
Genetica
Genetica
sommario: 1. Introduzione. 2. Il gene oggi. a) Sviluppo delle conoscenze. b) Ricerche sperimentali. c) Struttura e funzione del gene. d) Scoperte recenti. 3. Genetica parasessuale nei Mammiferi. a) Ibridazione cellulare somatica. b) Applicazioni dell'ibridazione alla biologia e alla medicina. c) Progressi tecnici recenti. 4. Una nuova classe di caratteri mendeliani: i frammenti di restrizione. a) Scoperta e utilizzazione di nuove varianti ereditarie. b) Attuale potenziale analitico del mendelismo. 5. Mappa molecolare del genoma umano e medicina preventiva. a) Sviluppo delle conoscenze. b) Mappa genetica totale del genoma umano. c) studi sul linkage umano e medicina preventiva. □ Bibliografia.
1. Introduzione
Ciò che ha sempre distinto la genetica dalle altre discipline biologiche sperimentali - dagli esperimenti dell'abate O. Mendel alla doppia elica di J. D. Watson e F. H. C. Crick - è stato il suo prescindere dallo studio delle strutture per dedicarsi all'analisi delle unità ereditabili, i geni, definiti in termini operativi piuttosto che meccanicistici. Per usare un'analogia corrente, è come dire che la genetica si è sempre interessata dei modelli di pianificazione (i blueprints) delle macchine viventi, lasciando la descrizione minuta delle parti e del loro assemblaggio all'anatomia, alla fisiologia e alla biochimica. Con la scoperta della struttura del DNA questo dualismo nella ricerca biologica sperimentale ha cessato di esistere. Il formidabile insieme di concetti derivati induttivamente dall'analisi genetica e il bagaglio sempre più dettagliato delle conoscenze biochimiche e fisiologiche sulle strutture cellulari e subcellulari hanno trovato il giusto punto d'incontro nell'analisi biochimica e molecolare dell'unità fondamentale dell'eredità, il gene. La descrizione della struttura del DNA segna dunque il superamento del dualismo genetica-biochimica e la nascita della disciplina cui è stato imposto il nome di ‛biologia molecolare'; questa, a seconda di come la si consideri, può essere definita come una rivitalizzazione della biochimica in termini genetici o della genetica in termini biochimici.
Com'è noto, nei vent'anni che sono succeduti alla descrizione della doppia elica da parte di Watson e Crick e alla formulazione del ‛dogma centrale' (DNA → RNA → proteina) l'arsenale del biologo si è arricchito di nuovi formidabili mezzi d'indagine, tra i quali i più significativi sono la scoperta di speciali proteine enzimatiche capaci di duplicare il DNA (DNA-polimerasi), di realizzare la trascrizione del messaggio genetico dal DNA all'RNA (RNA-polimerasi) e talvolta dall'RNA al DNA (transcriptasi inversa), di tagliare il DNA in minuti frammenti (endonucleasi di restrizione) e di ricucire insieme i frammenti di DNA separati (ligasi). Il lettore troverà dettagli su questi meccanismi molecolari nell'articolo biologia molecolare. Qui ci preme sottolineare che la disponibilità di queste speciali proteine enzimatiche è stata la condizione essenziale per lo sviluppo delle tecnologie del DNA ricombinante, ovvero per l'isolamento fisico del gene e il suo trasferimento da un organismo a un altro, spesso di specie, ordine o classe diversa. In effetti, il trasferimento più semplice è proprio quello dai Mammiferi ai Virus e ai Batteri, che, una volta modificati, permettono di replicare in laboratorio quantità illimitate del gene desiderato e del prodotto finale da esso controllato.
Le applicazioni industriali e mediche di queste tecnologie sono ormai argomento di cronaca nei quotidiani a grande tiratura, ma l'impatto che esse hanno avuto nella ricerca genetica può forse sfuggire ai non addetti ai lavori. Da Mendel ai nostri giorni i geni sono stati un'invenzione necessaria per spiegare l'esistenza di differenze alternative ed ereditabili osservate tra individui della stessa specie. D'ora in avanti diventerà invece sempre più frequente porsi il problema opposto: l'andare alla ricerca di una funzione specifica per ciascuna delle innumerevoli sequenze di DNA che si possono fisicamente isolare dagli organismi oggetto di studio. Questa sorta di ricerca genetica ‛a ritroso' sarebbe a dir poco arida negli organismi superiori, se non si fosse di recente scoperto che fenomeni parasessuali come la trasformazione, la trasfezione e la fusione di cellule somatiche (un tempo ritenuti appannaggio esclusivo dei microrganismi) possono essere indotti facilmente anche negli organismi superiori, Mammiferi inclusi, e utilizzati con successo per un'analisi genetica senza fecondazione sessuale.
Tuttavia andrebbe errato chi concludesse da tutto ciò che le strategie sperimentali della genetica mendeliana classica abbiano fatto il loro tempo. In realtà sta accadendo esattamente il contrario, perché il boom della biologia molecolare ha portato all'attenzione di ogni neofita quelle problematiche genetiche classiche che erano rimaste confinate entro la cerchia degli specialisti fino a pochi anni or sono, come la costruzione di mappe cromosomiche e genetiche (v. genetica e genetica: Citogenetica).
2. Il gene oggi
a) Sviluppo delle conoscenze
L'inizio degli anni quaranta segna la nascita della genetica molecolare e registra i primi passi sulla via dell'identificazione fisica del gene. Il certificato di nascita della genetica molecolare può essere considerato la prova data da O. T. Avery, M. McCarty e C. M. MacLeod (1944), e confermata da A. D. Hershey e M. Chase (1952), che il DNA è il vettore dell'eredità biologica. Nello stesso decennio G. W. Beadle ed E. L. Tatum (1941) riaffermano, in seguito ai loro studi su Neurospora, la validità dell'ipotesi ‟un gene - un enzima" (che il medico inglese A. Garrod aveva formulato nel 1908 per spiegare l'eredità degli errori congeniti del metabolismo nell'uomo), mentre J. Lederberg e Tatum (1946) e, in Italia, L. Cavalli Sforza dimostrano la sessualità (0vvero lo scambio di informazione genetica) anche nei Batteri, da sempre ritenuti l'esempio classico della riproduzione asessuata (v. genetica, cap. 4, §§ c-e).
Nei vent'anni successivi biologi molecolari e genetisti, lavorando principalmente su batteri e fagi, hanno imparato come l'informazione genetica sia immagazzinata e replicata nel DNA e hanno cominciato a capire in dettaglio i meccanismi molecolari che permettono la sintesi del prodotto finale dei geni: le proteine che caratterizzano la struttura delle singole cellule e degli organismi. Divenne così chiaro che, nei Batteri, un segmento ininterrotto di DNA rappresenta sempre un gene strutturale, che racchiude l'informazione genetica necessaria per la sintesi di una singola catena polipeptidica. Ciò vuol dire che, nei Batteri, a una sequenza lineare di nucleotidi nel DNA corrisponde sempre una sequenza lineare di amminoacidi, nella proteina finale, sintetizzati secondo le note regole del codice genetico (v. biologia molecolare; v. gene; v. genetica). Sulle prime si è pensato che questo fenomeno di colinearità fosse comune a tutti gli organismi, ma negli ultimi dieci anni si è scoperto che questa conclusione non è del tutto vera. Si sapeva da tempo che le cellule degli organismi superiori contengono una quantità di DNA di gran lunga maggiore di quella contenuta nell'organismo unicellulare dei Batteri. Nell'uomo, per esempio, ci sono circa 3 × 109 coppie di nucleotidi per nucleo cellulare aploide distribuiti su un insieme di 23 cromosomi diversi. Pertanto, se tutto questo DNA fosse preposto, come nei Batteri, al controllo della sintesi di proteine, ci si attenderebbe che il DNA presente in una cellula umana fosse capace di codificare, potenzialmente, circa tre milioni di proteine diverse, assumendo che un gene comprenda, in media, un migliaio di nucleotidi. Si è stimato invece che il numero totale di proteine diverse nei Mammiferi è compreso fra 30.000 e 150.000.
Per un certo tempo è sembrato che questa discrepanza fosse dovuta esclusivamente alla presenza di sequenze di DNA ripetitivo, detto anche ‛DNA egoista' perché non viene trascritto, quasi fosse un riempitivo per separare un gene strutturale dall'altro. Negli ultimissimi anni si è invece scoperto che il DNA egoista non è la sola differenza fondamentale tra le macchine genetiche degli organismi eucarioti e procarioti. Per esempio, si sapeva da tempo che le molecole di RNA direttamente prodotte attraverso la ‛trascrizione' dell'informazione genetica codificata nel DNA vengono modificate nel nucleo prima di essere trasferite nel citoplasma per la ‛traduzione' del messaggio genetico nelle corrispondenti proteine. Tuttavia si pensava che questa modificazione consistesse essenzialmente nell'accorciamento della molecola di RNA nucleare che veniva troncata nella sua porzione terminale per dar luogo alla formazione dell'RNA messaggero maturo. Le nostre cognizioni su questo interessante problema di biologia molecolare del gene sono rimaste nebulose fino a che non è stato possibile affrontare direttamente l'analisi molecolare di geni fisicamente isolati dall'insieme del genoma o sintetizzati in laboratorio partendo dall'RNA messaggero da essi prodotto. Questo tipo di studi è stato reso possibile dalla scoperta degli enzimi di restrizione (Arber, 1962; Nathans e Smith, 1965) e della ligasi (Gellert, 1967) e dalle tecniche di clonaggio del DNA sviluppate nei laboratori di Boyer, Cohen e Berg (1972-1973). Per dettagli esaurienti su questi importanti argomenti si rinvia il lettore all'aggiornamento dell'articolo biologia molecolare. Qui ci preme sottolineare che l'isolamento fisico dei geni, effettuato con le tecniche del DNA ricombinante, e la loro minuta analisi strutturale, resa possibile dall'impiego degli enzimi di restrizione, hanno chiaramente dimostrato che l'organizzazione del gene nei Mammiferi, Uccelli e Anfibi è fondamentalmente diversa da quella dei Batteri, in quanto la maggior parte dei geni eucariotici contiene delle interruzioni note agli specialisti col nome di ‛introni' o ‛sequenze intercalate'.
b) Ricerche sperimentali
Per dare un'idea delle strategie sperimentali usate per arrivare a queste conclusioni è utile descrivere in dettaglio l'elegante lavoro condotto sulla sintesi dell'ovalbumina del pollo dal gruppo di P. Chambon nel Laboratorio di genetica molecolare degli eucarioti di Strasburgo. L'ovalbumina è una proteina, costituita da 386 amminoacidi, che è sintetizzata dalle cellule ghiandolari dell'ovidotto della gallina al momento della deposizione delle uova. Il processo è controllato dagli ormoni sessuali femminili, in assenza dei quali il gene non è trascritto e la proteina non è sintetizzata.
Per comprendere il funzionamento di questo processo regolativo era necessario isolare il gene dell'ovalbumina dalle cellule che la producono e compararlo con lo stesso gene derivato da altri tessuti che non la producono. Approfittando del fatto che l'RNA messaggero dell'ovalbumina rappresenta il 50% dell'RNA prodotto dalle cellule dell'ovidotto della gallina, non fu difficile purificarlo in quantità adeguata. Si poté quindi stabilire che la molecola di RNA consta di una catena di 1.872 nucleotidi dei quali 1.158 includono l'informazione genetica necessaria per la sintesi dei 386 amminoacidi che compongono la proteina. A questo punto l'RNA messaggero dell'ovalbumina fu convertito nella copia complementare di DNA attraverso la transcriptasi inversa (un enzima virale che è capace di trasferire l'informazione genetica dall'RNA al DNA) e successivamente il DNA a singola catena fu convertito in DNA a doppia catena per mezzo dell'enzima DNA-polimerasi. Questo gene artificiale creato in laboratorio fu poi inserito in un plasmide (un vettore di derivazione batterica) e quindi clonato in E. coli per ottenere DNA in quantità sufficiente per studiarne la struttura con l'aiuto degli enzimi di restrizione. Il passo successivo consistette nel comparare la struttura del DNA derivato in laboratorio con quella del gene dell'ovalbumina isolato dalle cellule del pollo (DNA genomico). Fu così che apparve evidente la fondamentale differenza tra i due DNA, nel senso che il DNA genomico era molto più lungo di quello derivato in laboratorio.
L'esperimento dimostrava conclusivamente che una gran parte del DNA che costituisce il gene dell'ovalbumina non partecipa alla costruzione del prodotto finale del gene ovvero alla sintesi dell'ovalbumina. Le porzioni di DNA che non sono rappresentate nel prodotto finale sono appunto gli ‛introni', mentre quelle che vengono trascritte e tradotte sono gli ‛esoni'.
Dettagliate ricerche sull'argomento hanno consentito di stabilire che il gene dell'ovalbumina è costituito da 7.700 basi, con 7 introni inframmezzati agli esoni. In effetti la molecola di RNA inizialmente prodotta ha una dimensione analoga a quella del gene, ma per tappe successive (eliminazione prima di 5 introni e successivamente di altri 2) l'RNA messaggero si riduce fortemente, tanto che quello maturo consta solo di 1.872 nucleotidi. Questo processo è noto con il nome di splicing dell'RNA.
c) Struttura e funzione del gene
Si sa tuttora molto poco sul perché di questa differenza molecolare fra procarioti ed eucarioti. Che cosa sono gli introni? Geni nei geni? Sequenze di DNA parassita di origine esogena? Il prodotto di una ricombinazione genetica sbagliata?
Numerosi evoluzionisti pensano che quest'ultimo meccanismo abbia avuto un ruolo preminente nell'evoluzione degli introni. Essi suppongono che la maggior parte dei geni degli organismi superiori si sia evoluta attraverso la duplicazione di pochi geni ancestrali e che la maggior parte delle proteine oggi note sia derivata dalla duplicazione e dall'evoluzione dei pochi geni che inizialmente codificavano le poche proteine primordiali. In effetti non sono pochi gli esempi di famiglie multigeniche. Il caso delle globine alfa e beta è un esempio ben noto. Lo stesso dicasi dei geni delle catene pesanti delle immunoglobuline, la cui genetica molecolare costituisce uno dei più bei capitoli della biologia moderna.
Altri autori (J. Damell e W. F. Doolittle) pensano che la macchina per lo splicing dell'RNA esistesse in effetti prima della comparsa degli eucarioti, ma che sia stata perduta nel corso dell'evoluzione da parte dei procarioti oggi esistenti. Secondo questa teoria i procarioti non sarebbero quindi gli antenati degli eucarioti, ma i discendenti di organismi che hanno gradualmente eliminato dal loro genoma il DNA non codificante. Nelle cellule eucariotiche, dall'altra parte, il macchinario per lo splicing dell'RNA si è andato raffinando per generare nuove funzioni da funzioni vecchie, creando via via organismi più adatti all'ecosfera contemporanea.
Se l'evoluzione procede attraverso manipolazioni casuali del genoma, dice F. Jacob, il meccanismo dello splicing dell'RNA è il mezzo ideale per realizzarla. Con la riscoperta delle unità genetiche mobili (oggi chiamate ‛transposoni', ma in effetti identificate da oltre quarant'anni nel mais da Barbara McClintock, che le considerò alla stregua di mutazioni instabili con funzioni di controllo genico) viene fatto naturale di pensare che gli introni siano catapultati all'interno di geni strutturali proprio come dei transposoni per assumervi probabilmente una funzione regolativa (v. gene). Con questa prospettiva in mente, taluni ritengono che la presenza di introni in un gene strutturale aumenti la velocità con la quale sequenze codificanti possono ricombinare, generando in tal modo una maggiore variabilità genetica e quindi aumentando la potenzialità del genoma all'adattamento biologico.
Se le sequenze introniche non avessero un significato biologico positivo - sottolineano i sostenitori di questa teoria - perché mai esse si sarebbero conservate così stabili nel corso dell'evoluzione? La risposta più semplice - obbiettano gli increduli - potrebbe essere appunto che gli introni siano dei tratti di DNA senza significato, che la cellula degli eucarioti non ha ancora imparato a eliminare.
d) Scoperte recenti
Non si può concludere un sia pur breve aggiornamento sulla struttura e sulla funzione dei geni senza far riferimento a due delle più importanti scoperte dell'ultimo decennio: i mutanti omeotici e i protoncogeni (questi ultimi devono il loro nome alla ben nota analogia molecolare con i retrovirus oncogeni).
I protoncogeni esistono in tutti i Vertebrati, con una sorprendente omologia nelle sequenze nucleotidiche che li compongono, e recentemente è stato dimostrato che alcuni di essi codificano per proteine che controllano la crescita di particolari tipi cellulari (fattori di crescita) e che vengono prodotte - come c'era da attendersi - in piccole quantità o in eccesso a seconda che si tratti di tessuto normale o tumorale. Tra queste proteine specializzate vi sono il fattore di crescita delle piastrine (PDGF), analogo al prodotto dell'oncogene sis, e il fattore di crescita dei tessuti ectodermici (EGF), analogo all'oncogene erb. A tutt'oggi sono state individuate oltre una ventina di sequenze nucleotidiche che corrispondono alla definizione di protoncogene per via della loro omologia molecolare con un oncogene virale. Ognuna di queste sequenze ha una localizzazione cromosomica precisa, che è spesso molto prossima, quando non sovrapponibile, alla posizione di siti cromosomici definiti ‛fragili' per la facilità con cui vanno incontro a rotture cromatidiche indotte da sostanze clastogene. Le circostanze descritte e le correlazioni trovate finora tra certi tipi di tumori e il riordinamento cromosomico o strutturale di specifici protoncogeni (per esempio la traslocazione del protoncogene c-myc dal cromosoma 8 al 14 nel linfoma di Burkitt o dell'abl dal cromosoma 9 al 22 nella leucemia mieloide cronica) sono alla base dell'ipotesi che la trasformazione cellulare abbia inizio con un evento molecolare abnorme verificatosi in un protoncogene (v. Croce e Klein, 1985). Resta tuttavia ancora da stabilire se queste correlazioni siano espressione di eventi del tutto anormali o non piuttosto la deviazione patologica di un ruolo importante che i protoncogeni potrebbero avere nel normale meccanismo del differenziamento ontogenico. Se ciò fosse vero, sarebbe più appropriato parlare di ‛ontogeni'.
I mutanti omeotici sono quelli in cui una parte dell'organismo assume le sembianze di un'altra parte in conseguenza di una singola mutazione. Classico esempio di questo tipo di mutazioni è la mutazione dominante antennapoedia (Ant), in Drosophila melanogaster, che trasforma le antenne in arti. A tutt'oggi sono state individuate diverse mutazioni omeotiche che si raggruppano nel complesso antennapoedia (Ant-C) e nel complesso bi-thorax (BX-C). Entro questi due complessi omeotici i loci Antp e Ubx controllano la morfogenesi di specifici segmenti, mentre un terzo locus, il locus ftz, ne controlla il numero. Una scoperta inattesa è che tutti questi geni omeotici contengono una sequenza nucleotidica di circa 180 basi, che è presente in almeno sette copie nel genoma di Drosophila e che è stata ritrovata in un ampio intervallo di specie di Invertebrati e Vertebrati (topo e uomo compresi) con una omologia sorprendente. Queste sequenze di DNA, cui si è dato il nome di ‛scatole omeotiche' o homeo-boxes, sembrano essere assenti nelle specie animali a organizzazione non metamerica, il che sembra suggerire un loro preminente ruolo nel programma di sviluppo degli organismi a struttura metamerica. Usando dei probes (o sonde) molecolari derivati da homeo-boxes di Drosophila sono state di recente identificate almeno due homeo-boxes nel topo (localizzate sul cromosoma 6) e nell'uomo (localizzate sul cromosoma 17), con una percentuale di omologia del 75% al livello delle sequenze nucleotidiche e di oltre l'85% al livello della struttura primaria delle proteine da esse codificate. La dimostrazione ottenuta in Drosophila dell'associazione diretta tra homeo-boxes e i complessi di geni omeotici autorizza a speculare sulla possibilità di un loro analogo ruolo nello sviluppo dei vertebrati superiori a organizzazione metamerica.
3. Genetica parasessuale nei Mammiferi
a) Ibridazione cellulare somatica
Negli ultimi trentacinque anni si sono scoperti meccanismi diversi dalla riproduzione sessuata, ma come questa capaci di trasferire l'informazione genetica da cellule o organismi parentali a cellule o organismi filiali ibridi. Questi meccanismi, che vanno dalla trasformazione e dalla trasduzione nei Batteri e nei Virus ai cicli parasessuali dei Funghi procanoti ed eucarioti, sono serviti come modello per lo sviluppo di una genetica delle cellule somatiche nei Mammiferi e in particolare nell'uomo. La produzione di ibridi cellulari somatici attraverso la fusione spontanea o indotta di cellule somatiche derivate da organismi di specie, ordini e persino classi diverse è un'acquisizione degli anni sessanta. Nel decennio successivo quest'approccio sperimentale è stato utilizzato con particolare successo per la costruzione di mappe cromosomiche dell'uomo e di altri mammiferi, che non si prestano alla sperimentazione genetica classica. Com'è noto (v. genetica: Applicazioni della genetica), la mappatura cromosomica con questo approccio si basa sulla circostanza che il genoma di cellule ibride interspecifiche si riduce rapidamente nel corso delle prime divisioni cellulari con la perdita progressiva e preferenziale dei cromosomi di una soltanto delle due specie parentali. Gli ibridi tra cellule di Roditori (topo, ratto e criceto) e cellule umane perdono di solito i cromosomi umani, permettendo così di dedurre i gruppi di linkage (i geni dello stesso cromosoma) dalla correlazione positiva tra la ritenzione (e naturalmente la perdita) di particolari cromosomi e marcatori genetici umani. Va notato che - a differenza dalla sperimentazione genetica classica - questa metodologia parasessuale permette di mappare geni anche in assenza di varianti individuali nell'ambito della specie, in quanto, in conseguenza della distanza evolutiva tra le cellule parentali, gli ibridi interspecifici sono in effetti degli eterozigoti multipli per tutti i marcatori genetici che siano espressi nelle cellule coltivate in vitro. Inoltre essa permette di scoprire facilmente l'appartenenza allo stesso gruppo di linkage, anche per loci che alla meiosi segregano indipendentemente l'uno dall'altro per essere separati da una frequenza di crossing over del 50%. È stato proprio per quest'ultima prerogativa degli ibridi topo/uomo che si è creato il termine di ‛sintenia', un neologismo derivato dal greco per dire ‛sullo stesso nastro'. Ovviamente geni linked sono sempre ‛sintenici', mentre il contrario non è necessariamente vero. È comprensibile come le circostanze appena descritte facciano degli ibridi cellulari somatici interspecifici il mezzo elettivo d'indagine genetica - anche per le specie animali che si prestano alla sperimentazione genetica tradizionale - quando si vogliano mappare geni che non sono polimorfi o che si trovano a distanza genetica non misurabile.
b) Applicazioni dell'ibridazione alla biologia e alla medicina
La costruzione di mappe cromosomiche (di quella umana e della sua importanza in medicina si dirà più avanti: v. cap. 5) è senza dubbio la più nota ma certamente non l'unica nè la più significativa applicazione dell'ibridazione cellulare somatica alla biologia e alla medicina. In effetti i pionieri di questa metodologia sperimentale avevano piuttosto visto in essa l'occasione propizia per estendere ai Mammiferi, e in particolare all'uomo, quegli stessi test di complementazione genetica che avevano permesso di acquisire dati fondamentali sulla struttura fine e sul meccanismo d'azione dei geni nei microrganismi. Dalla fusione di cellule altamente differenziate con cellule indifferenziate B. Ephrussi (1973) derivò il concetto di ‛funzioni cellulari di lusso' (luxury functions) e di ‛funzioni ordinarie' (household functions); nel senso che queste ultime sono parte integrante del metabolismo cellulare in tutti i tipi di cellule e rimangono tali negli ibridi da essi derivati, mentre le prime sono il prodotto di cellule specializzate e vengono di solito soppresse dall'ibridazione con cellule indifferenziate o con cellule differenziate per una funzione di lusso di tipo diverso. È con l'uso di queste strategie sperimentali che nell'ultimo decennio sono stati dimostrati il comportamento recessivo della trasformazione maligna (Harris e Klein), l'esistenza di almeno cinque gruppi di complementazione per le mutazioni recessive responsabili dello xeroderma pigmentosum (Bootsma e altri), l'eterogeneità del fenomeno di scambio tra cromatidi fratelli, la possibilità di indurre l'espressione di funzioni di lusso (come la sintesi dell'albumina e delle aptoglobine umane) in ibridi interspecifici ottenuti dalla fusione di cellule di epatoma murino con fibroblasti o linfociti umani (Darlington e Ruddle) e, per chiudere l'elenco con l'evento più importante, la produzione illimitata di anticorpi monoclonali in ibridi intraspecifici derivati dalla fusione di cellule di mieloma murino e splenociti di topo immunizzato con l'antigene di scelta (Koler e Milstein, 1975). Quest'ultima applicazione della genetica parasessuale è stata giustamente premiata con l'attribuzione del premio Nobel ai suoi ideatori, per i contributi che la produzione di anticorpi monoclonali ha già arrecato, e promette di arrecare, alla ricerca biologica di base e alla medicina. Basti citare, ad esempio: a) il ruolo che gli anticorpi monoclonali hanno nell'armamentario del biologo molecolare quando si tratta di isolare dai poliribosomi l'RNA messaggero della proteina antigenica nascente da esso codificata; b) le innumerevoli applicazioni diagnostiche e le promettenti applicazioni terapeutiche riportate ormai quasi quotidianamente nella letteratura medica internazionale.
c) Progressi tecnici recenti
Negli ultimissimi anni la lista dei fenomeni parasessuali nelle cellule dei Mammiferi si è rapidamente accresciuta grazie a una serie di importanti eventi e progressi tecnici come: a) la possibilità di trasferire nelle cellule murine singoli cromosomi o frammenti di cromosomi isolati per arresto mitotico (McBride e Ozer, 1973; Willecke e Ruddle, 1975) o per micronucleazione (Ege e Ringertz, 1974; Fournier e Ruddle, 1977); b) l'estensione alle cellule eucariote (Bacchetti e Graham, 1977; Wigler e altri, 1977; Lin e altri, 1983) dei fenomeni di trasformazione o cotrasformazione da DNA (che per lungo tempo erano parsi appannaggio esclusivo degli organismi procarioti); c) la trasfezione di sequenze oncogeniche da cellule tumorali a cellule normali (R. A. Weinberg, 1984); d) il trasferimento di sequenze geniche di scelta da un tipo cellulare a un altro con l'uso di vettori virali opportunamente modificati (Gluzman e altri, 1984; Mulligan e altri, 1984).
4. Una nuova classe di caratteri mendeliani: i frammenti di restrizione
a) Scoperta e utilizzazione di nuove varianti ereditarie
L'applicazione delle tecniche del DNA ricombinante e degli enzimi di restrizione (v. tab. I) allo studio della struttura molecolare fine dei geni ha portato alla scoperta inattesa di una nuova e numerosa classe di varianti genetiche che si comportano come caratteri mendeliani classici sia nelle famiglie che nelle popolazioni. A scoperchiare il vaso di Pandora furono il dottor J. Sambrook e i suoi colleghi del Cold Spring Harbor Laboratory (1975), che utilizzarono per la prima volta questo nuovo tipo di varianti ereditarie per mappare, in Adenovirus, la posizione reciproca di due mutanti già noti e comparare la distanza genetica e la distanza fisica tra i loci corrispondenti in termini di frequenza di crossing over e, rispettivamente, del numero di nucleotidi interposti. Queste varianti sono note con l'acronimo RFLV (restriction fragment length variants, ‛varianti della lunghezza dei frammenti di restrizione'), mentre la coesistenza di più varianti dello stesso locus in una popolazione mendeliana rappresenta un classico esempio di polimorfismo genico o RFLP (restriction fragment length polymorphism, 'polimorfismo della lunghezza dei frammenti di restrizione'). La potenzialità di questa nuova classe di varianti genetiche nella diagnostica medica fu prontamente compresa da Kan e Dozy (1978), quando si accorsero che nell'87% degli afro-americani originari dell'Africa occidentale il gene per la falcemia (il mutante βS) è costantemente associato a un frammento di restrizione Hpa I di 13 kb invece del comune frammento di 7,6 kb che include la sequenza di DNA omologa al gene della globina β. Kan e Dozy si avvidero che questo polimorfismo Hpa I è la conseguenza di un'alterazione della sequenza nucleotidica a un sito di restrizione Hpa I che fiancheggia il gene strutturale della globina β a circa 5.000 nucleotidi dalla sua estremità 3′-terminale e quindi non ha nulla a che vedere con la mutazione falcemica. Ciò vuol dire che la summenzionata associazione tra le due mutazioni in Africa occidentale è evidentemente la conseguenza dell'estrema rarità con cui siti nucleotidici così vicini vanno incontro al fenomeno del crossing over, generando nel caso citato una distribuzione non casuale (linkage disequilibrium) tra le singole varianti Hpa I e i mutanti al locus β.

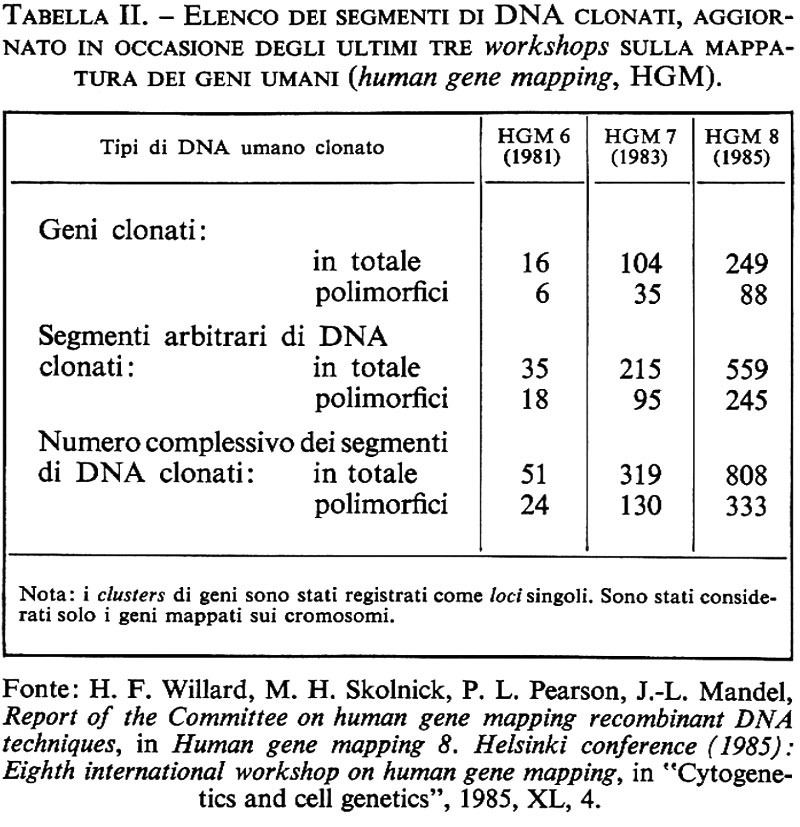
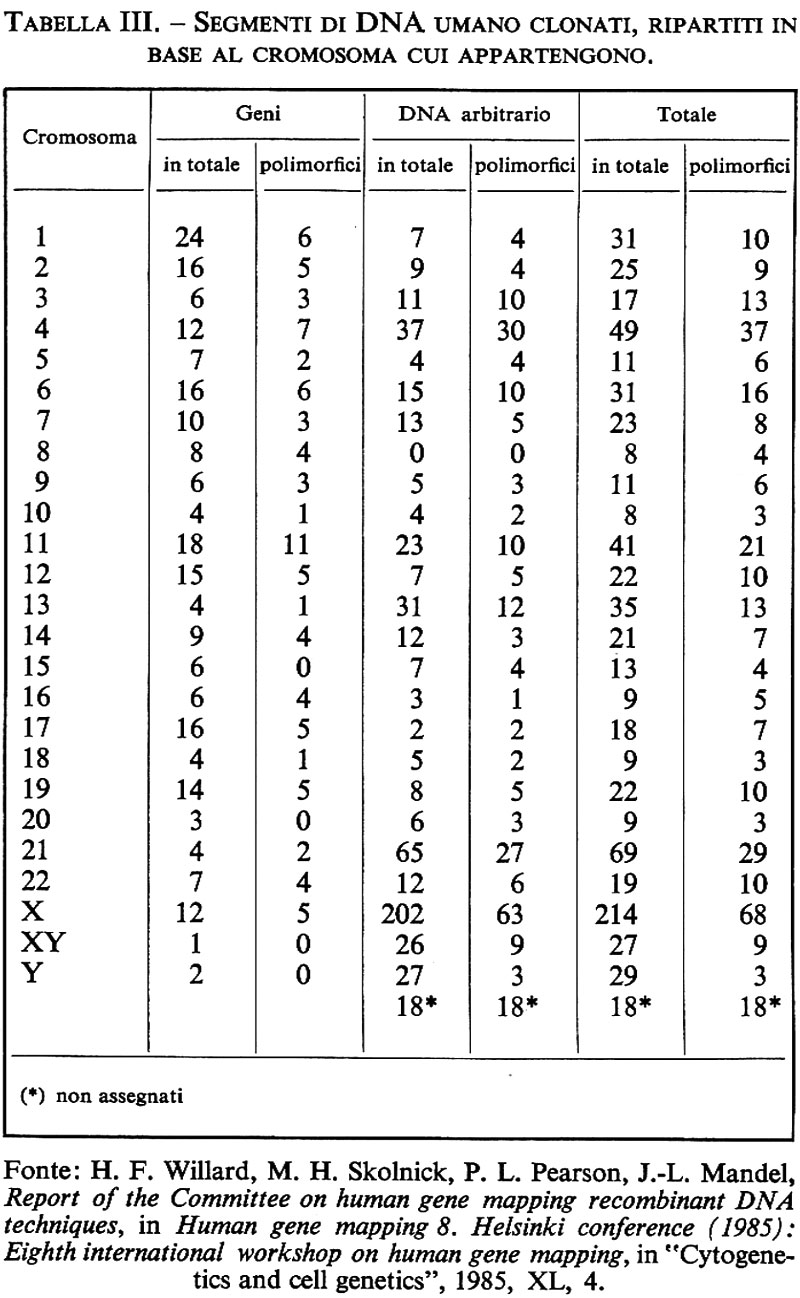
Nei pochi anni intercorsi dalla scoperta di questo primo esempio di RFLP umano comune, la lista dei polimorfismi mendeliani ai siti di restrizione è andata aumentando con una velocità imprevista, che tuttora non accenna a diminuire. È ormai ben stabilito che frammenti di DNA umano non ripetitivo (non importa che si tratti di geni isolati, di sequenze di cDNA ottenute per trascrizione inversa o semplicemente di sequenze genomiche arbitrarie) possono essere utilizzati come ‛sonde molecolari' per identificare RFLV con l'uso di uno o più enzimi di restrizione. Dai dati raccolti negli ultimi cinque anni sembra ormai chiaro che queste varianti genetiche ai siti di restrizione del DNA umano (la maggior parte delle quali non sono riconoscibili al livello di un prodotto genico finale) sono molto comuni nell'intero genoma e sono mantenute in una condizione di polimorfismo genetico in tutte le popolazioni finora esaminate. Tutto ciò suggerisce la conclusione che, al livello del DNA, il numero delle differenze ereditarie proprie della nostra specie ecceda probabilmente di molto il totale di 50.000 geni finora ritenuto il limite superiore del nostro carico genetico (genetic load). Vedremo subito quanto questa inattesa vendemmia di varianti mendeliane comuni abbia migliorato le prospettive di costruzione di una mappa totale del genoma umano e di una sua applicazione alla medicina diagnostica e preventiva. Per dare un'idea del ritmo con il quale procede l'accumulo di dati in questo campo basterà notare che all'epoca del sesto workshop sulla mappatura del genoma umano tenutosi a Oslo nell'estate del 1981 i geni umani donati erano 16 in tutto e i polimorfismi a siti di restrizione (RFLP) 24. Quattro anni dopo, all'ottavo workshop di Helsinki, il numero dei geni donati era già salito a 249 e quello degli RFLP a 333. Tra questi ultimi, 88 sono stati identificati con sonde molecolari contenenti delle sequenze geniche propriamente dette e 245 con sonde contenenti frammenti arbitrari di DNA genomico non ripetitivo (v. tab. II). Ormai non c'è cromosoma umano che non sia costellato da uno fino a diverse decine di polimorfismi ai siti di restrizione con localizzazione subregionale ben precisa (v. tab. III). I meccanismi molecolari che producono questo tipo di variazioni ereditarie sono presumibilmente gli stessi che danno luogo alla variabilità genetica in generale. Essi vanno pertanto dalle mutazioni puntiformi, con la sostituzione di una singola base nucleotidica, alla serie di più complesse modificazioni del DNA che possono derivare da un evento di crossing over impreciso, da una conversione genica o dall'inserzione/delezione di un transposone (v. biologia molecolare, suppl.). Il risultato finale è sempre lo stesso: la produzione di frammenti di restrizione di lunghezza diversa che vengono ereditati come caratteri mendeliani classici. Il già citato polimorfismo al sito di restrizione Hpa I nella regione che fiancheggia il lato 3′ del gene della globina β è probabilmente un esempio di variante del primo tipo. Il polimorfismo Dl4Sl trovato da Wyman e White (il primo a essere identificato con l'uso di un frammento arbitrario di DNA umano non ripetitivo) e quello trovato da Bell usando come sonda molecolare il gene dell'insulina sono invece esempi dell'ultimo tipo: in entrambi i casi, infatti, le singole varianti alleliche possono venire identificate con più di un enzima di restrizione, come ci si attende quando la variazione di lunghezza è appunto il risultato di una inserzione/delezione di un frammento di DNA tra due siti di restrizione inalterati. È interessante notare che finora i casi di RFLP più comuni e multiallelici (che sono ovviamente anche quelli che permettono di identificare il più elevato numero di eterozigoti) sono tutti dell'ultimo tipo e sono stati mappati in regioni genomiche che sembrano soggette a un'elevata frequenza di ricombinazione genetica e di riordinamenti cromosomici. Tale è il caso dei due ultimi esempi citati: l'RFLP Dl4Sl di Wyman e White, mappato nella regione 14q32 del cr0mosoma 14, che è anche il sito di rottura implicato in diversi casi di traslocazioni meiotiche o mitotiche (tra cui la più nota è quella associata al linfoma di Burkitt: v. neoplasie), e l'RFLP trovato da Bell con il gene dell'insulina, mappato nella regione 11p15 del cromosoma 11, contigua alla sede di delezioni e traslocazioni complesse associate al tumore di Wilms (v. neoplasie). Un terzo esempio dello stesso tipo è l'RFLP DXS52, recentemente identificato da J. L. Mandel con una sonda genomica arbitraria X-specifica, che, come i due precedenti, include numerosi alleli ed è stato mappato nella regione Xq28 in posizione molto prossima al cosiddetto ‛sito fragile' del cromosoma X.
b) Attuale potenziale analitico del mendelismo
È opinione generale che questa nuova classe di varianti ereditarie non abbia alcun significato adattativo, se non nel caso di quelle mutazioni che avvengono nell'ambito di sequenze nucleotidiche regolarmente trascritte, come la mutazione falcemica che altera la sequenza palindromica necessaria per la digestione con l'enzima Dde I. D'altra parte, la scoperta di questa enorme variabilità individuale, sia pure inespressa, ha accresciuto enormemente il potenziale analitico del mendelismo nella ricerca biologica di base e applicata. Per esempio, è intuitivo che le innumerevoli combinazioni aploidi identificabili ai diversi siti di restrizione polimorfici dei singoli cromosomi o di regioni cromosomiche (aplotipi di restrizione) offrono la possibilità di descrivere, con una minuziosità mai raggiunta prima, la costituzione genetica dei singoli doni cellulari, degli individui, della popolazioni e delle specie. Le applicazioni biologiche e mediche di questo nuovo bagaglio di conoscenze sul genoma umano sono già numerose: dagli studi molecolari sul meccanismo e sulla distribuzione della ricombinazione genetica, alla misura delle distanze genetiche intra- e interspecifiche, alla dimostrazione dell'evoluzione molecolare somatica che caratterizza importanti processi del differenziamento biologico normale e patologico, come la produzione di anticorpi e la trasformazione tumorale maligna. Tutte queste applicazioni sono condizionate da una premessa comune: la necessità di costruire una mappa molecolare totale del genoma umano. Le circostanze fin qui descritte suggeriscono che la realizzazione di tale obbiettivo non può essere lontana. Le ragioni che giustificano quest'ottimismo e le tanto discusse applicazioni della mappa genetica alla medicina diagnostica e preventiva sono descritte nel prossimo capitolo.
5. Mappa molecolare del genoma umano e medicina preventiva
a) Sviluppo delle conoscenze
La costruzione di una mappa genetica dei singoli cromosomi umani è proceduta a un ritmo esasperantemente lento dalla riscoperta delle leggi di Mendel fino a tempi molto recenti. Nel 1967 (l'anno della scoperta dell'ibrido cellulare instabile topo-uomo) gli esempi di linkage autosomico nell'uomo si potevano enumerare con le dita di una sola mano e per nessuno di essi si conosceva la localizzazione cromosomica. Alla stessa data gli esempi di caratteri legati al sesso (facilmente riconoscibili per via della loro caratteristica trasmissione da maschio a figlie femmine a nipoti maschi) superavano il centinaio, ma anche per essi mancavano dati precisi sulla localizzazione subregionale dei singoli geni e sulla loro distanza genetica misurata in base alla frequenza di scambio di materiale genetico tra i cromosomi omologhi nella regione che separa i loci a confronto.
I fattori principali di questo lento accumulo di dati sul linkage umano sono ben noti: la necessità di reperire nelle popolazioni naturali (invece che allestire in laboratorio come si fa in genetica sperimentale) gli incroci informativi, lo scarso numero di figli per famiglia, il lungo tempo di generazione, l'elevato numero di cromosomi proprio della specie e, soprattutto, la scarsezza di polimorfismi genici comuni e multiallelici che potessero servire da marcatori genetici per la ricerca sistematica di un rapporto di linkage sia tra di loro che con le mutazioni d'interesse medico.
Abbiamo già sottolineato quanto la scoperta dell'ibrido cellulare instabile del tipo topo-uomo e quella dei polimorfismi ai siti di restrizione abbiano improvvisamente cambiato la situazione, rendendo possibile la mappatura fisica dei geni (anche di quelli non polimorfici) per via sperimentale e fornendo quella profusione di marcatori mendeliani comuni e multiallelici che sono la condizione indispensabile per la costruzione di mappe genetiche.

La tab. IV riassume l'evoluzione delle conoscenze sul linkage umano dalla riscoperta delle leggi di Mendel al 10 dicembre 1985. Il vertiginoso aumento del numero dei geni mappati nel periodo successivo alla scoperta dell'ibrido topo-uomo (Weiss e Green, 1967) testimonia del ruolo determinante che quest'ultimo ha svolto nella costruzione delle mappe citogenetiche dei singoli cromosomi umani. Ad accelerare sensibilmente questo processo hanno contribuito altri importanti progressi e innovazioni metodologiche come: a) la capacità di riconoscere i singoli cromosomi umani e le loro alterazioni, con raffinate tecniche di bandeggio; b) la possibilità di stabilire direttamente la localizzazione di sequenze arbitrarie di DNA o di specifici geni - isolati con le tecniche del DNA ricombinante (v. biologia molecolare) - attraverso l'ibridazione molecolare in situ su cromosomi meta- o prometafasici; c) la disponibilità di anticorpi monoclonali per la selezione di ibridi ridotti con il cromosoma desiderato; d) in generale, l'insieme di quelle strategie sperimentali che permettono di accelerare la costruzione di ibridi cellulari somatici con una minima frazione di genoma umano, come i transgenoti di McBride e Ozer (1973) o gli ibridi microcellulari di Fournier e Ruddle (1977).
b) Mappa genetica totale del genoma umano
La base teorica che giustifica l'uso dei polimorfismi RFLP per la costruzione di una mappa genetica totale del genoma umano è stata discussa da Botstein e altri (v., 1980) subito dopo la descrizione del primo esempio di RFLP autosomico comune e multiallelico, scoperto - come si è già detto - da Wyman e White con l'uso di una sequenza arbitraria di DNA umano non ripetitivo derivata dalla ben nota prima libreria genomica umana costruita da Maniatis (v. biologia molecolare).
La lunghezza fisica del genoma aploide umano, misurata in nucleotidi, è di circa 3,5 miliardi di basi; la corrispondente lunghezza genetica, misurata in termini di ricombinazione o scambio tra cromosomi omologhi, è di 33 chiasmi (un chiasma = un crossing over) o, come si usa dire in linguaggio specialistico, di 3.300 centimorgan. Pertanto l'unità di ricombinazione genetica (1 centimorgan, ovvero l'1% di crossing over) sarebbe equivalente a una distanza fisica di un milione di nucleotidi: 3,5 × 109/3,3 × 103 ≃ 106 (Renwick, 1976). Tenendo per buone queste stime della lunghezza fisica e genetica del genoma umano totale e ricordando che la condizione per scoprire un linkage nei pedigrees umani è che la distanza genetica tra i loci sintenici non superi il 2o% di crossing over, si giunge alla conclusione che basterebbe saturare il genoma con un RFLP ogni 20 milioni di nucleotidi (ovvero con circa 200 RFLP, come si deduce dal rapporto 3,5 × 109/2 × 107 = 175), per riuscire a trovare sempre un linkage misurabile tra qualsivoglia caratteristica ereditaria normale o patologica e i due RFLP che fiancheggiano il locus corrispondente.
Naturalmente questa previsione di Botstein e colleghi si basa su una serie di premesse teoriche che potrebbero non essere interamente corrette. Per esempio, è possibile che la stima del numero totale di scambi meiotici per genoma, derivata dal numero dei chiasmi visibili, sia fortemente in difetto. Inoltre - come vedremo subito - sembra ormai accertato che anche nel genoma umano la frequenza di ricombinazione sia distribuita in modo tutt'altro che omogeneo. Queste due circostanze da sole fanno ritenere che il numero di RFLP necessari per la costruzione di una mappa totale del genoma umano debba essere in effetti di qualche ordine di grandezza più elevato di quello stimato. Inoltre è chiaro che, per essere utile, ciascun RFLP dovrebbe essere comune e multiallelico come gli RFLP identificati dalle sonde molecolari di Wyman e White (nel gergo degli specialisti il plasmide pAW101), di Bell (il gene donato dell'insulina) e di Mandel (il plasmide Stl4 derivato da una libreria genomica del cromosoma X) - perché ciò facilita il reperimento delle famighe più informative, che sono quelle che segregano per aplotipi di restrizione diversi ai loci sintenici dei quali si voglia misurare la distanza genetica.
c) Studi sul linkage umano e medicina preventiva
Il potenziale contributo degli studi sul linkage alla medicina diagnostica e preventiva era stato chiaramente percepito fin dal tempo della scoperta del primo linkage umano, tra emofilia e daltonismo, che Haldane e Smith (1947) avevano visto segregare nelle famiglie informative in modo non indipendente l'una dall'altro questo fatto offriva la possibilità di riconoscere le portatrici silenti del mutante recessivo letale attraverso la segregazione del gene per il daltonismo a esso strettamente linked. Per esempio, se era noto che una doppia eterozigote certa per l'emofilia e il daltonismo aveva ricevuto i due mutanti separatamente da ciascuno dei suoi genitori, si poteva escludere (con un rischio di errore pari alla frequenza di ricombinazione del 7%) la presenza del gene letale per l'emofilia in tutte le figlie che avessero già avuto un figlio daltonico (Haldane e Smith, 1947). Oggi sappiamo che esistono due diversi difetti della coagulazione del sangue, trasmessi con il meccanismo dell'eredità legata al sesso: quello relativo al fattore VIII (la cui deficienza caratterizza l'emofilia A) e quello relativo al fattore IX (emofilia B), che sono il risultato di mutazioni a loci separati. Corrispondentemente sappiamo dell'esistenza di due difetti della visione cromatica legati al sesso, il daltonismo deutan e il daltonismo protan, che sono anch'essi il risultato di mutazioni a loci distinti. Sappiamo inoltre che l'emofilia B segrega in maniera del tutto indipendente sia dall'emofilia A che dal daltonismo e che il linkage originariamente descritto da Haldane e Smith è in effetti un linkage molto più stretto (con frequenza di ricombinazione probabilmente inferiore all'1%) tra il locus per l'emofilia A e un cluster di quattro loci strettamente linked (noto come cluster G6PD), che include entrambi i loci del daltonismo, il locus per l'enzima glucosio-6-fosfato-deidrogenasi (G6PD) e quello per l'adrenoleucodistrofia (ALD). Queste circostanze sono state sfruttate per la classificazione delle portatrici eterozigoti del gene per l'emofilia A - e talora per la diagnosi prenatale dei pazienti - in quegli alberi genealogici che segregano allo stesso tempo per uno degli altri loci del cluster G6PD.
L. S. Penrose, direttore del Galton Laboratory di Londra quando questo era il quartier generale degli studiosi del linkage umano, non perdeva mai l'occasione per incoraggiare la ricerca di linkages tra malattie ereditarie recessive e polimorfismi genetici comuni, non soltanto per le applicazioni diagnostiche del tipo descritto sopra, ma anche per motivi di classificazione nosologica ovvero per dimostrare, con il linkage, l'eterogeneità genetica di sindromi ereditarie clinicamente simili. Classici esempi dell'uso del linkage a questo scopo sono la dimostrazione di due forme di ovalocitosi ereditaria attraverso il linkage stretto di una sola di esse con il fattore Rh e, molto recentemente, il riconoscimento di almeno due forme di ritardo mentale legato al sesso attraverso i loro diversi rapporti di linkage con i loci del cluster G6PD.
Le applicazioni mediche delle ricerche sul linkage sono rimaste per lungo tempo allo stadio di speculazione accademica, per via della bassa probabilità di reperire famiglie che segregassero per una determinata malattia ereditaria recessiva e, allo stesso tempo, per un marcatore genico comune strettamente linked a essa. In effetti, fino a poco tempo fa, l'unico esempio di linkage utilizzabile a scopo diagnostico era quello tra l'iperplasia surrenale congenita e il sistema HLA, in quanto quest'ultimo era il solo marcatore genico normale che segregasse in modo informativo in quasi tutte le famiglie.
L'inattesa scoperta dei polimorfismi RFLP a livello dei siti di azione degli enzimi di restrizione ha modificato enormemente la situazione, come si è detto nel capitolo precedente: si hanno ora fondate ragioni per ritenere che il reperimento di marcatori genici comuni e multiallelici, in qualsiasi regione del genoma umano, sia solo una questione di spesa e di qualche mese di lavoro intenso. Con questa idea in mente J. Gusella e i suoi collaboratori hanno ricercato e trovato nel giro di pochi mesi un linkage molto stretto tra la corea di Huntington (una malattia degenerativa del sistema nervoso centrale a insorgenza tardiva, determinata da un gene autosomico dominante) e un polimorfismo RFLP individuato con l'uso di una sequenza nucleotidica derivata a caso da una libreria genomica umana e successivamente mappata al braccio lungo del cromosoma 4. Usando strategie pressappoco analoghe sono stati di recente individuati nella regione subterminale del cromosoma X due RFLP comuni e multiallelici che segregano in una condizione di linkage quasi assoluto con l'emofilia A e presumibilmente con tutti gli altri loci del cluster G6PD. Naturalmente, quando questi polimorfismi a livello di siti di restrizione sono identificati con l'uso di geni (o frazioni di geni) donati in opportuni vettori, non c'è alcun dubbio che essi segreghino in un rapporto di linkage assoluto con i mutanti naturali degli stessi geni. La stessa cosa non si può dire per i casi di linkage tra malattie e RFLP identificati con sequenze arbitrarie di DNA genomico scelte a caso, sia pure seguendo il criterio della contiguità fisica (misurata con criteri citogenetici) tra la regione cromosomica omologa alla sonda molecolare e il locus genetico del mutante in esame. Per esempio, è noto che la distrofia muscolare legata al sesso di tipo Duchenne è il risultato di mutazioni al livello della regione intermedia del braccio corto del cromosoma X. In alcuni casi è stato possibile precisare che la mutazione consiste in una delezione minuta nella banda cromosomica Xp2l. L. Kunkel, del Massachusetts General Hospital, è riuscito a isolare delle sonde molecolari omologhe alla stessa regione deleta, con le quali egli ha poi identificato dei polimorfismi RFLP che, con sua profonda sorpresa, segregano in un rapporto di linkage tutt'altro che stretto con la mutazione Duchenne. Altro motivo di sorpresa è scaturito da recenti studi sui rapporti di linkage tra una serie di loci del braccio lungo dell'X distribuiti nella regione compresa tra la banda Xq26 e il telomero Xqter. Questa regione cromosomica è quella che include il cosiddetto sito fragile inducibile, Xq27.3, che è associato a una forma grave di ritardo mentale legato al sesso (la sindrome dell'X fragile). Dai dati sinora disponibili appare chiaro che geni localizzati al di sopra o al di sotto della regione Xq27.3 (per esempio il locus del fattore IX e quelli del cluster G6PD, che sono rispettivamente in posizione immediatamente prossimale e distale rispetto a essa) segregano indipendentemente nella discendenza di madri normali, ma mostrano un linkage misurabile in quella di madri eterozigoti per alcune delle mutazioni che determinano la sindrome dell'X fragile. L'insieme di queste osservazioni suggerisce l'ipotesi che la frequenza di ricombinazione meiotica tra loci dello stesso cromosoma sia determinata non soltanto dalla distanza fisica che li separa, ma anche dalla regione cromosomica nella quale essi sono localizzati e dal tipo di mutanti che si comparano. Se ciò fosse vero per tutto il genoma, com'è presumibile, gli RFLP richiesti per la costruzione di una mappa molecolare totale del genoma umano potrebbero essere molto più numerosi dei 200 previsti da Botstein e altri (v., 1980) e, in particolare, la mappatura di alcune regioni del genoma umano potrebbe richiedere l'impiego di strategie sperimentali diverse da quelle finora utilizzate.
Queste conclusioni raccomandano l'esercizio di una salutare prudenza nell'uso dei rapporti di linkage come criterio indiretto per la diagnosi dei portatori sani o malati di geni letali. Nonostante queste riserve, l'approccio molecolare alla diagnostica medica e alla medicina preventiva è ormai una realtà di fatto, come dimostra il già numeroso elenco di mutazioni per le quali è oggi possibile sia la diagnosi prenatale dello stato morboso che il riconoscimento degli eterozigoti sani.
BIBLIOGRAFIA
Botstein, D. e altri, Construction of a genetic map in man using restriction fragment length polymorphism, in ‟American journal of human genetics", 1980, XXXII, pp. 314-331.
Chambon, P., Geni discontinui, in ‟Le Sciene", 1981, XXVII, 155, pp. 16-27.
Croce, C., Klein, G., Traslocazioni cromosomiche e cancro umano, in ‟Le Scienze", 19885, XXXIV, 201, pp. 52-60.
Darnell, J. Jr., L'RNA, in ‟Le Scienze", 1985, XXXV, 208, pp. 40-51.
Doolittle, W. F., Sapienza, C., Selfish genes, the phenotype paradigm and genome evolution, in ‟Nature", 1980, CCLXXXIV, pp. 601-603.
Fedoroff, N.V., Elementi genetici trasponibili nel granturco, in ‟Le Scienze", 1984, XXXIII, 192, pp. 48-58.
Hunter, T., Le proteine degli oncogeni, in ‟Le Scienze", 1984, XXXIII, 194, pp. 56-71.
Kaufman, T. C., Abbott, M., Homeotic genes and the specification of segmental identity in the embryo and adult thorax of Drosophila melanogaster, in Molecular aspect of early development. Symposium of the American Society of Zoology, New York 1984.
Lewis, E. B., A gene complex controlling segmentation in Drosophila, in ‟Nature", 1978, CCLXXVI, pp. 565-570.
Mangiarotti, G., Dai geni agli organismi, Padova 1983.
Nicoletti, B., Chieffi, G., Baccetti, B. (a cura di), Biologia, Bologna 1984.
Siniscalco, M. e altri, Combination of old and new strategies for the molecular mapping of the human X-chromosome, in Human genetics. Part A. The unfolding genome, New York 1982, pp. 103-124.
Weinberg, R. A., Le molecole della vita, in ‟Le Scienze", 1985, XXXV, 208, pp. 16-26.