
HIV
HIV
Il 4 giugno 1981 lo scarno bollettino "Morbidity and Mortality Weekly Report" dei Centers for Disease Control di Atlanta segnalava un'inusuale serie di casi mortali di polmonite da Pneumocystis carinii, un agente normalmente innocuo, nella comunità di omosessuali maschi di Los Angeles. L'infezione mortale era associata a uno stato di profonda immunodeficienza caratterizzato soprattutto dalla scomparsa di una delle due classi principali di linfociti T identificabili dall'espressione alla loro superficie della proteina CD4. I linfociti CD4+ erano già noti per esercitare funzioni helper per altre cellule del sistema immunitario, quali i linfociti CD8+, specializzati nel riconoscere e uccidere cellule infettate da virus (CTL, Cytotoxic T lymphocyte) e i linfociti B, produttori di anticorpi (Ab). Tuttavia, la constatazione che, oltre alla popolazione omosessuale, sia tossicodipendenti eroinomani sia individui emofilici politrasfusi fossero colpiti da patologie analoghe ha rapidamente indirizzato a ipotizzare una causa infettiva e trasmissibile, per contatto sessuale e mediante trasfusione di sangue o emoderivati. Questo è l'inizio di venticinque anni di scoperte, progressi, ma anche delusioni nella sfida lanciata alla fine del secondo millennio da una nuova malattia: la sindrome da immunodeficienza acquisita o AIDS.
La causa di questa nuova patologia è stata individuata dopo un paio d'anni in un virus fino ad allora sconosciuto e ribattezzato HIV, ovvero 'virus dell'immunodeficienza (acquisita) umana'. Inoltre, non solo l'agente era sconosciuto, ma la sua natura di retrovirus, ovvero di un virus a RNA capace sia di produrre una copia di DNA uguale a sé stesso sia d'integrarsi in maniera permanente nel genoma delle cellule infettate, introduceva una classe di patogeni quasi ignota per la specie umana. In pochi anni, l'intero genoma di HIV è stato sequenziato e la sua strategia replicativa, basata su geni strutturali, regolatori e accessori, è stata compresa nei suoi aspetti fondamentali. Una nuova classe di farmaci, ossia gli antiretrovirali in grado d'inibire meccanismi fondamentali per la replicazione di HIV, è stata rapidamente scoperta e ha cambiato, dal 1995 in poi, il destino delle persone infettate da tale virus, prolungandone la vita per molti anni, in assenza di malattia.
Tuttavia, a oggi nessun individuo colpito è stato in grado, spontaneamente o in seguito alle terapie, di eliminare l'infezione dal proprio organismo (un retrovirus è per sempre!). Negli ultimi anni l'infezione si è trasmessa sempre di più per via eterosessuale, soprattutto in Africa, che da sola contribuisce per oltre il 50% della pandemia (oggi stimata in più di 43 milioni di casi). Nuove aree geografiche, soprattutto India, Sud-Est asiatico e Paesi dell'ex Unione Sovietica, sono colpite in modo esplosivo dall'infezione.
Purtroppo, la disponibilità di farmaci, almeno parzialmente efficaci nel frenare l'evoluzione della malattia, rimane ristretta a circa il 10% dell'umanità, ovvero ai Paesi a più alto reddito. In attesa di capire come e se sarà possibile sviluppare un vaccino in grado di prevenire l'infezione o di attenuarne la patogenicità (vaccino terapeutico), le speranze sono riposte nel breve-medio termine nell'identificazione di un microbicida, ovvero di una o più sostanze in grado d'inibire la trasmissione sessuale del virus.
Un virus, molti ceppi: l'origine di HIV
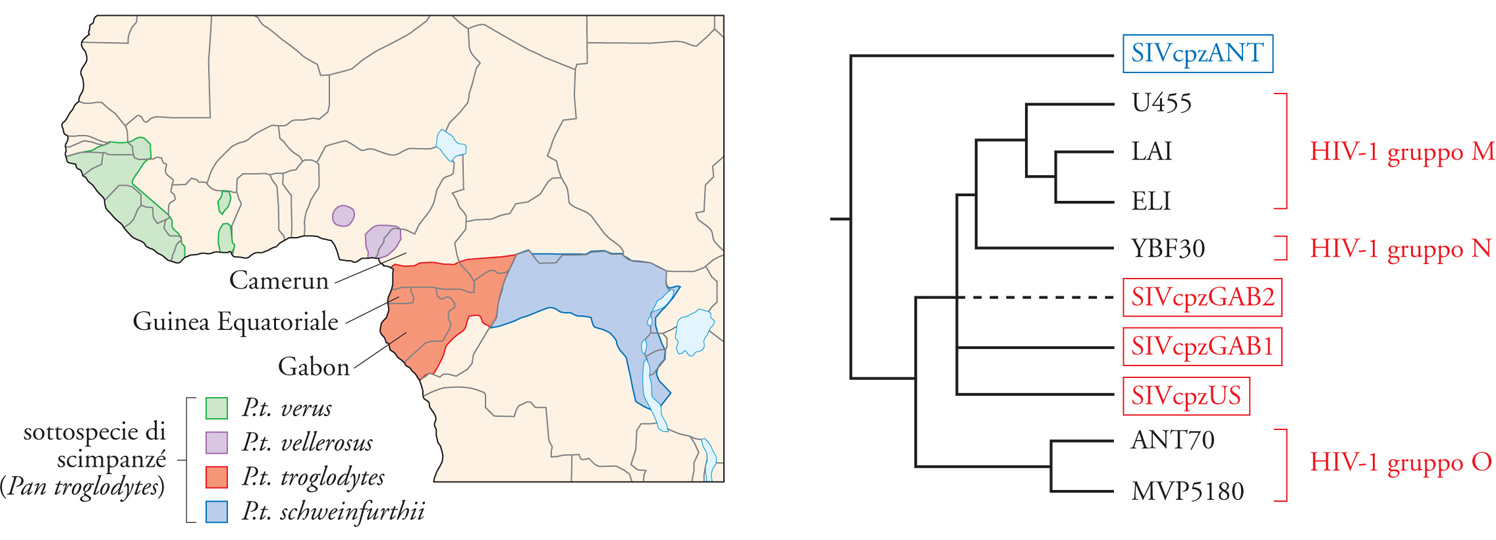
L'infezione da HIV è da considerarsi, in origine, una zoonosi, ossia un'infezione virale inizialmente presente in un'altra specie animale e trasmessa quindi all'uomo. Geograficamente, l'area in cui è stato possibile rintracciare i progenitori dell'attuale virus HIV-1 è in Africa centrale, in una regione che comprende Camerun e Gabon. L'ospite naturale di quest'infezione è rappresentato dallo scimpanzé, un primate non umano molto vicino evolutivamente all'uomo. È stato possibile calcolare che vi sono stati almeno tre episodi indipendenti di trasmissione dallo scimpanzé all'uomo, dando origine al cosiddetto 'gruppo M' (che comprende i sottogruppi classificati dalla lettera A alla H), al gruppo N e a quello O. Un virus simile, ma distinto da HIV-1, HIV-2, è invece presente, senza causare malattia, nelle scimmie verdi africane, dove prende il nome di Simian immunodeficiency virus (SIV) (fig. 2). Geograficamente, il SIV/HIV-2 è presente in zone distinte da quelle in cui è diffuso HIV-1, anche se la coinfezione è possibile e documentata, soprattutto in Costa d'Avorio e Guinea Bissau.
Com'è avvenuta la trasmissione dagli scimpanzé (o dalle scimmie verdi africane) all'uomo? Quasi sicuramente, come per molte zoonosi, dal frequente contatto tra la specie umana e questi animali, adottati come animali da compagnia, nonché dall'abitudine a consumarne la carne e quindi dal contatto con il loro sangue e i loro tessuti durante la macellazione. Il fatto che l'infezione da HIV sia stata originalmente trasmessa dall'animale all'uomo, tra il 1920 e il 1930, implica che nuove trasmissioni potrebbero ancora avvenire. All'interno del gruppo M sono contenuti i numerosi sottogruppi distribuiti geograficamente in modo diverso. Mentre nei Paesi occidentali l'epidemia è stata (e ancora è in buona parte) causata dal sottogruppo B, l'epidemia africana è soprattutto a carico dei sottogruppi A e C, come peraltro anche nel Sud-Est asiatico. Inoltre, la presenza di virus ricombinanti (Circulating recombinant forms, CRF) ha ormai assunto dimensioni comparabili a quelle dei sottogruppi originali.
Quest'osservazione ha portato alla rivalutazione del problema della superinfezione (due o più virus distinti che infettano lo stesso individuo in momenti successivi) e della coinfezione (due o più virus distinti che infettano un individuo nello stesso momento). Studi recenti dimostrano come la prima infezione non conferisca, al contrario di quanto creduto per anni, protezione per altre infezioni da HIV-1. A livello cellulare, la presenza simultanea di due (o più) genomi virali può portare alla ricombinazione dei geni e virus che si traduce nella generazione di una nuova specie virale chimerica. Nella generazione di una CRF è spesso riscontrabile la presenza di mutazioni che conferiscono resistenza a farmaci antiretrovirali. Tuttavia, complessivamente, non vi sono evidenze che le CRF abbiano compiuto un salto di qualità in termini di spettro d'infezione, tropismo cellulare o aggressività in vivo. Una delle differenze principali tra diversi sottogruppi riguarda l'interazione con i corecettori chemochinici di superficie. Una seconda differenza significativa riguarda la configurazione delle regioni regolatrici della trascrizione virale, con un'aumentata suscettibilità, almeno in vitro, a fattori di regolazione dell'ospite. Tuttavia, come già sottolineato, non vi sono evidenze che queste differenze si siano tradotte in un'aumentata capacità replicativa e patogenica del virus nella specie umana.
L'ingresso nella cellula bersaglio
Un virus non è patogenico se non è in grado d'infettare le cellule del proprio ospite, nonché di propagarsi efficientemente dopo la prima infezione. Al riguardo i virus utilizzano strategie diverse: alcuni infettano fondendo direttamente la propria membrana con quella della cellula bersaglio, mentre i virus dotati di mantello glicoproteico utilizzano molecole di superficie delle cellule dell'ospite come porte d'ingresso per iniziare il processo infettivo. A questa seconda categoria appartiene il virus HIV. Tuttavia, è bene sottolineare come, prima dell'interazione specifica col proprio recettore, HIV ‒ come altri virus dotati di mantello quali i virus erpetici ‒ si leghi a molecole zuccherine abbondantemente presenti alla superficie cellulare. È generalmente ritenuto che queste interazioni siano sostanzialmente basate sull'attrazione tra cariche elettrostatiche opposte, ma è possibile che intervengano meccanismi di riconoscimento più specifici. In generale, queste interazioni iniziali favoriscono l'incontro tra la molecola deputata al riconoscimento delle cellule bersaglio, la glicoproteina gp120 Env, e la molecola CD4, espressa da circa il 50% dei linfociti T, dalle cellule del sistema dei fagociti mononucleati (soprattutto monociti circolanti e macrofagi tessutali) e dalle cellule dendritiche immature (iDC). Queste ultime svolgono una funzione primaria in quanto deputate alla cattura di virus e antigeni alla superficie mucosa o cutanea per portarli nei linfonodi al fine di organizzare la risposta immunitaria primaria all'infezione. Oltre alla molecola CD4, le DC catturano il virus grazie a un altro recettore, detto Dendritic cell-specific ICAM3 grabbing nonintegrin (DC-SIGN), che, a differenza di CD4, non funge da porta d'ingresso per l'infezione, ma promuove la trasmissione di virus infettivo dalle DC ai linfociti T CD4+ che entrano in contatto con esse al fine d'innescare la risposta immunitaria specifica al virus stesso (fig. 3). È stato dimostrato di recente che proprio questi linfociti T CD4+ specifici per il virus HIV sono infettati e uccisi preferenzialmente in seguito all'infezione (che in questo modo mina alla base la possibilità di conservare una risposta immunitaria specifica contro di esso). A queste regole generali sfuggono gli astrociti del sistema nervoso centrale, che sono infettabili con modalità ancora sconosciute, forse attraverso il recettore al mannosio, in quanto negativi per espressione della molecola CD4.
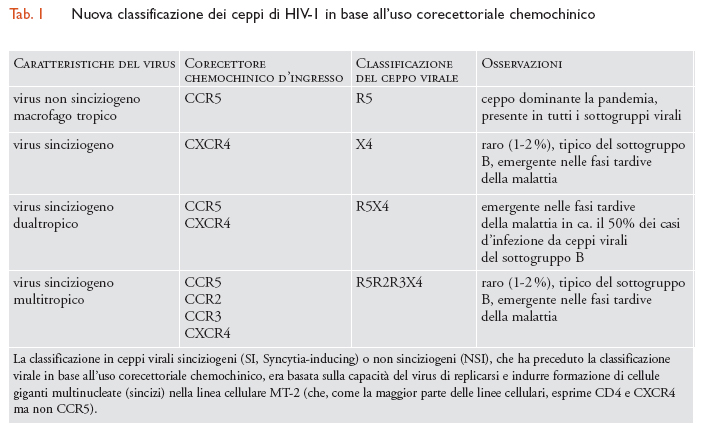
L'interazione tra un trimero di gp120 Env espresso sulla superficie della particella virale (virione) e CD4 induce un cambiamento conformazionale nella molecola del virus. Ciò risulta in due eventi importanti: l'esposizione di porzioni criptiche della molecola gp120 Env ‒ cosiddetti 'epitopi CD4-indotti', di grande rilevanza per lo sviluppo di anticorpi (Ab) neutralizzanti ‒ e l'interazione di questi col secondo recettore, o corecettore, indispensabile per l'infezione. È stato dimostrato che HIV è in grado di utilizzare i recettori chemochinici CCR5 o CXCR4, ma solamente dopo aver interagito con CD4, e che il loro legame permette l'entrata in azione della seconda glicoproteina del mantello virale, gp41, responsabile della fusione delle membrane del virus e della cellula bersaglio grazie a un movimento simile a un coltello a serramanico, innescato dall'interazione tra gp120 Env, CD4 e recettore chemochinico (tab.1). Uno dei farmaci di ultima generazione, un peptide denominato T-20, inibendo il brusco cambiamento conformazionale di gp41, funge da inibitore della fusione tra la membrana del virione e della cellula bersaglio ed è stato utilizzato con un certo successo in terapia (nonostante esso sia penalizzato dal dover essere somministrato per via endovenosa).
La definizione del meccanismo preciso d'infezione da parte di HIV ha permesso una migliore comprensione della storia naturale dell'infezione nonché della rilevanza specifica dei recettori coinvolti. La maggioranza dei ceppi virali responsabili della pandemia utilizza quasi esclusivamente CCR5 quale corecettore d'ingresso virale (ceppi R5), mentre i ceppi in grado di utilizzare CXCR4 emergono solo tardivamente in circa il 50% dei pazienti infettati da ceppi appartenenti al sottogruppo B e presenti nei Paesi occidentali. Inoltre, questi ceppi generalmente continuano a utilizzare CCR5 (ceppi dualtropici R5X4), mentre l'uso solitario di CXCR4 (ceppi X4) risulta abbastanza raro (1÷2%, secondo studi recenti). L'utilizzo di CXCR4 da parte del virus coincide con (ed è forse causato da) una fase di aumentata immunodeficienza e rappresenta un indice di progressione della malattia. Oltre al ruolo dominante nella pandemia mondiale, i ceppi R5 sono quasi universalmente trasmessi da individuo a individuo indipendentemente dalla modalità d'infezione (sessuale, per derivati ematici, da madre a bambino sia al parto che per allattamento) anche nei casi in cui colui o colei che trasmette è infettato/a da un ceppo prevalentemente dualtropico o X4. Tutto ciò suggerisce che l'utilizzo di CCR5 rappresenti un vantaggio per il virus rispetto all'utilizzo di CXCR4. La comprensione dei meccanismi che sottendono questo vantaggio potrebbe schiudere ulteriori informazioni utili allo sviluppo di nuovi farmaci e di un vaccino.
L'anatomia del virus HIV e il suo ciclo replicativo
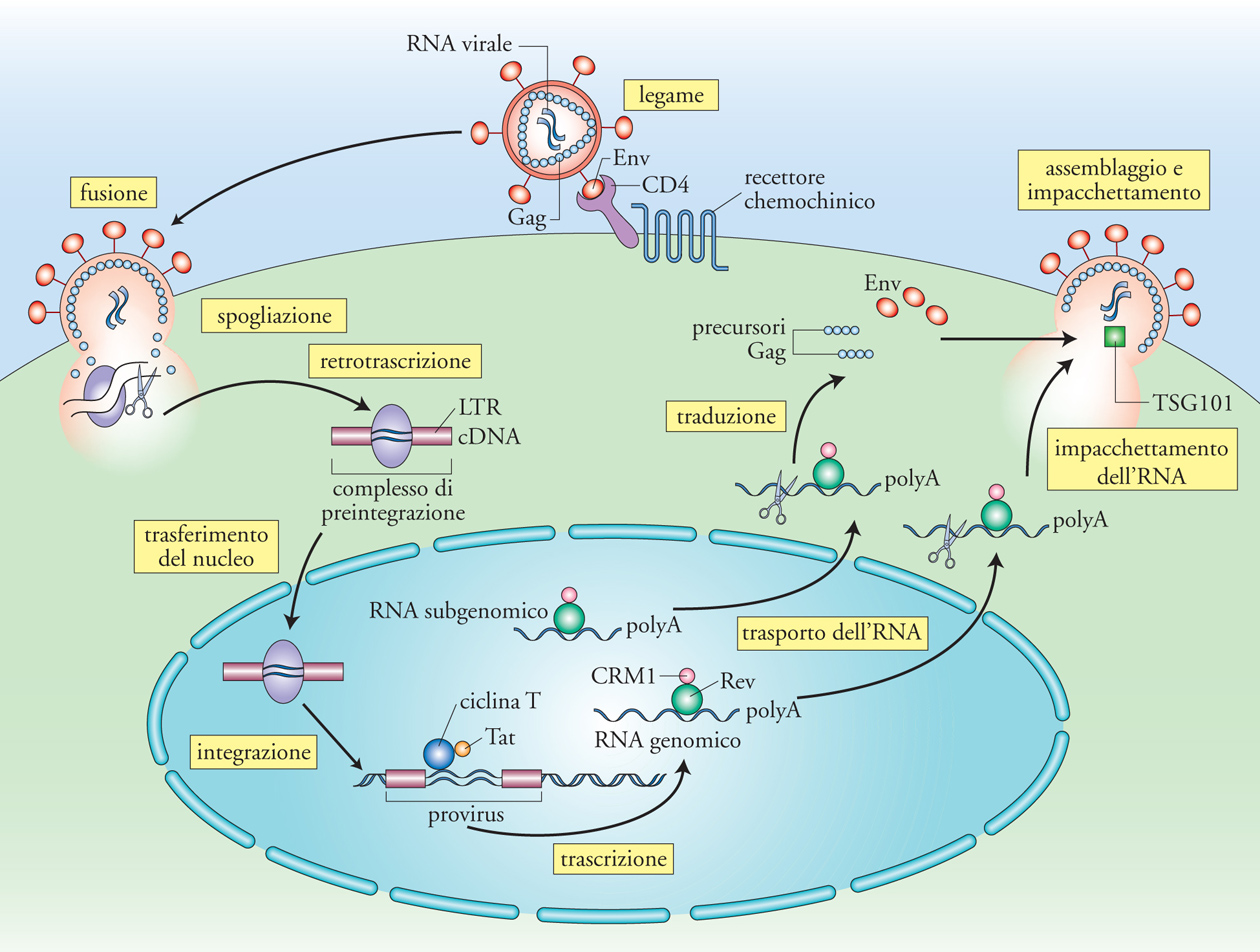
L'interazione tra le proteine del mantello virale (gp120 e gp41 Env) con la molecola CD4 e il recettore chemochinico nonché la fusione della membrana del virione e della cellula bersaglio rappresentano gli eventi infettanti fondamentali perché il virus possa compiere il suo ciclo replicativo. La conseguenza è l'iniezione del contenuto della particella virale all'interno del citoplasma della cellula umana. Entro poche ore ha inizio la fase detta di 'spogliazione' (uncoating), in quanto la proteina del core virionico p24 rimane dispersa nel citoplasma e l'RNA genomico inizia a essere retrotrascritto in cDNA nel processo di retrotrascrizione mediato dalla proteina virionica retrotrascriptasi (RT), una DNA-polimerasi RNA-dipendente. Si forma il cosiddetto 'complesso di preintegrazione' che migra dal citoplasma al nucleo.
Recenti studi hanno dimostrato l'importanza di fattori cellulari perché queste prime fasi possano compiersi. Alcuni fattori cellulari controllano l'efficienza di queste fasi precoci probabilmente interagendo con proteine Gag del core virionico entrante. Il fattore meglio caratterizzato a oggi, Tripartite motif protein 5-α (TRIM5-α) previene l'infezione xenotropica di HIV-1 in cellule di macaco, ma non sembra giocare un ruolo analogo nell'infezione interumana. Tuttavia, altri membri della famiglia TRIM sono stati recentemente implicati nella regolazione del processo d'infezione, sebbene il loro meccanismo d'azione non sia ancora chiarito.
Il processo di retrotrascrizione si compie mediante autentici salti tra una molecola e l'altra dei due genomi virali presenti all'interno di ciascun virione, per giungere alla sintesi di filamenti di DNA a doppia elica codificante i geni virali racchiusi da due sequenze identicamente ripetute al 5′ e al 3′, dette Long terminal repeat (LTR). Di tutte le sequenze sintetizzate, solo quelle lineari possono integrarsi nel genoma cellulare a opera dell'enzima virale integrasi, codificato anch'esso dal gene pol come l'enzima RT e l'RNAsi-H. Altre sequenze, caratterizzate da uno o due LTR, si racchiudono circolarmente su sé stesse e non possono quindi essere integrate. Nel momento in cui il DNA virale si integra in quello dell'ospite, il virus diviene un 'provirus' connotato da estrema stabilità, in contrasto con la variabilità e la breve vita che caratterizza la sua versione a RNA. La natura del provirus giustifica l'immagine per cui 'un retrovirus è per sempre': una volta integrato nel genoma cellulare, ivi rimarrà per la durata della vita di quella cellula e, in senso lato, di quell'organismo. Sebbene tradizionalmente il processo d'integrazione sia descritto come casuale, studi sofisticati hanno dimostrato che esistono 'punti caldi' del genoma in cui c'è una più alta frequenza di siti d'integrazione.
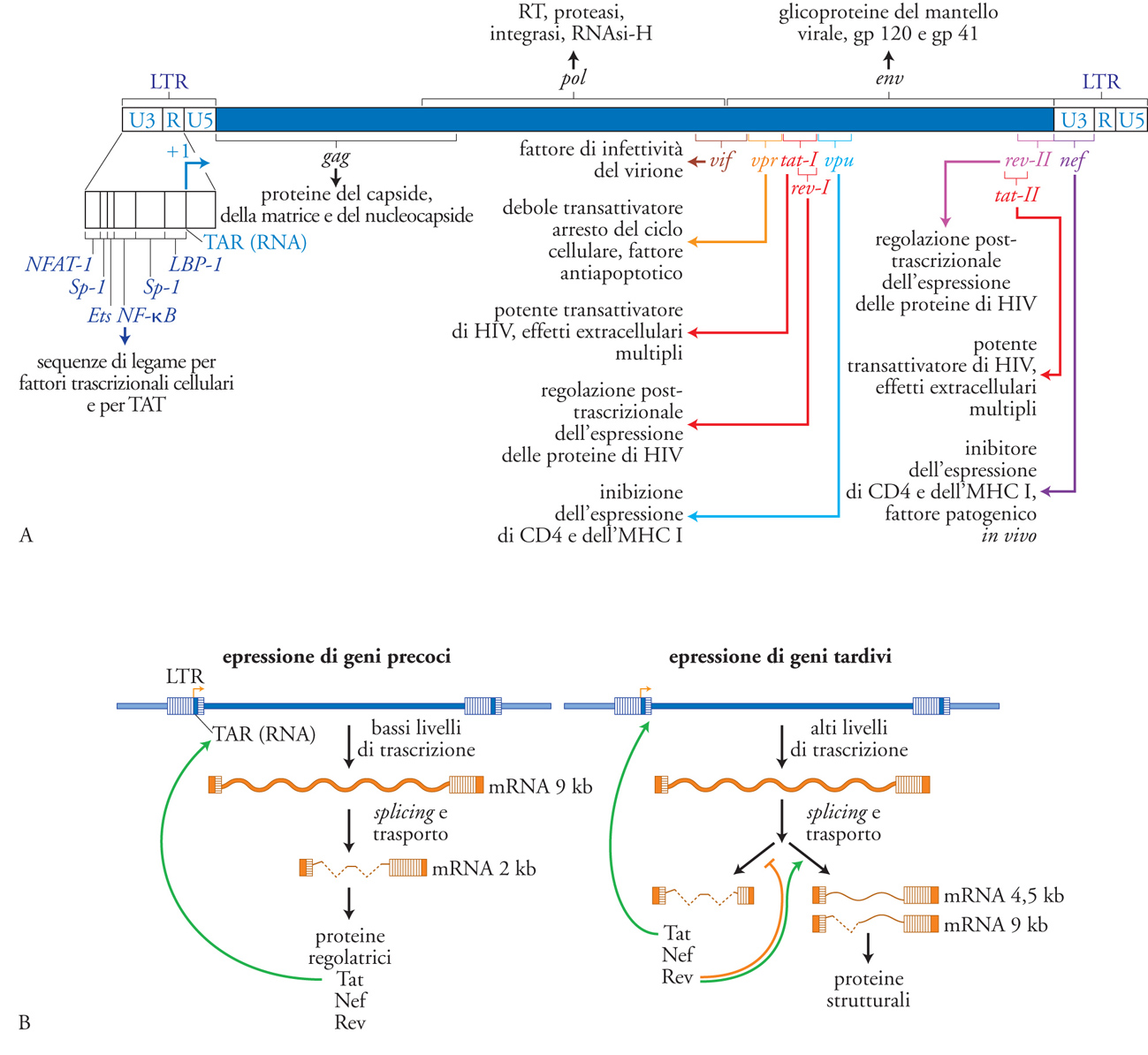
La struttura del provirus è rappresentata da due LTR identicamente ripetuti al 5′ e al 3′, racchiudenti la regione codificante i geni virali. Gli LTR sono convenzionalmente suddivisi nelle regioni U3, R e U5. U3 contiene una serie di sequenze di DNA in grado di legare fattori trascrizionali cellulari, quali NF-κB (attivato da molte citochine proinfiammatorie), NFAT (indotto dall'attivazione T linfocitaria), AP-1 (indotto da segnali in grado di attivare le chinasi ERK-1/-2 e quindi vari membri delle famiglie Jun e Fos) e Sp-1 (attivato costitutivamente) in grado d'innescare e regolare la trascrizione dei geni virali (operata dalla RNA-polimerasi II cellulare, RNA-pol II). Al confine tra U3 e R sta il sito d'inizio della trascrizione ('+1'), mentre R contiene la sequenza a RNA in grado di legare la proteina virale Tat, il maggiore transattivatore della trascrizione dei geni virali. Nella sequenza U5 risiedono invece importanti sequenze per la terminazione dei trascritti virali e l'integrazione del DNA provirale nel DNA della cellula ospite. Il genoma provirale convenzionalmente suddiviso in geni strutturali (gag, pol, env), in geni fondamentali per la replicazione virale, detti 'geni regolatori' (tat e rev), e in geni non indispensabili per la replicazione del virus (ma importanti per la sua patogenicità), denominati 'accessori' (nef, vpr, vif, vpu). Geni regolatori e accessori si concentrano nella porzione 3′ del genoma provirale (fig. 4).
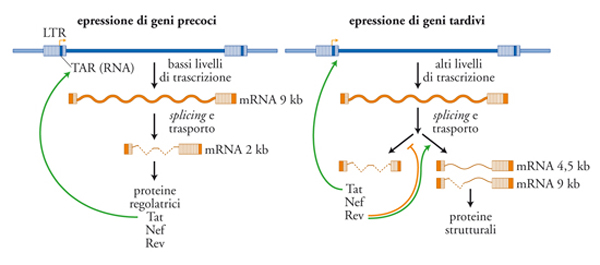
Tra i geni strutturali, gag codifica un precursore poliproteico (p55) che viene processato a opera di una proteasi virale (anch'essa specificata dal gene pol come RT, integrasi e RNAsi-H) in molti frammenti più piccoli, tra cui la matrice p17, e il capside (p24), ognuno con funzioni specifiche. L'espressione di proteine Gag è condizione necessaria e sufficiente per la produzione di una progenie virale da parte di una cellula infettata. L'inibizione della maturazione del precursore di Gag è il principio fondamentale degli agenti farmacologici che hanno segnato una tappa storica nella terapia anti-HIV: gli inibitori della proteasi (IP). I due geni regolatori essenziali tat e rev, assieme al gene accessorio nef, sono espressi precocemente in quanto derivano dalla traduzione di RNA messaggeri (mRNA) di 2 kb in seguito a un processo di splicing a partire dall'RNA di 9 kb derivante dalla trascrizione del genoma provirale. La proteina Tat agisce principalmente inibendo la preterminazione della trascrizione dei geni virali. L'enzima della cellula ospite (RNA-pol II) in assenza di Tat, inizia la trascrizione, ma dopo poche centinaia di basi si stacca 'abortendo' trascritti corti e non funzionali. Il meccanismo d'azione di Tat dipende dal suo legame specifico a una sequenza di RNA, definita TAR (Tat-binding region), localizzata in prossimità del sito d'inizio della trascrizione e formante una struttura secondaria caratterizzata da un gambo e da un'ansa. Tat esercita un classico effetto di feedback positivo sulla trascrizione di HIV: una volta sintetizzata nel citoplasma, essa è in grado di entrare nel nucleo grazie alla presenza di un segnale di localizzazione nucleare (NLS) e di potenziare la trascrizione di HIV che porterà alla sintesi di altra proteina Tat (fig. 5). Il legame di Tat a TAR catalizza l'assemblaggio di proteine cellulari implicate nel controllo della proliferazione cellulare, quali la ciclina T1 e la chinasi CDK9. Questo complesso, assemblato in prossimità del sito d'inizio della trascrizione, probabilmente interagisce con fattori trascrizionali cellulari, quali Sp-1 e NF-κB, che si legano al DNA dell'LTR in 5′ in prossimità del +1. Il tutto favorisce l'azione del complesso della RNA-pol II che sintetizza un nuovo RNA virale. Oltre a questi effetti intracellulari, la proteina Tat ha una 'seconda vita' extracellulare, dove può interagire con diversi recettori di superficie (quali il recettore per il Vascular endothelial growth factor, VEGF e il recettore chemochinico CXCR4) inducendo segnali di attivazione intracellulare, nonché essere catturata e internalizzata dalle cellule in prossimità del sito della sua produzione (ovvero delle cellule produttivamente infettate) ed esercitare effetti intracellulari di tipo infiammatorio.
L'azione protrascrizionale di Tat sarebbe sterile in assenza della proteina Rev, che è dotata di un NLS; una volta entrata nel nucleo Rev lega specificamente sequenze di RNA disperse nei vari geni di HIV, definite RRE (Rev responsive element), e inattiva con un meccanismo complesso sequenze inibitorie anch'esse disperse nel genoma virale. La conseguenza funzionale dell'interazione Rev/RRE si traduce nel permettere l'esporto dal nucleo al citoplasma di mRNA virali parzialmente spliced (di 4,5 kb e codificanti Env e altre proteine accessorie quali Vif e VpU), e dell'RNA genomico a lunghezza piena (9 kb) codificante sia Gag sia Pol che sarà assemblato in nuovi virioni. In assenza di Rev, si verifica un accumulo di mRNA brevi (2 kb) che vengono esportati dal nucleo al citoplasma per sintetizzare le proteine Nef, Tat e la stessa Rev (fig. 5). Quantitativamente, la maggioranza dei messaggeri codifica per Nef, una proteina non cruciale per la replicazione virale, ma di grande rilevanza per la patogenesi dell'infezione. Studi condotti sul modello d'infezione di macachi infettati con SIV, filogeneticamente quasi indistinguibile da HIV-2, hanno infatti dimostrato che l'assenza di nef si manifesta in un'infezione apatogenica. Inoltre, gli animali infettati con virus deleti in nef (Δnef) erano 'vaccinati' contro l'infezione da ceppi virali selvatici. Questi studi hanno trovato successivo riscontro in alcuni individui infettati da HIV-1 considerati Long term non progressor (LTNP), ovvero capaci di controllare spontaneamente l'infezione per diversi anni in assenza di progressione di malattia.
Tuttavia, studi successivi hanno dimostrato che macachi neonati sviluppavano AIDS quando infettati con SIVΔnef e che anche le scimmie adulte e molti LTNP con delezione in nef progredivano verso l'AIDS, sebbene in tempi più tardivi rispetto alla maggioranza dei soggetti infettati. La delezione di nef, quindi, non è sufficiente per ottenere un vaccino vivo e attenuato, anche se questo gene precoce virale gioca sicuramente un ruolo importante per le strategie patogenetiche del virus. È stato quindi dimostrato come uno dei principali effetti di Nef (come anche di Env e VpU), almeno in vitro, sia la regolazione negativa dell'espressione in membrana della molecola CD4 (il recettore primario di HIV) e di antigeni della classe I del Major histocompatibility complex (MHC), che giocano un ruolo fondamentale nel riconoscimento e nell'uccisione di cellule infettate da parte di CTL. Più recentemente è stato anche osservato che Nef è in grado di stimolare l'espressione di una molecola citotossica, Fas ligando (Fas-L), della famiglia del Tumor necrosis factor (TNF). Possiamo quindi inferire che Nef contribuisca significativamente allo spegnimento della risposta CTL specifica contro le cellule infettate da HIV, da un lato diminuendo l'espressione di molecole MHC di classe I, presentanti antigeni virali ai linfociti CD8+, che contengono la maggior parte dei CTL, dall'altro, uccidendo i CTL che riuscissero a riconoscere la cellula infettata attraverso l'interazione Fas-L (espresso dalla cellula infettata) e Fas (espresso costitutivamente dai CTL).
Oltre alla sintesi di proteine Gag, mediata da RNA di 9 kb, due copie di RNA genomico di 9 kb dovranno interagire con proteine Gag per essere incorporate nei virioni di nuova formazione. Quest'interazione avviene a livello citoplasmatico e richiede la presenza di ciclofillina A, la quale lega p24 Gag. È interessante notare come questo enzima cellulare sia il bersaglio farmacologico della ciclosporina A, un farmaco immunosoppressivo utilizzato nei pazienti sottoposti a trapianto d'organo. Ciclosporina A ha dimostrato in maniera consistente effetti antivirali selettivi per HIV-1, ma non per HIV-2/SIV, in quanto la ciclofillina non riconosce le loro proteine Gag. Sebbene questo farmaco non abbia dimostrato effetti antivirali in pazienti con infezione cronica da HIV-1, quando testato in uno studio su pazienti con infezione acuta ha dimostrato importanti effetti di rapido recupero del numero di linfociti CD4+ circolanti che rimanevano a livelli più alti nel tempo rispetto a controlli trattati con sola terapia antiretrovirale.
Due molecole di RNA genomico si appaiano e vengono incorporate nel core virionico in associazione con la RT. Le particelle gemmanti vengono rilasciate dalla cellula infettante dopo un ultimo processo di sigillatura della particella virale: una mutazione di una piccola proteina Gag-p6, impedisce quest'ultimo processo e crea l'immagine ultramicroscopica di cellule infettate dalla cui superficie protrudono catenelle formate da particelle virali incapaci di staccarsi tra loro e dalla superficie cellulare. Ciò sottolinea come ogni tappa del ciclo vitale di HIV sia oggetto di regolazione endogena (controllata dallo stesso virus) nonché esogena da parte dell'ospite. Un blocco del rilascio di particelle virali dalla superficie di cellule infettate è stato dimostrato in seguito a trattamento con interferone α (IFN-α), anche se, a differenza dei mutanti di Gag-p6, le particelle risultavano perfettamente sigillate; altre molecole, come IFN-γ, CCL2 e urochinasi, sono invece in grado di modulare il sito di produzione dei virioni dalla superficie cellulare a compartimenti vacuolari endocellulari, verosimilmente di natura endosomiale. La produzione di nuova progenie virale rappresenta quindi l'ultimo atto del ciclo vitale di HIV e la premessa per l'infezione di nuove cellule bersaglio.
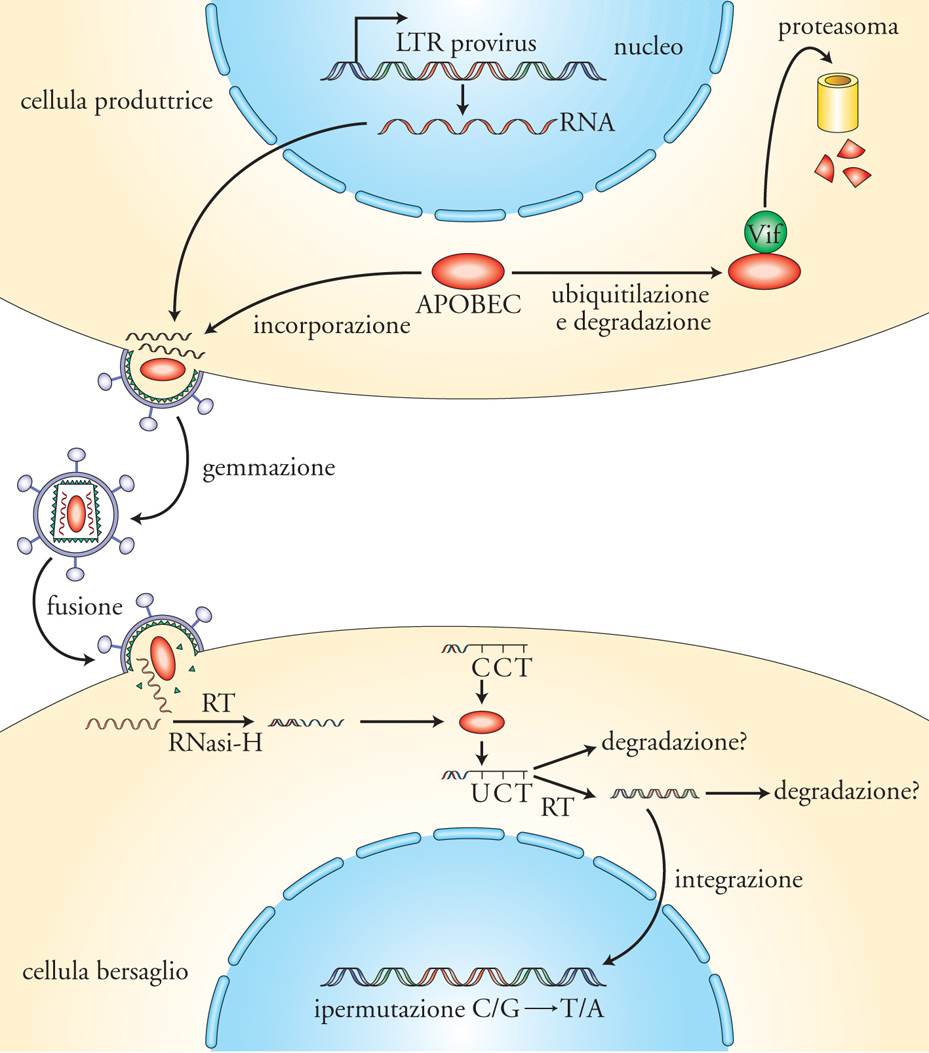
Negli ultimi anni è stata chiarita la funzione del gene accessorio vif codificante per la proteina definita Virion infectivity factor (Vif), originariamente descritta come influenzante l'infettività virale in quanto la sua eliminazione rendeva la replicazione di HIV meno efficiente. Si è notato in seguito che alcune linee cellulari CD4+ positive erano tuttavia in grado di sostenere efficientemente la replicazione di un virus difettivo per Vif (cellule permissive), a differenza dei linfociti T CD4+, non permissivi. È stato quindi scoperto un fattore cellulare espresso selettivamente nelle cellule non permissive e assente in quelle permissive la cui funzione è soppressa da Vif. Questo fattore è un membro della famiglia APOBEC-1 (APOlipoprotein B mRNA editing enzyme catalytic polipeptide). Il ruolo fisiologico di APOBEC-3G è di presiedere alla fedeltà del genoma legandosi ai DNA a singola elica. Il genoma virale, durante il processo di retrotrascrizione, si presenta transitoriamente come un DNA a singola elica e viene quindi legato da APOBEC-3G, il quale catalizza la reazione di deamminazione delle Citidine (C) trasformandole in Uridine (U). Conseguentemente, le U sono copiate in Adenine (A), invece che in Guanine (G), e questo processo di sostituzione sistematica di G con A (ipermutazione) porta all'introduzione di potenziali codoni di stop e cambiamenti di amminoacidi fondamentali per la funzionalità delle proteine virali con conseguente interruzione del ciclo replicativo virale. Il legame della proteina virale Vif ad APOBEC-3G (e APOBEC-3F) ne causa la degradazione a livello di proteosoma, permettendo quindi la replicazione virale in cellule altrimenti non permissive, per virus non dotati di Vif (fig. 6). Sebbene quest'elegante meccanismo sia stato dimostrato per il momento solo in vitro, è plausibile che esso sia funzionale anche negli individui infetti, come indicato dalla dimostrazione di ipermutazioni G>A in sequenze virali amplificate da cellule d'individui infettati caratterizzati da lunga sopravvivenza asintomatica. Inoltre, sono stati descritti polimorfismi nel gene codificante APOBEC-3G che potrebbero alterarne l'espressione, nonché varianti del gene vif incapaci d'inattivare APOBEC-3G. La funzionalità di APOBEC-3G nelle cellule bersaglio sembra inoltre regolata dal suo stato fisico di associazione/ dissociazione da un complesso ad alto peso molecolare contenente diverse proteine cellulari e molecole di RNA. L'attivazione cellulare mediante mitogeni o citochine causerebbe appunto il sequestro di APOBEC-3G in questo complesso permettendo al processo di retrotrascrizione di procedere efficientemente e, quindi, d'infettare stabilmente la cellula mediante l'integrazione di DNA virale nei cromosomi dell'ospite. Queste ultime scoperte sottolineano con forza il concetto di coevoluzione dell'uomo con i virus e altri agenti microbiologici. La scoperta e la definizione del meccanismo d'azione di questi meccanismi di difesa e di superamento delle difese dell'ospite non solo permetterà una miglior comprensione del rapporto tra uomo e HIV (e sicuramente altri virus) ma, con tutta probabilità, stimolerà la messa a punto di nuove strategie preventive e terapeutiche.
La storia naturale dell'infezione da HIV _e la risposta immunitaria dell'ospite
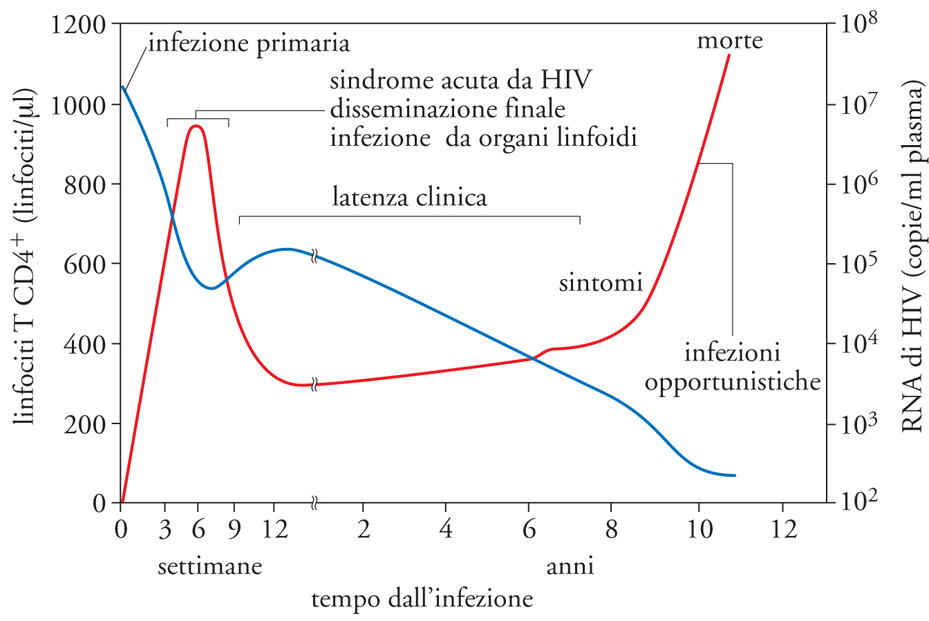
L'infezione da HIV, comunque contratta, è seguita dopo 1-2 settimane da segni e sintomi simil-influenzali, con ingrossamento doloroso dei linfonodi latero-cervicali, febbre, malessere e sudorazioni notturne. Questa sindrome acuta concomitante con l'infezione primaria da HIV coincide con la fase di disseminazione sistemica del virus e con l'infezione dei linfociti CD4+ presenti nei tessuti linfoidi quali tonsille e linfonodi, ma, soprattutto, nel tessuto linfoide associato al tratto digerente e perciò definito GALT (Gut-associated lymphoid tissue). L'infezione primaria termina entro 2-3 mesi dal momento dell'infezione con l'emergenza della risposta immunitaria specifica al virus, sfociando quindi nella successiva fase clinicamente asintomatica che, in assenza di terapie antiretrovirali, dura mediamente 8-10 anni (fig. 7).
Contrariamente al profondo stato d'immunodeficienza che caratterizza lo stadio di AIDS conclamato, che succede alla fase asintomatica dell'infezione, il virus induce una potente attivazione immunitaria che risulta nel parziale controllo della sua replicazione. Paradossalmente, proprio questo controllo parziale relativamente efficace per diversi anni è alla base della capacità del virus di diffondersi nella società. In una minoranza di casi (〈10%) la malattia ha un decorso acuto in individui definiti 'progressori rapidi', tra cui una percentuale significativa di bambini, che giungono a morte da AIDS entro 2-3 anni dall'infezione. Per contro, una percentuale ancora più bassa (〈5%) di individui convive (in alcuni casi da oltre 20 anni) con l'infezione senza aver mai assunto farmaci antiretrovirali, con un sistema immunitario relativamente intatto (come rivelato dal fatto che i loro linfociti T CD4+ circolanti sono >500 cellule/μl) e in buone condizioni di salute. Questi individui LTNP rappresentano il migliore modello naturale di convivenza relativamente 'pacifica' tra virus e ospite (come, evolutivamente, è accaduto per le scimmie africane nei confronti del SIV, molto simile al virus umano HIV-2).
Ancora prima della formazione di Ab specifici, le proteine del mantello virale sono in grado di attivare la cascata del complemento, il quale può rivestire le particelle virali. Tali complessi, come gli immunocomplessi formati da virioni e anticorpi specifici che si formeranno dopo qualche settimana, vengono intrappolati nelle maglie della rete formata dalle cellule follicolari dendritiche (FDC, Follicular dendric cell) residenti nei centri germinativi linfonodali la cui funzione è di conservare una 'banca' di antigeni e di virus in configurazione nativa (cioè intatti), o, come nel caso di HIV, particelle virali intere. A questa fase ne segue una atrofica con conseguente distruzione della rete delle FDC, vero e proprio scheletro portante dei centri germinativi, verosimilmente a causa di un attacco 'autoimmune' da parte di linfociti T citotossici CD8+, che infiltrano i centri germinativi linfonodali (quadro istopatologico assolutamente tipico dell'infezione da HIV).
La transizione tra fase acuta e fase asintomatica, verosimilmente a causa dell'insorgenza della risposta immunitaria, soprattutto T linfocitaria, comporta un abbassamento della viremia plasmatica (ovvero del numero di particelle virali presenti in circolo, ognuna delle quali contenente due copie di genoma virale) raggiungendo, in ogni individuo, un punto d'equilibrio tra produzione ed eliminazione virale, definito set point, a circa 6 mesi dall'infezione acuta, che permane relativamente costante negli anni durante la fase asintomatica d'infezione (fig. 7). Il valore della viremia, come quello dei linfociti T CD4+ circolanti, rappresenta un parametro fondamentale di progressione della malattia, e quindi di prognosi, come è stato stabilito sia da studi retrospettivi che prospettici. Basandosi sul concetto di set point, D.D. Ho e G. Shaw hanno potuto definire che ogni giorno vengono prodotti circa 1010 nuovi virioni e che la loro fonte deriva principalmente (per più del 90%) da linfociti T CD4+ che vengono infettati e rapidamente distrutti quale conseguenza dell'infezione. Tra i risultati di queste osservazioni c'è anche la possibilità che varianti virali farmacoresistenti siano generate spontaneamente dagli errori della RT (la quale non ha la funzione di correzione di errori di trascrizione delle più evolute polimerasi cellulari), evento molto probabile quando la terapia antiretrovirale viene assunta, per motivi diversi, in modo subottimale, come in caso di monoterapia con azidotimidina (AZT, il primo farmaco antiretrovirale scoperto a metà degli anni Ottanta) o altri farmaci.
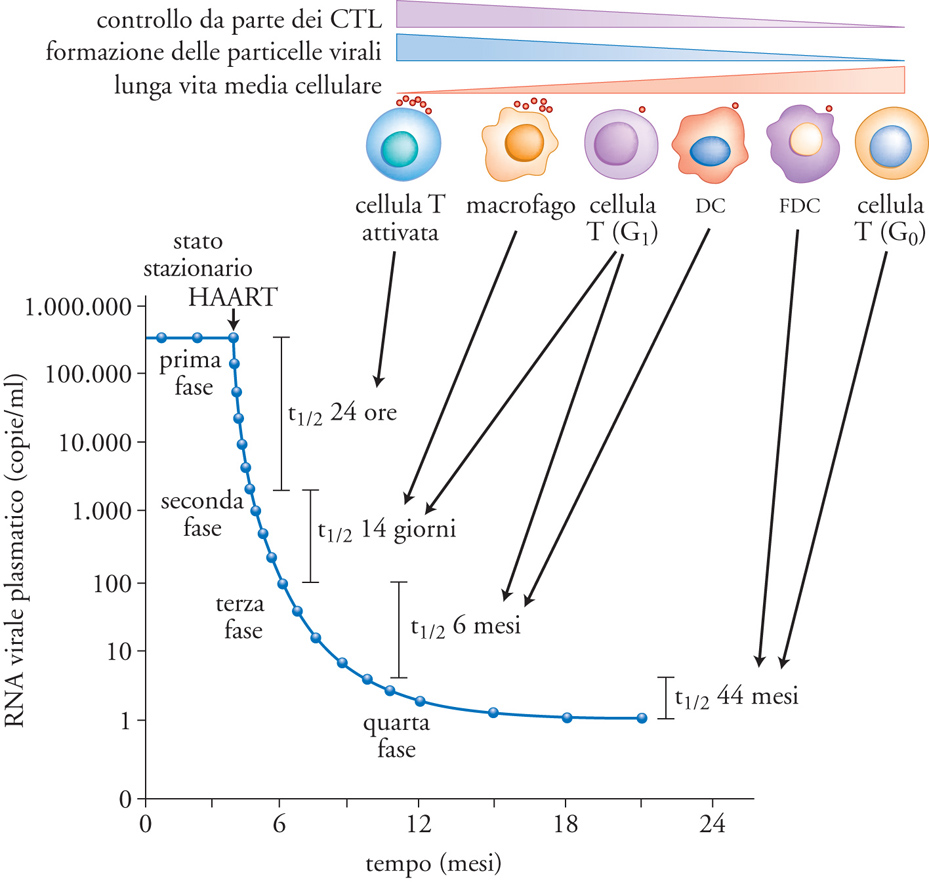
La scoperta della seconda classe di inibitori dell'infezione, ovvero gli IP, ha permesso la formulazione dell'HAART (Highly active antiretroviral therapy) in grado di sopprimere quasi interamente la capacità replicativa del virus (per lo meno a livello di viremia plasmatica) e ha giustificato una fase di comprensibile ottimismo nella possibilità di eradicare l'infezione, cioè di curare definitivamente la malattia in individui mantenuti costantemente in presenza di dosi massimali di farmaci antiretrovirali. Purtroppo, l'ottimismo è svanito rapidamente quando alla sospensione della terapia in questi soggetti, pur dopo anni di 'completa' soppressione della replicazione virale, è corrisposta una rapida (poche settimane) ripresa della stessa, generalmente a livelli simili al set point precedente l'inizio della terapia. In questi individui è stata dimostrata la presenza di virus infettivo in una piccola percentuale di linfociti T CD4+ definiti 'linfociti memoria', in base all'espressione di alcuni marcatori di superficie, ovvero cellule che avevano già incontrato il virus e che si trovavano funzionalmente in stato quiescente (resting). Queste cellule, note per la loro lunga vita media (si suppone anche di decenni), rappresentano quindi le 'vestali' dell'infezione nella fase in cui questa è soppressa dalla potente terapia farmacologica di combinazione e permettono la rapida ripresa del processo di propagazione del virus nel momento in cui essa viene interrotta (fig. 8).
Inoltre, altre cellule, quali i monociti circolanti del sangue e i macrofagi dei diversi organi e tessuti, possono svolgere un ruolo analogo a quello dei linfociti T memoria. Queste osservazioni non devono essere interpretate come la dimostrazione del fallimento delle correnti terapie antiretrovirali, il cui avvento ha letteralmente cambiato la storia naturale della malattia, trasformandola in una malattia cronica o comunque prolungando di diversi anni la vita media delle persone infettate dal virus. Tuttavia, tali terapie mettono in evidenza la difficoltà di colpire farmacologicamente un virus che si nasconde, letteralmente, nel nostro DNA per 'saltar fuori' nel momento in cui esistono ‒ dal punto di vista del virus ‒ le condizioni favorevoli per propagarsi (ovvero, quando i livelli di farmaci sono bassi o assenti, quando varianti virali farmacoresistenti si sono accumulate oltre un certo livello nell'organismo, oppure quando il sistema immunitario perde ogni capacità di contenimento dell'infezione).
Le terapie correnti e imminenti
La scoperta della natura retrovirale dell'HIV ha anticipato, stimolandola, solo di qualche anno la scoperta di una nuova classe di farmaci: i farmaci antiretrovirali. Si potrebbe addirittura sostenere che, con l'eccezione dell'interferone e di pochi altri farmaci (amantadina, rimantadina e tetracicline), i farmaci antivirali in genere sono nati con l'infezione da HIV. Il primo farmaco antiretrovirale, AZT, era un farmaco già presente e non utilizzato per motivi di tossicità e scarsa efficacia. AZT si sostituisce alla timidina durante il processo di retrotrascrizione e causa la terminazione prematura della catena nascente di DNA virale; la sua efficacia dipende dalla relativa affinità per l'enzima virale, circa 200 volte superiore a quella per le polimerasi cellulari. AZT rappresenta quindi il capostipite della categoria di farmaci nota come inibitori della retrotrascrittasi (RT inhibitors, RTi), categoria oggi estremamente ricca e diversificata (suddivisa principalmente in farmaci a struttura nucleosidica e non nucleosidica). Tuttavia, la monoterapia con AZT, o con altri farmaci antiretrovirali, non è sufficiente a inibire significativamente e per lungo tempo la replicazione virale e causa invariabilmente l'insorgenza di varianti virali portatrici di mutazioni di resistenza al farmaco. Per contro, la monoterapia, con AZT o nevirapina, un altro RTi, somministrata a donne infettate nell'ultimo trimestre di gravidanza e/o al momento del parto, ha significativamente ridotto la frequenza di trasmissione dell'infezione da madre a bambino (da 20÷30% in alcune aree a meno dell'1%).
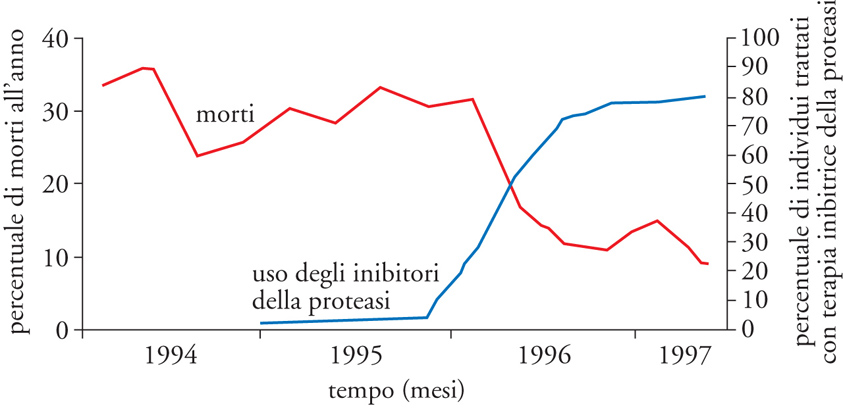
Dopo gli RTi, una nuova classe di farmaci anti-HIV si è affacciata a metà degli anni Novanta: gli IP. La proteasi del virus è fondamentale per tagliare la poliproteina Gag (p55) nelle sue componenti mature, quali p24, p17 e p6; la sua inibizione determina l'impossibilità di generare nuovi virioni infettanti e causa quindi l'aborto dell'infezione: la cellula infettata rimane tale, ma non è in grado di propagare ad altre cellule il virus. Come per gli RTi, anche per i diversi IP la monoterapia ha efficacia limitata e porta rapidamente alla selezione di varianti virali resistenti (spesso in modo crociato ai diversi IP). Tuttavia, la combinazione di farmaci RTi e IP ha dimostrato grande sinergia sia in vitro che in vivo, dando vita ai cosiddetti protocolli HAART, che hanno radicalmente cambiato l'aspettativa di vita delle persone infettate da HIV (fig. 9).
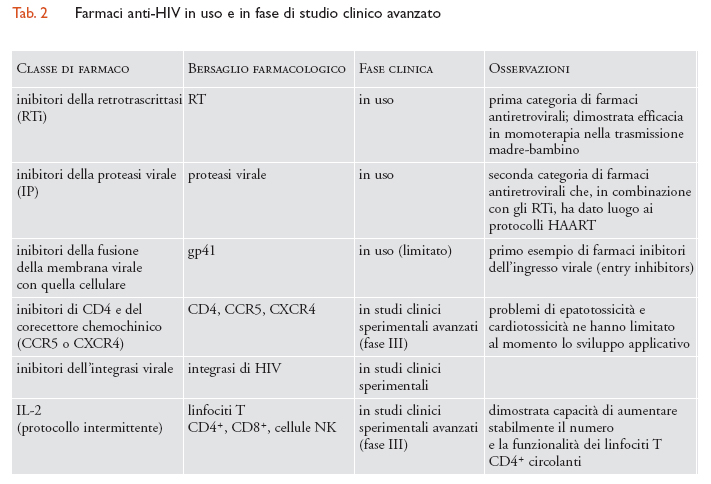
La comprensione del fine meccanismo d'ingresso del virus nelle cellule CD4+ ha inoltre portato alla scoperta di una terza categoria di farmaci: gli inibitori dell'ingresso e della fusione del virus con la cellula bersaglio. Il primo tra i nuovi farmaci che ha dimostrato efficacia clinica in questa categoria è stato un peptide (T-20) in grado d'inibire il processo di fusione mediato dalla glicoproteina virale gp41 Env. Purtroppo, il fatto di dover essere somministrato per iniezione ha fortemente limitato l'uso di questo farmaco su ampia scala. Altre molecole hanno dimostrato la potenzialità di inibire l'infezione mediante legame al recettore primario CD4 e ai corecettori CCR5 e CXCR4; oltre all'emergenza di resistenze virali (le quali peraltro dimostrano che i farmaci in questione hanno causato un effetto significativo sulle quasispecie virali infettanti), sono emersi problemi di tossicità che al momento hanno frenato l'utilizzo di queste molecole al di fuori di protocolli strettamente sperimentali. Inoltre, sono attesi a breve inibitori dell'integrasi, enzima chiave del ciclo vitale di HIV che media appunto l'integrazione dei genomi lineari a doppia elica di DNA nei cromosomi della cellula ospite. Non è difficile prevedere che nei prossimi anni la panoplia dei farmaci antiretrovirali si amplierà e permetterà una gestione sempre più articolata del paziente infettato, anche se, contemporaneamente, si complicheranno ulteriormente i problemi legati alle varianti virali resistenti ai farmaci antivirali (tab. 2).
Oltre ai classici farmaci antivirali, un fronte importante di sviluppo riguarda la possibilità di ricostituire le difese immunitarie compromesse, ovvero agire sull'ospite invece che sul virus. Quest'idea è stata discussa e perseguita sperimentalmente sin dagli anni Ottanta, ma solo nell'ultimo decennio si è concretizzata in potenziali schemi di trattamento clinico. L'esempio meglio definito è sicuramente la somministrazione intermittente di interleuchina-2 (IL-2), una citochina che svolge molte funzioni regolatrici sul sistema immunitario e induce un'importante risposta infiammatoria se somministrata ad alti dosaggi. L'idea di ricostituire le difese immunitarie è nata con la definizione dell'AIDS quale patologia caratterizzata da grave immunodeficienza. Tra gli elementi di questa immunodeficienza, un profondo difetto nella produzione di IL-2, da cui l'idea di una terapia sostitutiva, effettivamente sperimentata fin dagli anni Ottanta. Si è dovuto attendere però fino al 1995 per capire quale fosse la 'giusta' modalità di somministrazione della citochina, basata sullo schema di cicli intermittenti. L'infusione continua di IL-2, generalmente a dosaggi 〈1 milione di unità internazionali (MUI)/die non dà effetti collaterali significativi ed è stato dimostrato aumentare l'attività di cellule natural killer; tuttavia, ciò non ha avuto finora chiari risvolti terapeutici per questa patologia. Per contro, la somministrazione di IL-2 a dosaggi variabili tra i 5 e i 15 MUI/die (tipicamente in cicli di 5 giorni di somministrazione intervallati da 4-8 settimane tra loro) causa un aumento progressivo e selettivo di linfociti T CD4+ circolanti nella maggior parte degli individui, senza interferire negativamente sull'effetto di terapie antiretrovirali concomitanti. I meccanismi d'azione sottostanti a questo effetto ricostitutivo sull'indicatore principale dell'infezione da HIV (la caduta progressiva dei linfociti T CD4+) sono sostanzialmente ignoti e sono in corso studi clinici controllati di fase III, finalizzati a dimostrare se l'aumento del numero di linfociti T CD4+ circolanti si traduca in un'attuale prevenzione dell'insorgenza dell'AIDS e della mortalità a essa associata. Alcune molecole, quali GM-CSF e altre citochine, sono in studio per il loro ruolo di stimolazione immunologica e non è difficile prevedere che il futuro della terapia 'di mantenimento' dell'infezione da HIV sarà basato sulla somministrazione di formulazioni semplificate di farmaci antiretrovirali (1-2 compresse al giorno) associate a periodici trattamenti d'immunostimolazione specifica (vaccini terapeutici) o non specifica (come qui esemplificato dall'uso clinico sperimentale di IL-2).
L'avvento della terapia antiretrovirale di combinazione ha causato un significativo aumento (calcolato mediamente in 13 anni) della vita media dell'individuo infettato da HIV e, in generale, della sua qualità di vita. Tuttavia, sono emersi anche importanti problemi sia d'intolleranza ai farmaci con conseguenti interruzioni più o meno programmate della loro assunzione, sia di resistenze farmacologiche crociate. L'aumento della vita media delle persone infettate da HIV ha determinato un significativo cambiamento delle cause di mortalità di queste persone. Attualmente, nei Paesi occidentali, è più frequente che una persona sieropositiva muoia a causa di una concomitante epatite virale (di tipo B o C), di un tumore o per l'aumentata frequenza di patologia cardiovascolare (incluso l'infarto del miocardio), che per la progressione dell'AIDS e l'insorgenza di infezioni opportunistiche. In altre parole, l'infezione da HIV ha cambiato 'faccia' (nei Paesi dove è diffusa l'HAART) e l'emergenza di queste nuove gravi patologie suggerisce che esistano aspetti patogenetici, sia di natura genetica che epigenetica, non apprezzabili in assenza di farmaci antiretrovirali.
La prevenzione dell'infezione da HIV e della sua progressione: vaccini e microbicidi
Riusciremo mai ad avere una strategia efficace per prevenire l'infezione da HIV o limitarne la progressione di malattia una volta contratta (a esclusione rispettivamente della campagna d'informazione e della terapia antiretrovirale)? Perché non abbiamo ancora un vaccino in grado di prevenire l'infezione, mentre abbiamo fatto progressi enormi nella terapia della malattia 'inventando' una nuova categoria di farmaci? Potremmo cominciare a sottolineare come l'infezione da HIV rappresenti la prima infezione di proporzioni planetarie causata da un retrovirus, ovvero da un virus che basa la sua strategia riproduttiva sulla sua capacità d'integrarsi stabilmente nel genoma della cellula infettata. Inoltre, HIV è caratterizzato da un'importante variabilità genetica che si manifesta durante la sua replicazione a causa della trascrittasi inversa deficitaria nell'attività di riconoscimento e riparazione dei propri errori. La variabilità interindividuale è calcolata intorno al 5÷10%, mentre la variabilità tra i sottogruppi del gruppo M può variare tra il 15 e il 35%. Queste sono verosimilmente le ragioni principali per cui non sono noti casi di eradicazione spontanea dell'infezione, né in individui LTNP né in soggetti aderenti per anni ai migliori protocolli HAART con perfetto controllo della replicazione virale (almeno a livello ematico, che riflette però anche in buona misura quanto accade nei distretti linfoidi periferici). In generale si potrebbe quindi concludere che l'eliminazione dell'infezione virale, una volta acquisita, non appartiene al nostro repertorio di risposte immunitarie, sia innate che adattative (Ab, CTL).
Ne consegue che il semplice potenziamento quantitativo della nostra risposta immunitaria al virus (l'obiettivo dei cosiddetti 'vaccini terapeutici' da somministrarsi a individui già infettati) potrebbe non essere sufficiente alla sua eliminazione. Ma quale potrebbe essere la causa del fallimento (a oggi) di tutte le strategie preventive di vaccinazione? A questo proposito va chiarito che i vaccini preventivi sono quelli in grado d'indurre Ab neutralizzanti capaci di legarsi specificamente al patogeno e di causarne o la lisi diretta o la sua eliminazione per fagocitosi da parte di macrofagi e altre cellule competenti. La risposta CTL, per contro, tende a 'controllare' i livelli di propagazione del patogeno eliminando quelle cellule che esprimono peptidi nel contesto di antigeni della classe I dell'MHC. In base alla convinzione, a sua volta supportata da evidenze sperimentali, che la risposta Ab al virus fosse di scarsa rilevanza o addirittura dannosa, si è investito negli ultimi anni nel cercare d'indurre risposte CTL e CD4+ (helper, necessarie al mantenimento nel tempo di una risposta specifica, sia anticorpale che CTL) anche sulla base di suggestivi modelli sperimentali d'infezione di macachi da parte di SIV e di osservazioni cliniche su individui con infezione acuta primaria. Purtroppo queste strategie non hanno, al momento, portato a evidenze convincenti di efficacia preventiva, mentre i risultati in chiave vaccinale terapeutica sono controversi e di non facile interpretazione.
Notizie più ottimistiche vengono dallo studio della risposta anticorpale. Alcuni Ab monoclonali hanno dimostrato la capacità di neutralizzare in vitro il virus con un ampio spettro d'azione (la maggior parte degli Ab neutralizzanti l'HIV ha basso potere neutralizzante e limitato al virus che li ha generati). Questi Ab sono diretti contro le componenti gp120 e gp41 del mantello virale (regola pressoché assoluta per gli Ab neutralizzanti) e la loro definizione strutturale sta portando nuove conoscenze che potrebbero permettere il disegno razionale d'immunogeni e la selezione di Ab sempre più efficaci. Tuttavia, l'aspetto più incoraggiante è che studi d'immunizzazione passiva con cocktail di questi Ab hanno dimostrato capacità protettive rispetto all'infezione nel macaco e di poter interferire, sebbene transientemente, sulla replicazione di HIV in individui infettati. Questo significa avere 'la prova di principio' che è possibile indurre una risposta Ab protettiva contro l'infezione e/o influire per questa via sulla storia naturale della malattia. Un aspetto recentemente emerso in questo contesto è la dimostrazione che alcuni Ab neutralizzanti diretti contro la glicoproteina gp41 Env riconoscono anche costituenti lipidici della membrana plasmatica della cellula ospite. Quest'osservazione ha innescato una profonda discussione, tuttora in corso, sul ruolo delle componenti non virali nella generazione di una risposta anticorpale neutralizzante, tenendo presente la possibilità d'indurre risposte francamente autoimmunitarie. Il risultato di maggior rilievo è quello di aver individuato nuovi filoni di sviluppo della ricerca miranti all'induzione di una risposta immunitaria almeno parzialmente protettiva.
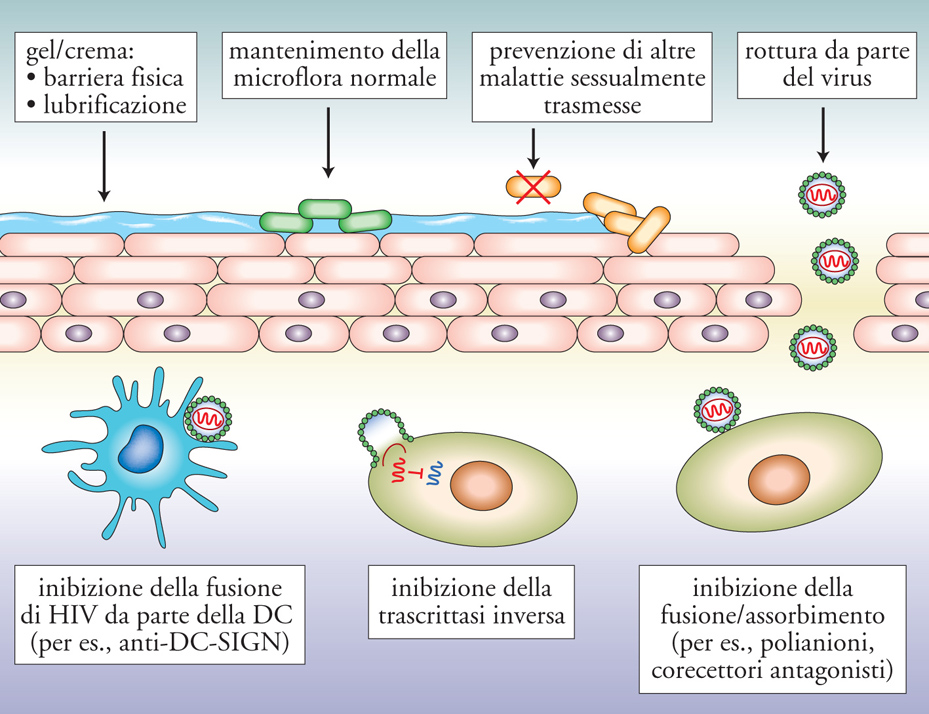
Se la strada verso un vaccino si presenta oggi ancora lunga, tortuosa e d'incerto successo, un grande impegno si è concentrato negli ultimi anni nella ricerca di un microbicida almeno parzialmente efficace. Per microbicida s'intende qualsiasi sostanza, o combinazione di sostanze, che impedisca l'infezione mucosale da parte di HIV (o altri patogeni). Le sostanze sotto intenso scrutinio comprendono: farmaci antiretrovirali, Ab neutralizzanti, polisaccaridi complessi, chemochine modificate, strategie basate sull'innesto di una flora batterica modificata e in grado di rilasciare sostanze anti-HIV e sul principio dell'interferenza di RNA brevi (Small interference RNA, siRNA, fig. 10). Una sostanza largamente utilizzata come spermicida, il nonossinolo-9, è stata il primo candidato microbicida testato in studi clinici di fase III. Questi studi hanno dimostrato non solo l'inefficacia preventiva di questa sostanza, ma addirittura un'aumentata incidenza d'infezione nelle donne che l'assumevano. Ciò era causato dalle proprietà esfolianti e infiammatorie della sostanza, che favoriscono sia il richiamo di cellule bersaglio per HIV (linfociti CD4+ e macrofagi) a livello di mucosa vaginale, sia la promozione della replicazione virale in cellule infettate. Quest'esperienza ha quindi insegnato inequivocabilmente che un microbicida innanzitutto non deve indurre reazioni infiammatorie a livello locale.
L'identificazione di un microbicida che possa almeno parzialmente ridurre la trasmissione per via sessuale del virus ha una grandissima importanza per i Paesi in via di sviluppo (l'Africa subsahariana innanzitutto), dove la trasmissione eterosessuale rappresenta la modalità dominante di propagazione dell'infezione. In queste realtà, spesso non toccate dai farmaci antiretrovirali, le donne non hanno voce in capitolo sulla loro vita sessuale e spesso l'uso del profilattico (come la pratica dell'astinenza) ha uno spettro d'azione molto limitato. In questo scenario, è recentemente emerso che la pratica della circoncisione può portare fino al 70% di riduzione della probabilità di trasmissione dell'HIV. Il dato è impressionante ‒ e importanti studi presto lo confermeranno (o ridimensioneranno) ‒ e potrebbe portare a una diffusione di questa misura igienico-sanitaria, peraltro non priva di controindicazioni e rischi, quale strategia preventiva anti-HIV (ed efficace contro altre infezioni sessualmente trasmesse, come per esempio quella da virus umano del papilloma, HPV, correlato al cancro del pene e della cervice uterina). Non è difficile ipotizzare che la definizione di misure generali di prevenzione, quali l'uso di profilattici sia maschili che femminili e la pratica della circoncisione, assieme alla scoperta di uno o più microbicidi efficaci e alla costante educazione sessuale preventiva almeno parzialmente riesca a contenere la propagazione dell'infezione. Ciò rappresenta un importante risultato intermedio in attesa della scoperta di una strategia vaccinale preventiva, nonché della soluzione del problema complesso dell'esportazione delle terapie virali di combinazione nei Paesi a basso reddito che, da soli, rappresentano il 90% dell'epidemia dell'infezione oggi.
Bibliografia
Alfano, Poli 2005: Alfano, Massimo - Poli, Guido, Role of cytokines and chemokines in the regulation of innate immunity and HIV infection, "Molecular immunology", 42, 2005, pp. 161-82.
Berger 1998: Berger, Edward A. e altri, A new classification for HIV-1, "Nature", 1998, 391, p. 240.
Chiu 2005: Chiu, Yi-Lin, HIV Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells, "Nature", 435, 2005, pp. 108-114.
Emery 2000: Emery, Sean e altri, Pooled analysis of 3 randomized, controlled trials of interleukin-2 therapy in adult human immunodeficiency virus type 1 disease, "Journal of infectious diseases", 182, 2000, pp. 428-434.
Ferrantelli 2004: Ferrantelli, Flavia e altri, Complete protection of neonatal rhesus macaques against oral exposure to pathogenic simian-human immunodeficiency virus by human anti-HIV monoclonal antibodies, "Journal of infectious diseases", 189, 2004, pp. 2167-2173.
Gao 1999: Gao, Feng e altri, Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes, "Nature", 397, 1999, pp. 436-441.
Geijtenbeek 2000: Geijtenbeek, Teunis B.H. e altri, DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells, "Cell", 100, 2000, pp. 587-597; 491-494.
Haynes 2005: Haynes, Barton F. e altri, Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies, "Science", 308, 2005, pp. 1906-1908.
Ho 1995: Ho, David D. e altri, Rapid turn-over of plasma virions and CD4 lymphocytes in HIV-1 infection, "Nature", 373, 1995, pp. 123-126.
Hudson 2005: Hudson, Christopher P., Zidovudine monotherapy and the prevention of mother-to-child HIV-1 transmission, "Lancet infectious diseases", 5, 2005, p. 68.
Lazzarin 2003: Lazzarin, Adriano e altri, Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia, "New England journal of medicine", 348, 2003, pp. 2186-2195.
Mattapallil 2005: Mattapallil, Joseph J. e altri, Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection, "Nature", 434, 2005, 1093-1097.
Pantaleo 1993: Pantaleo, Giuseppe e altri, HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease, "Nature", 362, 1993, pp. 355-358.
Potterat 2006: Potterat, John J. e altri, The protective effect of male circumcision as a faith lift for the troubled paradigm of HIV epidemiology in Sub-Saharan Africa, "PLoS medicine", 3, 2006, p. e64 (author reply e67).
Rizzardi 2002: Rizzardi, G. Paolo e altri, Treatment of primary HIV-1 infection with cyclosporin A coupled with highly active antiretroviral therapy, "Journal of clinical investigation", 109, 2002, pp. 681-688.
Schröder 2002: Schröder, Astrid e altri, HIV-1 integration in the human genome favors active genes and local hotspots, "Cell", 110, 2002, pp. 521-529.
Shattock, Moore 2003: Shattock, Robin J. - Moore, John P., Inhibiting sexual transmission of HIV-1 infection, "Nature reviews. Microbiology", 1, 2003, pp. 25-34.
Sheehy 2003: Sheehy, Anne M. - Gaddis, Nathan C. - Malim, Michael H., The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif, "Nature medicine", 9, 2003, pp. 1404-1407.
Stevenson 2003: Stevenson, Mario, HIV-1 pathogenesis, "Nature medicine", 2003, 9, pp. 853-860.
Stremlau 2004: Stremlau, Matthew e altri, The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys, "Nature", 427, 2004, pp. 848-853.
Towers 2003: Towers, Grey J. e altri, Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors, "Nature medicine", 9, 2003, pp. 1138-1143.
Trkola 2005: Trkola, Alexandra e altri, Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies, "Nature medicine", 11, 2005, pp. 615-622.
Wei 1995: Wei, Xiping e altri, Viral dynamics in human immunodeficiency virus type 1 infection, "Nature", 373, 1995, pp. 117-122.