DNA di alcune migliaia di basi a monte del gene per la globina β (fig. 7). Tale regione importante per l'espressione dei geni, è stata definita regione di controllo del locus (LCR, Locus Control Region). Esperimenti in vivo con topi transgenici hanno confermato l'importanza di questa regione: quando il gene della globina β è stato inserito nel topo insieme al tratto di DNA situato a 20 Kb a monte della sequenza dei geni della famiglia β, la quasi totalità dei topi transgenici ottenuti ha espresso il gene per la globina umana, con un'efficienza superiore a quella del gene murino. Il gene viene, inoltre, espresso esclusivamente nelle cellule eritroidi e la sua attività è indipendente dalla posizione in cui esso si integra nel genoma dell'ospite. Nella LCR è presente un tipo di cromatina a struttura particolare, eccezionalmente sensibile alla DNasi, enzima in grado di tagliare il DNA. È stato osservato che regioni geniche attive mostrano una sensibilità alla DNasi dieci volte superiore rispetto ai tratti vicini, geneticamente silenti, e che vi sono inoltre delle posizioni lungo la cromatina che sono mille volte più sensibili di quanto non sia la cromatina ordinaria. Questi siti ipersensibili alla DNasi (DHS, DNA Hypersensitive Sites) si trovano nella regione del promotore e dell'enhancer di tutti i geni attivi; è pertanto evidente che, con l'induzione dell'attività di un gene, si forma un DHS nelle sue sequenze regolatrici. Una causa possibile di questo evento è la separazione o l'allontanamento di un promotore compattato sul nucleosoma da parte di una proteina attivatrice. Tale evento può essere dovuto a un'azione diretta della proteina attivatrice o, in alternativa, a una sua azione indiretta tesa a facilitare il legame dei fattori di trascrizione con il DNA. Le ricerche sui geni delle globine hanno portato all'identificazione di 5 zone DHS a monte della famiglia delle globine β e di una sesta zona disposta a 20 Kb dopo l'ultimo gene del gruppo. Negli individui normali, dopo la nascita, spariscono i DHS davanti all'emoglobina ε e γ. Si può affermare pertanto che i DHS sono gli elementi attivi della regione di controllo del locus e hanno due precipue funzioni: allentamento dello stato condensato della cromatina e attivazione della trascrizione attraverso un'azione sui promotori dei singoli geni.
Acetilazione degli istoni e metilazione del DNA. - Nelle cellule di mammifero le modificazioni epigenetiche di maggior frequenza sono, a livello della cromatina, l'acetilazione e la deacetilazione degli istoni e, a livello del DNA, la metilazione della citosina. Si è osservato che l'acetilazione degli istoni rende più efficiente il legame con i fattori di trascrizione. L'istone H4 viene acetilato prima di assemblarsi nel nucleosoma; la deacetilazione di H4 invece si verifica in coincidenza del legame di H1 al nucleosoma e il conseguente compattamento della cromatina di ordine superiore. La cromatina trascrizionalmente attiva presenta sempre un alto livello di acetilazione degli istoni rispetto a quella inattiva, e l'istone H4 è acetilato in quelle regioni cromosomiche (bande R) che si pensa contengano la maggior parte dei geni housekeeping.
Nel genoma dei vertebrati sono presenti sequenze nucleotidiche GpC (guanina, citosina) in larga parte metilate sui residui di citosina (60-90%). Le sequenze GpC hanno una frequenza più bassa rispetto a quella che ci si potrebbe attendere in termini statistici in relazione alla composizione percentuale in basi del DNA, e questo ha una spiegazione in termini evolutivi. La bassa frequenza di GC costituisce una protezione dalle mutazioni, in quanto la deaminazione idrolitica spontanea della metil-citosina porta alla formazione di timina, che non viene eliminata dai sistemi di riparazione della cellula. La deaminazione della citosina non metilata porta alla formazione di una base errata, l'uracile, che viene invece riconosciuto ed eliminato dall'enzima della riparazione del DNA, uracilsintetasi. La metilazione della citosina, che avviene, durante la replicazione del DNA, sul nuovo filamento appena sintetizzato a opera di un enzima, la metiltrasferasi, si è dimostrata necessaria al corretto sviluppo dei mammiferi. Questa reazione costituisce, infatti, la maggiore modificazione nel DNA dei vertebrati, anche se non è presente in altri organismi quali la Drosophila melanogaster, i nematodi o i lieviti. Isole GpC, ossia tratti di genoma nei quali le sequenze GpC sono dieci volte più frequenti, si trovano all'estremità 5´ dei geni. Nei geni housekeeping le isole GpC non sono mai metilate. Nei geni tessuto-specifici la metilazione varia con l'attività del gene: le isole GpC davanti ai geni attivi sono ipometilate, mentre quelle davanti ai geni silenti sono ipermetilate.
Varie osservazioni hanno permesso di avanzare ipotesi sulla funzione della metilazione, anche se i numerosi dati raccolti non consentono ancora di formulare un'interpretazione univoca: per es., la metilazione potrebbe determinare una maggiore affinità di legame con l'istone H1, che si trova di preferenza nei tratti di cromatina ipermetilati ed è responsabile dello stato fortemente condensato della cromatina, oppure essa potrebbe ridurre le interazioni fra i fattori di trascrizione e i siti specifici sul DNA, dato che essi contengono spesso siti GC.
Gli studi sulla regolazione epigenetica attualmente più interessanti sono l'effetto di posizione, la compensazione di dosaggio e l'imprinting genomico. Le analogie fra di essi costituiscono interessanti campi di indagine genetica, che forniranno spiegazioni sul ruolo della cromatina nell'attivazione e nel silenziamento dei geni.
Effetto di posizione. - Nell'effetto di posizione l'attività di un gene dipende dalla posizione che esso occupa nel genoma e riflette i diversi stati della cromatina presente in posizioni diverse del genoma e la tendenza di questi stati a diffondersi interessando le zone vicine. Nel lievito Saccharomyces cerevisiae, per es., l'espressione di alcuni geni dipende dalla loro posizione sul cromosoma: se, sperimentalmente, vengono spostati dalla localizzazione originaria e riposizionati vicino ai telomeri, situati all'estremità dei cromosomi, la lo-ro trascrizione viene spenta, anche se la cellula contiene tutte le proteine necessarie perché essa possa verificarsi. Il silenziamento si estende per 10.000 coppie di nucleotidi, si applica ad altri geni, si indebolisce gradualmente con l'allontanarsi dei geni dai telomeri ed è un fenomeno che una volta stabilito è ereditabile. Nella regolazione di questo fenomeno sono coinvolti un gruppo di geni chiamati SIR (Silent Information Regulator), ma soprattutto si è osservato che il DNA vicino ai telomeri è compattato in una forma di cromatina inaccessibile alle proteine che legano il DNA. All'effetto di posizione è dovuta anche la variegatura del colore degli occhi della Drosophila, descritta da H.J. Muller per la prima volta nel 1930.
Nella Drosophila è stato anche osservato un tipo di silenziamento locale diverso da quello che si verifica con la giustapposizione con la eterocromatina. Sono stati infatti identificati dei geni chiamati Polycomb (Pc) che, se mutati, non permettono l'espressione di alcuni geni omeotici coinvolti nello sviluppo dell'insetto (v. biologia dello sviluppo, in questa Appendice). Negli anni Novanta si è scoperto che le proteine da essi codificate costituiscono un complesso formato da più subunità, chiamato Pc-G (Polycomb-Group), che sono in grado di inibire l'espressione di specifici geni bersaglio inducendo la formazione di tratti di cromatina silente. In questi è presente una sequenza conservata di 48 amminoacidi chiamata cromodominio, associata a una proteina non istonica chiamata HP1. Dato che le proteine Pc-G si legano a sequenze trascrizionalmente represse e non a quelle attivamente trascritte, è stato ipotizzato che il complesso Pc-G modifichi attivamente la struttura della cromatina. Ad esso è sempre collegata una molecola di RNA che non viene tradotta. Molecole simili devono avere un ruolo nella regolazione epigenetica, in quanto si trovano sia nell'imprinting sia nel processo di inattivazione del cromosoma X dei mammiferi. Studi sul sistema Polycomb sono in corso e sono di grande interesse in quanto aprono nuove strade per la conoscenza di modelli di regolazione dei geni.
Compensazione di dosaggio. - Il termine si riferisce a quei processi mediante i quali, nelle femmine, i geni legati al cromosoma X hanno lo stesso livello di espressione rispetto ai maschi, anche se i maschi hanno un solo cromosoma X. Per ottenere questo scopo le strategie sono diverse. Nella Drosophila, i prodotti dei geni legati al cromosoma X nel maschio sono espressi in quantità doppia, mentre nel nematode Caenorhabditis elegans il prodotto dei geni del cromosoma X in individui XX viene dimezzato. Nei mammiferi (topo, uomo) si è evoluto un meccanismo mediante il quale uno dei due cromosomi X della femmina viene quasi completamente inattivato in modo permanente. Malgrado queste diversità, il meccanismo cromosomico usato per controllare l'espressione dei geni del cromosoma X è sorprendentemente lo stesso nell'uomo, nel verme e nella mosca.
Nell'embrione umano l'inattivazione si verifica allo stadio di blastocisti quando l'embrione è costituito da circa 200-400 cellule. Il cromosoma eterocromatico inattivo è visibile al microscopio ottico durante l'interfase. Esso costituisce una struttura distinta, nota come corpo di Barr, posta vicino alla membrana nucleare che si replica tardivamente, alla fine della fase S del ciclo cellulare. Poiché lo stato inattivo del cromosoma viene ereditato fedelmente e l'inattivazione si verifica casualmente, ogni femmina è un mosaico formato da una miscela di cloni di cellule, nelle quali è attivo il cromosoma X ereditato dal padre e da una miscela di cloni di cellule in cui è attivo il cromosoma X ereditato dalla madre. Sul cromosoma X è stato localizzato un centro di inattivazione unico, XIC nell'uomo e Xic nel topo (X-Inactivation Center), dal quale la condensazione della cromatina lungo il DNA si diffonde linearmente, tanto che frammenti spezzati del cromosoma X non subiscono inattivazione a meno che non contengano questo centro. Il cambiamento non è comunque permanente in quanto il cromosoma X condensato viene riattivato durante la formazione delle cellule germinali della femmina. Nella zona XIC è stato mappato un gene chiamato XIST (X-Inactive Specific Transcription), candidato a essere il gene implicato nel controllo dell'inattivazione del cromosoma X. Il gene trascrive un RNA di 17 kb che non viene tradotto e che è stato trovato specificamente associato al corpo di Barr. Alcune osservazioni suggeriscono tuttavia che XIST non è assolutamente richiesto per il mantenimento dello stato eterocromatico, pertanto il ruolo di XIST potrebbe essere confinato nei primi stadi del processo di inattivazione. Studiando la compensazione di dosaggio in altri organismi, le maggiori analogie si osservano nell'alterazione della struttura della cromatina. Nei mammiferi, per il cromosoma X inattivo, è ben documentata l'alterata morfologia in interfase, la ridotta accessibilità alle nucleasi, la mancata acetilazione dell'istone H4 e l'ipermetilazione delle isole GpC; nella Drosophila sono ben documentati l'iperacetilazione del cromosoma X attivo e il maggiore volume del cromosoma X attivo nelle ghiandole salivari del maschio.
Imprinting genomico
Si parla generalmente di imprinting genomico per indicare la situazione nella quale alleli ereditati dal padre vengono espressi in maniera diversa da quelli corrispondenti ereditati dalla madre. Sulla base dei principi mendeliani, dato che ciascun genitore fornisce alla progenie una quantità aploide di cromosomi, si è creduto per lungo tempo che essi dessero un uguale contributo genetico, cioè che l'espressione di un certo carattere non dipendesse dal sesso del genitore dal quale esso derivava. Un classico esperimento di embriologia ha dimostrato, in maniera convincente, che il genoma materno e quello paterno hanno invece funzioni differenti nel corso dello sviluppo.
Poiché nelle uova fecondate di topo è facile distinguere il pronucleo maschile da quello femminile, è possibile, con la microchirurgia, eseguire trapianti mediante i quali il pronucleo maschile viene scambiato con quello femminile. Come conseguenza di vari trapianti reciproci, si producono tre diversi tipi di uova fecondate: uova che contengono genomi sia paterni sia materni, uova che contengono solo genomi paterni, dette androgeniche, e uova che contengono solo genomi materni, dette ginogeniche. Embrioni completi si sviluppano solo dal primo tipo di uova; non si sviluppano mai embrioni dalle uova androgeniche o ginogeniche. I risultati indicano che la manipolazione in vitro di per sé non è la causa del mancato sviluppo dell'embrione, ma che, per completare lo sviluppo, sono realmente necessari sia il pronucleo materno sia quello paterno. Evidentemente entrambi i genomi danno contributi funzionalmente distinti all'embriogenesi. Altre osservazioni hanno fornito dettagli interessanti: cellule con patrimonio esclusivamente materno determinano l'iniziale sviluppo della cosiddetta massa cellulare interna, dalla quale, in condizioni normali, si sviluppa il vero e proprio embrione; cellule invece con assetto esclusivamente paterno formano principalmente il trofoectoderma extraembrionale, dal quale si sviluppa la placenta. Da tutte queste osservazioni si deduce che almeno alcuni geni sono espressi in maniera differente sui cromosomi paterni rispetto ai loro equivalenti sui materni. Numerose evidenze dimostrano che l'espressione differenziale dei geni nel fenomeno dell'imprinting è collegata alla metilazione del DNA. In modelli sperimentali, quali topi transgenici in cui è stata inattivata la DNA metiltrasferasi, si è osservata la morte precoce degli embrioni, come conseguenza della riduzione del 95% della metilazione. Questo esperimento, pur essendo molto significativo, tuttavia non dimostra se la metilazione è la causa dell'imprinting o se semplicemente mantiene lo stato imprinted del gene, una volta che il fenomeno si sia verificato. Le ricerche compiute sul genoma murino hanno dimostrato che esistono almeno 10 regioni distribuite su sei diversi cromosomi nelle quali gli alleli paterni e materni vengono espressi differentemente. Un gruppo di geni imprinted molto studiato è quello che si trova sul cromosoma 7: Mash2, H19 e Igf2r vengono espressi solo se si trovano sul cromosoma materno, mentre i geni Ins2 e Igf2 (Insulin like growth factor type 2) vengono espressi solo se si trovano sul cromosoma paterno. Questi ultimi codificano fattori di crescita, mentre H19 trascrive un RNA messaggero non codificante. L'espressione correlata dei geni H19 e Igf2, localizzati sullo stesso cromosoma alla distanza di 90 kb, ha suggerito ad alcuni ricercatori un probabile modello chiamato di espressione-competizione, che potrebbe spiegare in parte la complessità del fenomeno dell'imprinting anche per altri geni: sul cromosoma paterno il gene H19 non viene espresso, dato che il suo promotore è metilato già a livello della spermatogenesi e gli elementi regolatori (enhancer) sono in grado di attivare solo il gene Igf2. Viceversa, sul cromosoma materno il promotore del gene H19 è libero e si trova pertanto sotto l'influsso di elementi enhancer che attivano il gene, il cui prodotto (RNA) reprime Igf2.
L'imprinting condiziona l'espressione di geni anche nel genoma umano. Le ricerche si riferiscono soprattutto all'analisi molecolare di malattie genetiche e dimostrano che l'espressione fenotipica di alcune malattie è diversa a seconda che esse siano ereditate dal padre o dalla madre. Le maggiori differenze cliniche si notano nelle due sindromi ereditarie note come sindrome di Prader-Willi e sindrome di Angelman. Nella sindrome di Prader-Willi, caratterizzata da obesità, bassa statura, ipogonadismo, ritardo mentale e ipotonia, si osserva frequentemente una delezione di un tratto del braccio lungo del cromosoma 15. Il cromosoma 15 deleto è ereditato dal padre, pertanto l'individuo affetto dalla malattia ha, per quel tratto di cromosoma, informazioni genetiche esclusivamente materne. È stato dimostrato che, se la delezione dello stesso tratto di cromosoma 15 è ereditata dalla madre, i pazienti presentano invece la sindrome di Angelman, completamente diversa dalla precedente, caratterizzata da movimenti atassici, simmetrici e ripetitivi, attacchi convulsivi e accessi improvvisi di risa. Questo suggerisce l'esistenza di due geni, con espressione l'uno paterna e l'altro materna, che codificano distinte funzioni. R.D. Nicholls e altri (1989) hanno osservato che alcuni pazienti con sindrome di Prader-Willi avevano un paio di cromosomi 15 normali, ma che entrambi erano di origine paterna. Questa situazione inusuale, chiamata disomia uniparentale, dimostra che, per uno sviluppo normale, i geni di questa regione cromosomica devono essere ereditati in doppia copia ma che ciascun membro della coppia deve derivare da un genitore diverso. Una spiegazione verosimile è che in quest'ultimo caso si sia inizialmente verificata una trisomia del cromosoma 15, a seguito di un'anomalia della meiosi, poi compensata dalla perdita di uno dei cromosomi soprannumerari.
Analizzando il locus della delezione sul cromosoma 15 si è scoperto che la delezione interessa una serie di geni: alcuni trascrivono RNA non codificanti mentre il gene SNRPN codifica la proteina (SmN) costituente fondamentale di una delle particelle snRNP (small nuclear RiboNucleoProtein) coinvolte nello splicing di RNA messaggeri specifici nel cervello. Questa scoperta suggerisce una forte correlazione fra la mutazione nel gene SNRPN e i seri disturbi comportamentali degli individui affetti da questa sindrome. È stato identificato, 450 kb a valle del gene SNRPN, il gene responsabile della sindrome di Angelman, UBE3A, che codifica una ligasi per l'ubiquitina, proteina coinvolta nella degradazione delle proteine nella cellula. Nel 1997, da parte di U. Albrecht e collaboratori, è stata dimostrata l'espressione materna del gene nelle cellule del cervello. Dato che alcuni geni, distanti diverse migliaia di kilobasi dalle zone di DNA coinvolte nelle due sindromi, subiscono ancora l'influsso dell'imprinting genomico, si pensa che vi possano essere due lunghe regioni di controllo dell'imprinting che interagiscono fra di loro; a queste è stato dato il nome di PWS-ICR (Prader Willi Syndrome Imprinting Control Region) e AS-ICR (Angelman Syndrome Imprinting Control Region). L'inattivazione di un tratto così lungo dipende probabilmente da RNA non tradotti comparabili al RNA H19 del cromosoma 7 del topo.
Gli studi sulla base molecolare dell'imprinting sono in pieno sviluppo ed è difficile trarre conclusioni univoche dai numerosi dati sperimentali che si stanno accumulando, anche se si possono immaginare alcuni scenari, nei quali si muoverà la ricerca futura, basati sulle seguenti osservazioni: a) tratti di genoma suscettibili di imprinting possiedono un centro di inattivazione che è attivo durante la maturazione delle cellule germinali e agisce attraverso una metilazione di dinucleotidi CpG, ma non è ancora ben conosciuto il processo mediante il quale l'imprinting compare nella linea germinale e si mantiene nell'embriogenesi e nello sviluppo postnatale; b) sotto l'influsso del centro di inattivazione viene modificato un intero dominio cromatinico, con il risultato che il dispositivo di trascrizione non ha più accesso ai geni interessati, come avviene nella regione di controllo di locus (LCR) della famiglia genica delle globine. Non si sa tuttavia come un centro di inattivazione localizzato possa influenzare la struttura di un lungo tratto di cromatina; c) l'imprinting è presente nei mammiferi, ma non negli altri vertebrati e ci si domanda pertanto quale possa essere il suo vantaggio evolutivo. L'imprinting sembra costituire una specie di meccanismo di difesa dalla partenogenesi, ma risulta difficile capire come questa possa essere una funzione selezionata e non piuttosto un effetto collaterale. Sembra più probabile, invece, che sia il controllo della crescita del feto sia le relazioni materno-fetali siano le forze selettive che hanno agito nell'espressione dell'imprinting nei mammiferi, lo sviluppo dei quali è essenzialmente caratterizzato dalla presenza della placenta, che permette le interazioni fra l'embrione e la madre. È già noto, per es., che nel topo i geni paterni Igf2 e Ins2 aumentano la crescita del feto, mentre i geni materni come H19 la riducono. Particolarmente interessanti sono inoltre le analogie che l'imprinting presenta con l'inattivazione del cromosoma X. Esse si possono così riassumere: a) il meccanismo inattiva uno dei due cromosomi di una cellula diploide; b) l'inattivazione avviene specificamente nel cromosoma X paterno a livello del tessuto placentale ed è invece casuale nei tessuti embrionali; c) il prodotto di XIST, come quello di alcuni geni coinvolti nell'imprinting del topo, è un RNA non codificante, che esplica la sua azione sui geni localizzati sullo stesso cromosoma; d) la metilazione, essenziale per l'imprinting, è anche il meccanismo che regola in particolare l'espressione di XIST.
Eredità citoplasmatica
La trasmissione ereditaria dei geni presenti in organelli citoplasmatici quali i mitocondri non segue le leggi di Mendel e viene chiamata eredità citoplasmatica o extranucleare. Nell'eredità citoplasmatica i determinanti genetici in grado di duplicarsi e trasmettersi alle generazioni successive non sono situati nei cromosomi nucleari, ma nel cromosoma dei mitocondri o dei cloroplasti. La variegatura delle foglie nella bella di notte (Mirabilis yalapa) costituisce un tipico esempio di eredità citoplasmatica nelle piante, dovuta all'informazione genetica contenuta nei cloroplasti. Questo tipo di eredità è stato ben studiato anche nelle cellule di lievito: accoppiando due cellule aploidi di lievito, una delle quali presenta una mutazione del DNA mitocondriale, si forma una cellula diploide (zigote), nella quale sono presenti sia i mitocondri selvatici sia quelli mutati. Al momento della riproduzione per gemmazione la cellula figlia riceverà un numero limitato di mitocondri e dopo numerose gemmazioni successive, casualmente, potranno segregare nei nuovi organismi o tutti i mitocondri mutati o tutti i mitocondri di tipo selvatico. Tale processo, chiamato segregazione citoplasmatica, forma una progenie di lievito diploide che contiene un solo tipo di DNA mitocondriale. Negli animali superiori, all'atto della formazione dello zigote, la cellula uovo passa allo zigote molto più citoplasma di quanto non faccia lo spermatozoo e gli spermatozoi della maggior parte delle cellule animali contengono alcune centinaia di mitocondri, mentre le cellule uovo ne contengono centinaia di migliaia. Anche se alcuni mitocondri dello spermatozoo casualmente dovessero finire nello zigote durante la fecondazione, la maggior parte del genoma mitocondriale è ereditato quindi per linea femminile. È stato dimostrato che la progenie di incroci fra ratti che possiedono due tipi di DNA mitocondriale lievemente diversi contiene solo DNA mitocondriale di tipo materno. In questo caso si può parlare di eredità uniparentale o più precisamente materna.
I geni nei mitocondri
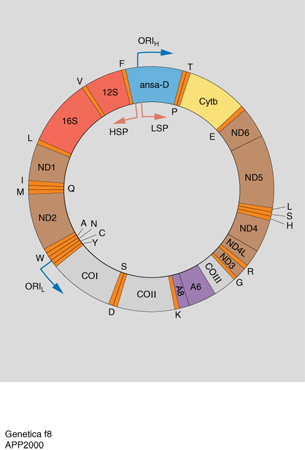
Il DNA mitocondriale (DNAmt) è solitamente una molecola di DNA circolare, eccetto che in alcuni protisti nei quali è lineare. Le sue dimensioni variano a seconda delle specie (da circa 15.000 nei mammiferi a circa 2 milioni di paia di basi nelle piante) e le variazioni possono essere notevoli anche all'interno della stessa specie. Poiché sia i DNAmt più corti sia quelli di maggiori dimensioni codificano gli stessi prodotti genici, probabilmente la maggior parte del DNA in eccesso è coinvolto in riarrangiamenti non funzionali. I DNAmt meglio studiati sono quelli del lievito e dell'uomo. Entrambi sono stati sequenziati: il DNAmt del lievito è costituito da circa 70.000 coppie di basi, ma solo un terzo codifica proteine, mentre quello dell'uomo ne contiene 16.569. Probabilmente il DNA in eccesso nei mitocondri di lievito e delle piante costituisce un 'DNA spazzatura', di poca utilità per l'organismo. Nel DNA mitocondriale si possono distinguere i due filamenti in relazione alla loro differente composizione delle basi: un filamento pesante (H, heavy ) e un filamento leggero (L, light; fig. 8). La maggior parte dei geni si trova sul filamento H: 2 geni per il RNA ribosomiale, 14 geni per il RNA transfer e 12 geni che codificano proteine; sul filamento L si trovano 8 geni per il RNA transfer e un gene che codifica una proteina. La presenza di geni per il RNAt e il RNAr dimostra che i mitocondri possiedono un loro sistema di traduzione anche se le relative componenti proteiche sono codificate da geni nucleari per le proteine ribosomiali, le RNA polimerasi, i loro fattori di trascrizione. La notevole simbiosi molecolare fra DNA mitocondriale e nucleare è anche aumentata dal fatto che proteine che agiscono di concerto fra loro, quali quelle della catena respiratoria, vengono codificate in parte dal DNA mitocondriale e in parte dal DNA nucleare.
Espressione dei geni mitocondriali. - Gli schemi di trascrizione dei geni mitocondriali sono piuttosto variabili; nell'uomo vengono trascritti entrambi i filamenti di DNAmt aumentando così la capacità di codificare di questa molecola. A differenza dei genomi nucleari, le sequenze del genoma mitocondriale umano sono quasi tutte codificanti e non contengono introni; molti geni risultano inoltre separati nel genoma da geni che codificano RNAt e non da spaziatori non codificanti come nel lievito. Dopo la trascrizione, i RNAt vengono separati dalle sequenze che codificano proteine. Nella mappa genica del DNA mitocondriale è presente infatti una regione indicata come ansa D (displacement). In questa regione si osservano tre filamenti di DNA: è presente infatti un frammento di DNA di circa 700 paia di basi che si lega al filamento L e allontana da questo il tratto corrispondente del filamento H. Nella regione dell'ansa D si trovano due promotori, uno per il filamento H e l'altro per il filamento R. Questo è un raro esempio di trascrizione simmetrica nella quale si formano due molecole di RNA pari alla lunghezza dell'intero genoma mitocondriale. I due trascritti vengono tagliati da nucleasi durante la loro sintesi: si formano così i diversi tipi di RNA del filamento H, mentre viene degradata quasi completamente la porzione L degli acidi ribonucleici. Quelli mitocondriali presentano inoltre alcune differenze rispetto a quelli nucleari: all'estremità 5´ della molecola non hanno né il cappuccio di 7-metil guanosina né sequenze non codificanti, ma iniziano direttamente con il codone di inizio AUG (Adenina Uracile Guanina). Per la sintesi proteica mitocondriale sono inoltre sufficienti solo 22 RNAt, perché le normali regole di appaiamento codone-anticodone sono attenuate nei mitocondri, così che molte molecole di RNAt riconoscono qualunque nucleotide in terza posizione del codone.
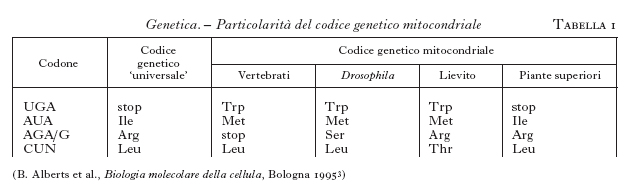
Il codice genetico dei mitocondri. - Un aspetto interessante del genoma mitocondriale è emerso quando le proteine codificate dal DNAmt sono state sequenziate e confrontate con le sequenze geniche (1979): come indicato nella tab. 1, il codice genetico mitocondriale differisce in piccola parte da quello universale. Si può osservare che nei mitocondri delle piante non vi è alcuna differenza rispetto al codice genetico 'universale', mentre negli altri eucarioti compaiono variazioni diverse. La spiegazione delle diversità nel codice genetico mitocondriale si può far risalire a fenomeni di deriva genetica casuale. Dal punto di vista evolutivo, infatti, il piccolo numero di proteine codificate dal genoma dei mitocondri rende tollerabile un cambiamento occasionale del significato di un codone raro, mentre lo stesso cambiamento, in un genoma più grande, altererebbe la funzione di molte proteine, distruggendo la cellula.
Filogenesi dei mitocondri. - Il DNA mitocondriale è una molecola di elezione per studi evolutivi di filogenesi molecolare: è stato infatti dimostrato che il ritmo di sostituzione di sequenze nucleotidiche nell'evoluzione dei genomi mitocondriali è 10 volte maggiore rispetto a quello dei genomi nucleari, forse a causa di una ridotta fedeltà nella replicazione del DNA o di una carenza di processi riparativi. Il ritmo relativamente alto di mutazione dei geni mitocondriali è pertanto utile per stimare le date di eventi evolutivi relativamente recenti. Inoltre, il genoma mitocondriale è ereditato per via materna e solo la cellula uovo contribuisce con del citoplasma allo zigote. Come conseguenza il DNAmt non va incontro a meiosi e non presenta processi di ricombinazione; gli individui che condividono le stesse sequenze di DNAmt devono quindi avere un comune antenato femminile. L'esempio più affascinante dello studio del DNAmt nelle ricerche filogenetiche giunge dai dati raccolti da A.C. Wilson e collaboratori (1984) che consentono l'identificazione di una progenitrice per il genere umano. Il confronto fra le sequenze di vari tratti di DNAmt in individui provenienti da diverse regioni geografiche ha reso infatti possibile la ricostruzione di un albero filogenetico. Supponendo che la velocità di variazione delle sequenze sia costante e dovuta solo a mutazioni, il numero delle differenze riscontrate fra i vari gruppi e fra gli individui dello stesso gruppo geografico può essere utilizzato per dedurre linee evolutive e il periodo in cui sono vissuti i comuni antenati. Su questa base è stato possibile calcolare che l'Eva mitocondriale visse in Africa circa 200.000 anni fa. In questi calcoli bisogna tenere conto del differente contributo dato al genoma mitocondriale rispetto a quello nucleare da parte dei progenitori. Per es., mentre solo la bisnonna materna fornisce tutto il DNAmt, ogni bisnonno paterno o materno fornisce un ottavo al genoma nucleare della discendenza. Inoltre, mediante la reazione a catena della polimerasi (PCR, Polimerase Chain Reaction), che amplifica anche piccole quantità di DNA, è stato recentemente dimostrato che anche gli spermatozoi cedono mitocondri all'uovo fecondato: pertanto la popolazione da cui proviene il DNAmt è più grande di quanto inizialmente assunto. Questo significa che la fonte del DNA mitocondriale attuale potrebbe essere più ravvicinata nel tempo e alcuni studi suggeriscono che la comparsa dell'Homo sapiens in Africa sia avvenuta 130.000 anni fa.
Mediante l'analisi del DNA mitocondriale sono state anche ricostruite le migrazioni delle popolazioni umane, stabilendo quali terre sono state colonizzate per prime, in quanto una maggiore variabilità nelle sequenze nei DNA mitocondriali di un continente è un segno di maggiore antichità. La massima variabilità è presente nelle popolazioni africane, che sono quindi le più antiche, seguite da quelle asiatiche, europee e indigene americane.
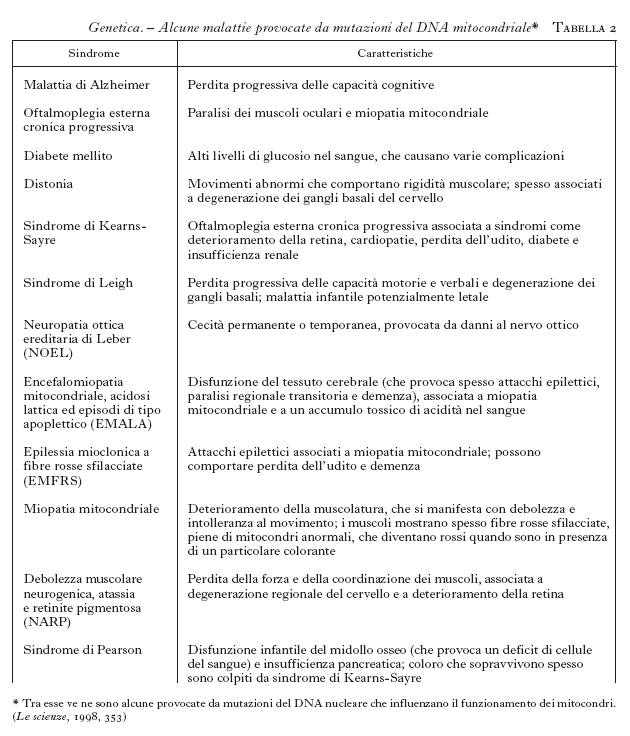
Malattie dovute a mutazioni del DNA mitocondriale. - Nella specie umana, i DNA mitocondriali di individui diversi possono differire fra loro per circa 70 posizioni per l'intera molecola. Queste differenze riguardano per lo più la terza posizione dei codoni, per cui non ci sono particolari conseguenze per la funzione del genoma. Si stima che la frequenza delle sostituzioni nucleotidiche nel DNA mitocondriale sia 10-20 volte più alta rispetto alle sostituzioni che interessano il DNA nucleare. Un certo numero di malattie genetiche umane, tuttavia, è dovuto a mutazioni di geni localizzati sul DNA mitocondriale. I difetti del DNA mitocondriale che causano patologie sono spesso ereditari, ma possono anche comparire spontaneamente in una cellula uovo o nelle prime fasi di sviluppo dell'embrione, e poi distribuirsi in tutto l'organismo durante lo sviluppo. Le patologie mitocondriali interessano soprattutto il cervello, il cuore, il muscolo, il fegato, la parte endocrina del pancreas, cioè i tessuti maggiormente interessati alla fosforilazione ossidativa, processo che fornisce energia alla cellula e che si attua nei mitocondri. Un fattore di complicazione nell'eredità delle malattie mitocondriali consiste nell'eterogeneità genetica dei mitocondri di uno stesso individuo, dovuta al fatto che in un uovo umano vi possono essere sia mitocondri normali sia mitocondri alterati. Questa eterogeneità viene chiamata eteroplasmia. Le cellule che derivano dalla segmentazione dell'uovo eteroplasmico erediteranno percentuali diverse di mitocondri con DNA mutato o normale. In base alle leggi della probabilità, via via che le cellule continuano a riprodursi, le popolazioni di DNA mitocondriale nelle cellule figlie tenderanno a uniformarsi (omoplasmia), diventando o prevalentemente normali o prevalentemente mutate. Come regola, una mutazione del DNA mitocondriale, che comporta una grave riduzione della produzione di energia della cellula, risulta eteroplasmica, in quanto l'omoplasmio sarebbe necessariamente incompatibile con la vita, mentre malattie meno gravi possono derivare da mutazioni sia eteroplasmiche che omoplasmiche. L'eteroplasmia influenza la localizzazione e la rapidità con la quale una mutazione si diffonde in una popolazione di cellule in divisione; individui nati con mutazioni nel DNA mitocondriale spesso si ammalano con una latenza di molti anni e le loro condizioni peggiorano col passare del tempo. La frazione di molecole di DNA mitocondriale mutante varia quindi nella stessa cellula, nei tessuti dello stesso individuo nel corso dello sviluppo ed è anche variabile nei diversi individui entro lo stesso albero genealogico. Un'altra fonte di variabilità è dovuta al fatto che le componenti della catena respiratoria codificate dai geni nucleari non provengono dagli stessi geni in tutti i tessuti. È stata stabilita l'esistenza di forme alternative (isoforme) di molte proteine tessuto-specifiche, che potrebbero avere un effetto significativo sull'espressione di una data mutazione mitocondriale in tessuti differenti o in differenti stadi di sviluppo: pertanto i fattori che influenzano l'eredità e le funzioni mitocondriali sono molteplici. La tab. 2 elenca alcune malattie provocate da mutazioni del DNA mitocondriale. Le mutazioni conosciute che determinano malattie nell'uomo sono mutazioni puntiformi nei geni che codificano proteine o RNA mitocondriali, o ampie deiezioni e duplicazioni del DNA che coinvolgono molti geni. Per es., nel 90% dei casi di neuropatia ottica ereditaria di Leber sono state descritte mutazioni nei geni mitocondriali, che codificano proteine coinvolte nelle prime fasi di trasporto di elettroni nella fosforilazione ossidativa. Le alterazioni di molecole di RNAt, per es., nell'epilessia mioclonica a fibre rosse sfilacciate e nell'encefalomiopatia mitocondriale, determinano l'alterazione della sintesi di molte proteine mitocondriali differenti e di conseguenza alterazioni nella sintesi di ATP. Queste mutazioni possono essere fatali in età giovanile, ma alcune fanno sentire i loro effetti solo in età avanzata: il 5% degli europei che nella vecchiaia manifesta il morbo di Alzheimer presenta mutazioni nel gene che codifica RNAt per la glutammina. Nella sindrome di Kearns-Sayre sono state descritte invece ampie delezioni del DNA mitocondriale localizzate in posizioni diverse. Tutte le delezioni coinvolgono diversi geni mitocondriali, ma l'unica caratteristica comune è che non sono mai delete le origini di replicazione del DNA. La maggior parte dei casi di malattie umane associate ad ampie delezioni del DNA mitocondriale è sporadica e non ereditaria. Presumibilmente infatti le delezioni si verificano in tempi e localizzazioni casuali durante lo sviluppo, con conseguente variabilità per quello che riguarda i tessuti affetti, l'età di insorgenza dei sintomi e la loro gravità. È invece altamente improbabile che si verifichino nella cellula uovo in quanto, con le successive replicazioni cellulari, si formerebbe una quantità di DNAmt così danneggiata da causare la morte della cellula. Si sono trovate delezioni del DNA mitocondriale anche in individui morti per varie cause, tra cui il morbo di Parkinson. Numerosi studi inoltre suggeriscono che le mutazioni che si accumulano con l'età nel DNAmt della muscolatura scheletrica o cardiaca, della cute e di altri tessuti costituiscano fattori chiave nel processo dell'invecchiamento.
bibliografia
R. Knippers, Molekulare Genetik, Stuttgart 1971, 1997⁷ (trad. it. Bologna 1998).
T. Maniatis, E. Fritsch, J. Lauer et al., The molecular genetics in human hemoglobins, in Annual review of genetics, 1980, pp. 145-78.
D.J. Weatherall, The new genetics and clinical practice, London 1982, 1985² (trad. it. Bologna 1990).
B. Alberts, D. Bray, J. Lewis et al., Molecular biology of the cell, New York 1983 (trad. it. Bologna 1983, 1995³).
A.C. Wilson et al., Polymorphic sites and the mechanism in human mitochondrial DNA, in Genetics, 1984, 106, pp. 479-99.
R.D. Nicholls, J.H. Knoll, M.G. Butler et al., Genetic imprinting suggested by maternal heterodisomy in non deletion Prader-Willi syndrome, in Nature, 1989, 342, pp. 281-85.
A. Efstratiadis, Parental imprinting of autosomal mammalian genes, in Current opinion in genetics & development, 1994, 4, pp. 265-80.
B.D. Hendrich, F.W. Huntington, Epigenetic regulation of gene expression: the effect of altered chromatin structure from yeast to mammals, in Human molecular genetics, 1995, 4, pp. 1765-77.
U. Albrecht, J.S. Sutcliffe, B.M. Cattanach et al., Imprinted expression of the murine Angelman syndrome gene Ube3a, in hippocampal and Purkinje neurons, in Nature genetics, 1997, 17, pp. 75-78.
M. Lalande, Parental imprinting and human desease, in Annual review of genetics 1997, pp. 73-95.
D.C. Wallace, Il ruolo del DNA mitocondriale nelle malattie degenerative, in Le Scienze, 1998, 353, pp. 40-48.