
Parkinson, malattia di
Parkinson, malattia di
Quella di Parkinson è una delle più frequenti malattie degenerative del sistema nervoso centrale, e si caratterizza per la presenza della triade sintomatologica costituita da tremore, rigidità e acinesia. Alcuni studiosi di storia della medicina ne attribuiscono una prima grossolana descrizione a Leonardo da Vinci: il genio rilevava acutamente i movimenti tremolanti, involontari e irrefrenabili dei pazienti, e li descriveva paragonandoli ai brividi e ai tremori mostrati da chi abbia molto freddo.
Tuttavia, fu soltanto nel 1817 che James Parkinson, medico e letterato inglese, in una monografia che sarebbe diventata una pietra miliare della medicina, descrisse in modo magistrale la malattia con il nome di 'paralisi agitante'. Con la sua accurata analisi, egli ebbe il merito e la capacità di raggruppare un insieme di sintomi che, a una prima osservazione, potevano apparire privi di collegamento e poco rilevanti. Fu un altro grande personaggio della storia della medicina, Jean-Martin Charcot, che, critico rispetto al termine paralisi, avendo osservato che i pazienti erano in grado di esprimere una buona forza muscolare e avendo preso in considerazione altri sintomi quali la rigidità e la micrografia, suggerì di chiamare la malattia con il nome del medico che per primo l'aveva descritta, e da allora essa è universalmente conosciuta come 'malattia di Parkinson' (MP).
Dopo questi primi studi, che erano basati esclusivamente sulla descrizione clinica, molti sono stati i contributi che hanno ulteriormente caratterizzato e definito la MP, e alcuni di essi costituiscono le pietre miliari della conoscenza che ne abbiamo attualmente. Nel 1912 Frederick H. Lewy individuò nella substantia nigra e nel nucleo dorsale del vago le caratteristiche inclusioni ialine citoplasmatiche, che da lui presero il nome e che in seguito, nel 1965, sarebbero state caratterizzate dal punto di vista strutturale da Philip E. Duffy e Virginia M. Tennyson. Constantin Trétiakoff nel 1919 e Constantin Von Economo nel 1920 posero l'accento sul possibile coinvolgimento della sostanza nera, in relazione alla grave moria di cellule neuronali che colpiva questo importante centro dei nuclei della base; in seguito, J.G. Greenfield e F.D. Bosanquet evidenziarono per primi dettagliatamente nella sostanza nera la perdita neuronale e i corpi di Lewy.
Agli inizi degli anni Sessanta, O. Bertel, Herbert Ehringer e Oleh Hornykiewicz individuarono in pazienti affetti da MP una netta riduzione del contenuto di un neurotrasmettitore, la dopammina, nelle cellule cerebrali e in particolare in quelle dello striato. Nel 1962 Hornykiewicz, Walter Birkmayer e André Barbeau utilizzarono in quegli stessi pazienti la levodopa (L-DOPA), un precursore della dopammina, riuscendo a ottenere una risposta clinica sbalorditiva. Nel 1969 lo stesso Birkmayer associò alla levodopa un inibitore periferico della dopa-decarbossilasi, riuscendo a osservare che ciò consentiva di impiegare dosi minori di levodopa e di ridurre dunque in maniera drastica gli effetti collaterali periferici. Queste costituiscono le tappe fondamentali nella storia della terapia della MP.
Nei primi anni Ottanta un'osservazione casuale ha permesso di compiere un ulteriore passo avanti nella comprensione dei meccanismi patogenetici di questa malattia: sette tossicodipendenti americani hanno presentato una sintomatologia analoga a quella della MP dopo essersi iniettati per via endovenosa un derivato sintetico dell'eroina. Si è scoperto che la droga era contaminata da una sostanza tossica denominata MPTP (1-metil-4-fenil-1,2,3,6-tetraidropiridina), la quale produceva gli stessi sintomi una volta iniettata negli animali da esperimento. Queste osservazioni hanno reso disponibile un modello sperimentale di malattia la cui importanza si è dimostrata essenziale per la ricerca futura sulla MP.
Fattori di rischio
Nonostante sia sempre stata prestata una grande attenzione ai fattori potenzialmente implicati nella patogenesi della malattia di Parkinson, ancora oggi è impossibile identificare quali individui siano effettivamente a rischio. Per quanto concerne le variabili ambientali, grande interesse ‒ specialmente dopo che, nel 1982, fu segnalata l'insorgenza di una sindrome parkinsoniana da MPTP (1-metil-4-fenil-1,2,3,6-tetraidropiridina) ‒ hanno ricevuto le sostanze tossiche, in particolare quelle contenute negli erbicidi e nei pesticidi: il più indiziato è il paraquat, proprio per via delle sue marcate somiglianze con la MPTP. Numerose ricerche epidemiologiche hanno evidenziato che negli ambienti rurali esiste una maggiore predisposizione a sviluppare la MP, suggerendo che pesticidi, erbicidi e acque contaminate possano incrementare il rischio.
Dopo un'attenta analisi, Jay M. Gorell e i suoi collaboratori hanno appurato che il rischio di sviluppare la MP aumentava in agricoltori che fossero stati esposti in maniera cronica per più di 10 anni a vari pesticidi, erbicidi e fungicidi, ma non hanno potuto stabilire se tale aumento fosse associato a uno specifico gruppo di sostanze. La letteratura riporta alcune segnalazioni sporadiche di manifestazioni parkinsoniane comparse dopo un'esposizione prolungata e costante a un fungicida (maneb) o a pesticidi quali il paraquat e il rotenone. Il fumo di sigaretta, al contrario, sembrerebbe costituire un fattore protettivo, ma a tale proposito i dati sono contrastanti. Diversi studi hanno focalizzato l'attenzione su una dieta povera di sostanze antiossidanti, che potrebbe predisporre il soggetto a risentire maggiormente di una vasta gamma di fattori esogeni ed endogeni, mentre l'effetto opposto potrebbe derivare da una dieta ricca di antiossidanti come la vitamina E. Fra gli altri possibili fattori di rischio è stata individuata l'esposizione a metalli pesanti, come per esempio il manganese. Infine, è stato postulato che alcuni tratti premorbosi di personalità, quali la depressione e l'anedonia, predispongano a un rischio maggiore di sviluppare la MP. Non è possibile, tuttavia, stabilire se tali tratti costituiscano un fattore di rischio o invece una conseguenza di un deficit precoce di dopammina.
Genetica
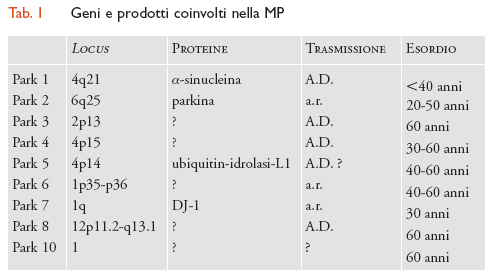
Fino a poco tempo fa la MP era considerata il prototipo delle malattie sporadiche non ereditarie. I grandi progressi compiuti in campo genetico hanno però permesso di rivedere tale posizione e, attualmente, ai fattori ereditari è riconosciuto un importante ruolo patogenetico. Diversi studi epidemiologici hanno infatti evidenziato una maggiore prevalenza della MP in soggetti con familiari già affetti. Sono state inoltre identificate forme ereditarie con modalità di trasmissione autosomica dominante (A.D.) e recessiva (a.r.). Per esempio, nel 1997 è stata identificata una mutazione del gene dell'α-sinucleina in una famiglia italiana e in tre famiglie di origine greca affette da una forma di MP a trasmissione autosomica dominante. I geni implicati in queste forme familiari codificano per proteine che sembrerebbero rivestire un ruolo importante nell'eziopatogenesi della malattia, e sono quindi risultati di importanza cruciale per la comprensione della forma considerata idiopatica. Allo stato attuale si conoscono 9 geni coinvolti nella MP (tab. 1).
Patologia
La principale caratteristica patologica della malattia di Parkinson è costituita dalla perdita dei neuroni dopamminergici contenenti neuromelanina (neuroni pigmentati) della pars compacta della substantia nigra mesencefalica. La degenerazione neuronale non è tuttavia limitata alla substantia nigra, ma si può estendere fino a interessare le cellule colinergiche del nucleo di Meynert e i neuroni dell'ipotalamo, del locus coeruleus, della corteccia entorinale, del giro del cingolo, del bulbo olfattivo e dei gangli simpatici e parasimpatici. È importante osservare che la perdita dei neuroni dopamminergici della pars compacta della substantia nigra non è casuale né ubiquitaria: nelle fasi iniziali della malattia essa interessa unicamente la regione ventrolaterale, per poi estendersi a quella medioventrale e infine a quella dorsale; la sintomatologia clinica compare soltanto quando è scomparso oltre il 70% delle cellule della regione ventrolaterale. La perdita neuronale fisiologica del soggetto anziano sano segue invece una topografia opposta.
Un'altra caratteristica della MP è la presenza di neuriti distrofici, denominati 'neuriti di Lewy', e di inclusioni intracitoplasmatiche eosinofile, conosciute con il termine di corpi di Lewy. Tali inclusioni hanno una forma sferoidale e sono composte da prodotti di degradazione delle proteine, degli acidi grassi, della sfingomielina e dei polisaccaridi. Si trovano tipicamente a livello della substantia nigra, ma con una certa frequenza si possono riscontrare anche nel locus coeruleus, nella corteccia cerebrale, nel nucleo basale di Meynert, nell'ipotalamo e nei gangli simpatici del sistema nervoso autonomo. I corpi di Lewy sono un reperto costante e costituiscono quindi un marker anatomopatologico specifico della MP idiopatica. Uno dei loro maggiori componenti è l'α-sinucleina, una proteina di 15-20 kDa che viene normalmente espressa nei terminali nervosi e che sembrerebbe avere un ruolo nel ricambio vescicolare a livello sinaptico e nella plasticità neuronale. Un'alterazione della sua struttura quaternaria potrebbe determinare la sua precipitazione, con conseguente formazione di aggregati insolubili. È dunque interessante notare che pesticidi quali il rotenone e il paraquat possono determinare modificazioni strutturali e favorire l'aggregazione dell'α-sinucleina in vitro.
I meccanismi responsabili della morte dei neuroni dopamminergici della substantia nigra nella MP sono ancora oggetto di speculazione: le ipotesi proposte sono numerose, ma quella più attuale fa riferimento al fenomeno dell'apoptosi (morte cellulare programmata). Altri fattori implicati nella progressiva degenerazione neuronale sono una disfunzione del sistema mitocondriale, lo stress ossidativo e l'eccitotossicità. Nella substantia nigra dei pazienti affetti da MP è stato osservato un deficit del complesso I della catena respiratoria, con conseguente ridotta sintesi di ATP. Inoltre, a livello dello stesso complesso mitocondriale, studi su modelli animali hanno riscontrato la neurotossicità dell'MPP, che è un metabolita dell'MPTP. Per quanto concerne lo stress ossidativo, nel corso del metabolismo neuronale fisiologico si formano i radicali liberi, che sono in grado di aggredire i lipidi, le proteine di membrana, il DNA e altre strutture molecolari, generando ulteriori radicali liberi e portando infine alla morte cellulare. L'aumento dei radicali liberi intracellulari potrebbe essere secondario a un eccesso di produzione oppure a una diminuzione di efficacia del processo di eliminazione. È stato postulato che nella MP sia presente una quantità eccessiva di fattori ossidanti fin dalle prime fasi di malattia. Il terzo meccanismo che potrebbe essere implicato nella neurodegenerazione è quello dell'eccitotossicità. In particolare, la morte cellulare potrebbe essere una conseguenza di un eccesso di glutammato e di una maggiore permeabilità cellulare al calcio mediata dall'attivazione dei recettori glutammatergici NMDA (N-metil-d-aspartato). L'idea di un possibile ruolo eccitotossico del glutammato nasce dal fatto che i neuroni dopamminergici nigrostriatali sono ricchi di recettori NMDA e ricevono numerose afferenze glutammatergiche dalla corteccia cerebrale e dal nucleo subtalamico, e dal fatto che il pretrattamento con antagonisti dei recettori NMDA sembra ridurre, in animali da esperimento, il danno causato alle cellule dopamminergiche da tossine mitocondriali.
Fisiopatologia
I gangli o nuclei della base, in particolare il nucleo caudato e il putamen (che insieme formano lo striato), il globo pallido, la substantia nigra, il nucleo subtalamico e il nucleo rosso, costituiscono il sistema extrapiramidale. Tale circuito neuronale lavora di concerto con la corteccia cerebrale e con il talamo, ed è deputato alla pianificazione e all'attuazione del programma motorio. In condizioni fisiologiche, lo striato riceve afferenze corticali eccitatorie prevalentemente glutammatergiche, e invia efferenze inibitorie GABA-ergiche al globo pallido interno e quindi al talamo attraverso due vie, una diretta e una indiretta. La prima raggiunge direttamente il pallido interno, mentre la seconda presenta due stazioni intermedie, il pallido esterno e il nucleo subtalamico, formato da neuroni eccitatori glutammatergici. Le due vie sono entrambe modulate dalla dopammina, che agisce su distinti recettori localizzati sui neuroni GABA-ergici striatali: essa esercita un effetto eccitatorio sulla via diretta, mediante l'interazione con i recettori D1 localizzati sui neuroni che contengono sostanza P e dinorfina, mentre inibisce la via indiretta attraverso i recettori della classe D2 localizzati sulle cellule che contengono encefaline. In condizioni normali, per pianificare ed eseguire un determinato movimento volontario è necessario attivare la parte della corteccia in cui quel gesto è contenuto nella cosiddetta 'memoria procedurale', e questo potrebbe essere il ruolo della via diretta; è anche necessario, tuttavia, inibire nel contempo altre aree cerebrali, che potrebbero attivare gesti interferenti od opposti, ed è qui che potrebbe entrare in gioco la via indiretta. I gangli della base sembrano dunque esercitare una specifica funzione di controllo motorio sulla corteccia, che svolgerebbero mediante l'interazione con il talamo.
Clinica
La storia della malattia riconosce una fase presintomatica che precede di alcuni anni l'esordio sintomatologico propriamente detto. Le manifestazioni riscontrabili in questa fase sono aspecifiche e di difficile interpretazione: il paziente può lamentare dolori a carico del rachide, una facile affaticabilità, una riduzione del tono dell'umore. La fase sintomatica esordisce generalmente verso i 50-60 anni di età, con disturbi motori tipicamente monolaterali. Dopo alcuni anni i disturbi tendono a generalizzarsi, e vi si associano manifestazioni non motorie. Infine, la malattia evolve verso uno scadimento delle condizioni fisiche generali. La fase terminale vede il progressivo peggioramento delle funzioni motorie, fino alla perdita completa dell'autonomia. In base alla risposta alla terapia con la levodopa è possibile distinguere un periodo definito 'luna di miele', durante il quale il paziente beneficia di uno stato di benessere dopo l'assunzione farmacologica. Trascorso qualche anno, tuttavia, si assiste a una progressiva riduzione della risposta e alla comparsa delle prime complicanze motorie indotte dalla terapia, fino alla fase di scadimento in cui queste ultime diventano sempre più invalidanti. L'evoluzione è estremamente variabile e non prevedibile, sebbene si abbia una prognosi migliore nelle forme prevalentemente tremorigene.
Una delle manifestazioni cardinali della malattia di Parkinson è il tremore, che è anche una delle cause principali che induce il paziente a effettuare una visita medica. Si tratta di un'oscillazione ritmica involontaria, più o meno regolare, di una parte del corpo attorno a un punto fisso, generalmente su di un solo piano. Nella MP la forma tipica di tremore compare a riposo ed è ritmica, con una frequenza che si aggira attorno ai 4 cicli al secondo. All'esordio interessa generalmente una mano, cui conferisce un movimento tipico, di flesso-pronazione del carpo, adduzione e abduzione del pollice e flesso-estensione dell'indice, che viene anche chiamato 'contare monete' o 'far pillole'; successivamente si può estendere al braccio e al piede omolaterali e, quindi, controlaterali. Possono essere interessate anche altre parti del corpo, tra cui la mandibola e le palpebre. Il tremore presenta notevoli fluttuazioni di ampiezza: esso risulta infatti accentuato dallo stress o dall'esecuzione di compiti mentali, mentre si allevia fino a scomparire durante il sonno e con il rilassamento.
Il secondo segno cardinale della MP è la rigidità, che è l'espressione di un aumento del tono di tutti i gruppi muscolari, sia agonisti sia antagonisti. Ciò si apprezza alla mobilizzazione passiva lenta di un arto, sotto forma di una resistenza costante che perdura per tutto il movimento sia a carico dei muscoli flessori sia di quelli estensori. Una caratteristica che gli è associata è il segno della ruota dentata, che si esprime come un cedimento a scatti della rigidità durante la mobilizzazione. Con il progredire della malattia, il prevalere del tono dei muscoli flessori su quello degli estensori fa assumere al paziente una postura denominata 'camptocormica', con il capo e il tronco in flessione e il centro di gravità spostato in avanti, gli avambracci in semi-flesso-pronazione, e le anche e le ginocchia lievemente flesse.
La terza manifestazione caratteristica, considerata la più invalidante, è l'acinesia, che si esprime come difficoltà nell'avviare e nell'eseguire i movimenti. La bradicinesia, che gli è strettamente associata, consiste invece nella riduzione dei movimenti in ampiezza. Questi problemi si manifestano in diversi modi e, come la rigidità, tendono a evolvere nel tempo. In particolare, si osserva una riduzione dell'ammiccamento, il volto diventa scarsamente espressivo, i movimenti pendolari della deambulazione si fanno meno ampi, l'andatura procede a piccoli passi, e il paziente è incerto nell'iniziare a camminare. I disturbi nella deambulazione possono evolvere nel cosiddetto fenomeno del freezing (o 'congelamento'), ossia in una difficoltà a iniziare la marcia in cui il paziente ha tipicamente la sensazione di avere i piedi attaccati al pavimento e necessita di compiere diversi passi sul posto senza riuscire ad avanzare. Tale difficoltà si ripresenta anche nell'inversione del senso di marcia, e quando è necessario passare attraverso spazi stretti. La scrittura si modifica, facendosi minuta benché sempre comprensibile (micrografia); la deglutizione diventa meno efficace, con conseguente ristagno di saliva nella bocca. La festinazione è un'accelerazione progressiva dei movimenti sequenziali, che si manifesta durante la deambulazione con la tendenza ad aumentare la velocità, come a rincorrere il proprio centro di gravità. Anche la voce si modifica, divenendo flebile e monotona, e per effetto della festinazione la pronuncia delle parole diventa veloce e a volte incomprensibile. È infine frequente la comparsa di un'instabilità posturale, le cui cause vanno ricercate nella riduzione dei riflessi posturali associata alla rigidità e all'acinesia.

Alla triade patognomonica della MP si possono associare nel tempo altre manifestazioni cliniche (tab. 2). Comuni sono i disturbi neurovegetativi, come l'ipotensione ortostatica, le disfunzioni urinarie e sessuali e i sintomi sensoriali quali algie (di tipo urente o trafittivo) o parestesie. Importanti fenomeni associati sono, inoltre, i disturbi comportamentali e il deterioramento cognitivo. Fra i primi, quelli che più frequentemente si accompagnano alla MP sono la depressione, l'apatia, l'ansia, le psicosi e le allucinazioni (queste ultime sono generalmente determinate dalla terapia antiparkinsoniana). La prevalenza della depressione varia dal 3 al 70% nei diversi studi. Nel 43% dei pazienti la depressione si presenta prima dell'esordio della MP. Fra i fattori di rischio per la depressione sono stati osservati un esordio precoce della MP, la presenza di instabilità posturale, una storia passata di depressione, elevati livelli di ansia e il sesso femminile. Circa il 40% dei pazienti presenta demenza, con un'incidenza che aumenta progressivamente dopo la sesta decade; essa si esprime come una sindrome disesecutiva, ovvero come una perdita dell'abilità di pianificare, organizzare e regolare un programma motorio. I fattori di rischio che maggiormente si associano alla demenza nella MP sono: età di esordio della malattia, depressione, alterazioni neurologiche atipiche quali precoci segni autonomici, modesta risposta alla terapia dopamminergica e sintomi simmetrici.

Accanto alla costellazione sintomatologica descritta, nel corso degli anni compaiono le manifestazioni motorie − di natura acinetica, ovvero le fluttuazioni, e di natura ipercinetica, ovvero le discinesie − legate al trattamento farmacologico (tab. 3). Quelle di natura acinetica, comunemente denominate 'fluttuazioni motorie', vengono distinte a loro volta in prevedibili e imprevedibili, in base alla loro correlazione con l'assunzione della levodopa. Le prime fluttuazioni motorie a comparire sono quelle prevedibili, che comprendono il deterioramento di fine dose, o wearing off, il ritardo della risposta, o delayed on, e la perdita di risposta, o no on. Le fluttuazioni motorie prevedibili si manifestano come periodi di acinesia per ricomparsa precoce dei sintomi parkinsoniani in stretta dipendenza cronologica dall'assunzione farmacologica. L'acinesia di fine dose, in particolare, consiste nella ripresa della sintomatologia prima dell'assunzione successiva della levodopa, e rappresenta la forma più comune di fluttuazione motoria. Al termine dell'effetto della dose precedente, il paziente riferisce una ripresa dei segni e dei sintomi parkinsoniani quali la bradicinesia, il tremore, la rigidità o la difficoltà ad alzarsi dalla sedia. Durante il periodo off (di blocco motorio) può comparire anche una sintomatologia sensoriale, che comporta dolore o parestesie, o comportamentale, consistente in depressione, ansia, disforia o attacchi di panico. L'acinesia al risveglio e quella notturna sono anch'esse espressione del deterioramento di fine dose. Le fluttuazioni imprevedibili, ovvero i fenomeni on-off o yo-yoing, compaiono in una fase successiva della progressione della malattia e non sono correlate alla somministrazione farmacologica. Prediligono il periodo postprandiale o comunque degli orari fissi, e consistono in un rapido passaggio, nell'arco di pochi minuti, da uno stato di motilità a uno di acinesia.
Le discinesie comprendono movimenti coreici, balistici, distonici e mioclonici, che interessano il capo, il tronco, gli arti e, occasionalmente, la muscolatura respiratoria. Le prime a comparire si evidenziano durante la fase on in cui la levodopa presenta un maggiore livello plasmatico (discinesia di picco dose). Le discinesie indotte da levodopa interessano in genere il piede omolaterale all'emicorpo maggiormente colpito dalla sintomatologia parkinsoniana. La variante distonica delle discinesie, che si esprime sotto forma di crampi dolorosi al piede durante il wearing off notturno o diurno, compare quando la levodopa raggiunge i livelli plasmatici più alti o quelli più bassi (distonia di picco, di fine dose, del mattino).
La frequenza delle complicanze motorie indotte dalla levodopa dipende da diverse variabili, tra cui l'età di esordio della malattia e di introduzione della terapia farmacologica, la durata del trattamento e la dose totale giornaliera. In particolare, tali manifestazioni sono correlate all'uso cronico di levodopa, e in termini fisiopatologici si ritiene che le fluttuazioni motorie siano associate a una stimolazione pulsatile dei recettori dopamminergici indotta dal farmaco. Molti pazienti esposti ad alti dosaggi non manifestano complicanze motorie, al contrario di altri che, pur assumendo basse dosi giornaliere, presentano discinesie dopo pochi anni di trattamento. Una spiegazione potrebbe essere ricercata nella variabilità individuale determinata da una predisposizione genetica.
Diagnosi


Nell'accezione comune la diagnosi della MP è considerata semplice. In realtà, fino a un decennio fa solo il 75% delle diagnosi cliniche trovava una conferma autoptica. La difficoltà nasce dal fatto che la MP può essere confusa con altre sindromi parkinsoniane secondarie e atipiche (tab. 4). Oggi, grazie all'introduzione di criteri diagnostici più accurati e delle moderne tecniche di neuroimaging, la conferma autoptica ha raggiunto delle percentuali superiori. I criteri diagnostici proposti dalla United Kingdom Parkinson's Disease Society Brain Bank, successivamente revisionati da Douglas J. Gelb e dai suoi collaboratori, distinguono la malattia in probabile, possibile e certa sulla base del riscontro dei suoi segni tipici e dell'esclusione dei segni atipici che orientano verso diagnosi alternative (tab. 5). Vengono presi in considerazione segni motori cardinali quali il tremore a riposo, la rigidità, la bradicinesia e l'esordio asimmetrico. Di importanza non trascurabile sono inoltre la vistosa risposta alla levodopa e la presenza di discinesie da essa indotte nelle fasi avanzate di malattia.
I segni atipici che orientano verso una diagnosi alternativa sono la precoce instabilità posturale, il freezing precoce, la scarsa risposta alla levodopa, le allucinazioni non legate all'assunzione della terapia e la paralisi verticale dello sguardo. È fondamentale una attenta raccolta anamnestica circa l'eventuale assunzione di farmaci o sostanze tossiche in grado di determinare un parkinsonismo secondario, la modalità di esordio e di evoluzione dei sintomi, e l'eventuale familiarità per tremore o patologie dell'andatura. Grande importanza riveste, inoltre, un accurato esame obiettivo neurologico volto alla ricerca dei segni tipici e atipici. Recentemente alcuni autori hanno focalizzato l'attenzione su alcuni quadri clinici che possono venire confusi con la MP in fase precoce per il riscontro di tremore, rigidità, acinesia o disturbi dell'andatura. Importante è anche l'esclusione, tramite indagini ematobiochimiche e genetiche, di altre patologie, come il morbo di Wilson, la sindrome di Fahr secondaria a ipoparatiroidismo, la neuroacantocitosi, la malattia di Huntington o la distonia dopasensibile. Un utile ausilio per la diagnosi sono le tecniche di neuroimaging (tomografia computerizzata e risonanza magnetica), non tanto per il riscontro dei segni caratteristici della MP quanto per la possibilità di individuare eventuali lesioni secondarie di natura vascolare, neoplastica o infiammatoria, oppure anomalie tipiche di parkinsonismi su base degenerativa.
Recentemente sono state introdotte metodiche di neuroimaging funzionale (come la PET, Positron emission tomography, e la SPECT, Single photon emission computerized tomography) che sono in grado di riconoscere la perdita neuronale dopamminergica del sistema nigrostriatale, discriminando così la MP dalle patologie in cui questa non si verifica, quali il tremore essenziale, i parkinsonismi vascolari e quelli iatrogeni (per lo più da farmaci). Più complessa è la discriminazione dai parkinsonismi atipici (atrofia multisistemica, paralisi sopranucleare progressiva, degenerazione corticobasale, demenza a corpi di Lewy) che sono anch'essi caratterizzati da alterazioni nigrostriatali. La PET è considerata la metodica ottimale, per via dell'alta risoluzione delle sue immagini tridimensionali, tuttavia essa non è attualmente di uso comune a causa dei costi elevati. Grazie alla PET è possibile valutare lo stato del tessuto sottostante. In particolare, nei pazienti affetti da MP si osserva una ridotta captazione della levodopa marcata con 18F a livello del putamen, espressione di una perdita di terminali nervosi dopamminergici. La metodica funzionale più comunemente utilizzata, per via dei costi inferiori rispetto alla precedente, è la SPECT. In questo caso il ligando radiomarcato (il più utilizzato è il 123β-CIT) si lega in maniera selettiva al trasportatore dopamminergico (DAT) che è localizzato a livello presinaptico delle terminazioni nervose nigrostriatali. Anche in questo caso, nella MP si osserva una ridotta captazione particolarmente in riferimento al putamen. L'associazione di traccianti in grado di legarsi selettivamente al recettore striatale D2 (123I-iodobenzamide) può essere utile per la discriminazione della MP dai parkinsonismi atipici in cui si osserva anche una degenerazione dei terminali postsinaptici.
Terapia
Per il trattamento della malattia di Parkinson sono disponibili diverse possibilità terapeutiche. Il farmaco considerato più efficace è la levodopa, precursore della dopammina. La sua efficacia è stata dimostrata per la prima volta nel 1961. Pochi anni dopo essa è stata associata a inibitori delle decarbossilasi periferiche (benserazide e carbidopa), enzimi in grado di ridurre il suo metabolismo periferico, aumentandone, di conseguenza, la disponibilità a livello encefalico, e riducendo gli effetti collaterali a livello periferico. Ciò ha permesso che la levodopa venisse impiegata su larga scala a dosaggi notevolmente inferiori rispetto a quelli precedentemente utilizzati. Tale farmaco determina un miglioramento della sintomatologia con particolare riguardo alla rigidità e all'acinesia. Sebbene efficace contro i disturbi motori, il suo utilizzo nelle prime fasi della malattia è stato messo in discussione in quanto associato alla comparsa più precoce di complicanze quali fluttuazioni motorie e discinesie.
I possibili effetti collaterali della levodopa comprendono disturbi di natura psichiatrica come le psicosi, disturbi gastrointestinali quali nausea e vomito, l'ipotensione ortostatica e, raramente, il glaucoma ad angolo chiuso. Un'alternativa terapeutica nelle prime fasi della malattia è offerta dai dopammino-agonisti, che sono in grado di interagire con i recettori dopamminergici a livello striatale. Sebbene di minor efficacia rispetto alla levodopa, essi sembrerebbero determinare minori complicanze motorie a lungo termine. Altri farmaci utilizzati, anche se con minore frequenza per la possibile presenza di effetti collaterali severi, sono gli anticolinergici (indicati in particolare per alleviare la componente tremorigena), gli inibitori delle MAO-B (monoamminossidasi di tipo B) e l'amantadina. Con il progredire della malattia il trattamento diventa sempre più complesso per via delle complicanze indotte dall'uso prolungato di levodopa.
Le strategie terapeutiche utilizzate per ridurre le fluttuazioni motorie sono numerose, ma ancora non completamente soddisfacenti. Come primo passo è utile ottimizzare l'assorbimento e il trasporto di levodopa mediante l'introduzione di una dieta povera di proteine (per ridurre la competizione a livello gastrico). Una seconda possibilità è quella di stabilizzare i livelli plasmatici di levodopa frazionando le dosi giornaliere o introducendo gli inibitori delle COMT (Catecol-o-metiltransferasi). Può essere anche utile l'associazione con dopammino-agonisti e, al fine di aumentare la concentrazione della dopammina a livello striatale, possono essere aggiunti alla terapia gli inibitori delle MAO. Studi recenti hanno confermato l'efficacia dell'infusione continua di apomorfina per ridurre le fluttuazioni motorie, e della sua infusione intermittente per il trattamento dei blocchi motori.
Anche per il trattamento delle discinesie sono disponibili diverse strategie terapeutiche finalizzate a ottimizzare la risposta. L'obiettivo è quello di mantenere costanti nel tempo i livelli plasmatici di levodopa e di ridurne l'assunzione giornaliera. I tentativi consistono in frazionamenti delle dosi giornaliere di levodopa o in una sua riduzione e associazione con dopammino-agonisti e inibitori delle COMT. Utile, soprattutto per il trattamento delle discinesie di picco, è l'introduzione dell'amantadina. In caso di fallimento della terapia convenzionale, possono essere anche prese in considerazione tecniche di chirurgia stereotassica che sono in grado di intervenire sulla modulazione del circuito dei nuclei della base. In particolare, la stimolazione cerebrale profonda (DBS, Deep brain stimulation) utilizza una stimolazione elettrica cronica ad alta frequenza che inibisce l'attività elettrica del nucleo subtalamico. Tale metodica è efficace sulle complicanze motorie da uso cronico di levodopa, quali le fluttuazioni motorie e le discinesie, ma non sulle complicanze psichiatriche. Sono ancora in corso di studio tecniche di trapianto allogenico di cellule mesencefaliche fetali e approcci di terapia genica.
Bibliografia
Adler 2005: Adler, Charles H., Nonmotor complications in Parkinson's disease, "Movement disorders", 20 (suppl. 11), 2005, pp. 23-29.
Emre 2003: Emre, Murat, Dementia associated with Parkinson's disease, "Lancet neurology", 2, 2003, pp. 229-237.
Fall 2003: Fall, Per-Arne e altri, Survival time, mortality, and cause of death in elderly patients with Parkinson's disease: a 9-year follow-up, "Movement disorders", 18, 2003, pp. 1312-1316.
Ferraz 1988: Ferraz, Henrique B. e altri, Chronic exposure ___to the fungicide maneb may produce symptoms and signs of CNS manganese intoxication, "Neurology", 38, 1988, pp. 550-553.
Forno 1996: Forno, Lysia S., Neuropathology of Parkinson's disease, "Journal of neuropathology and experimental neurology", 55, 1996, pp. 259-272.
Gelb 1999: Gelb, Douglas J. - Oliver, Eugene - Gilmen, Sid, Diagnostic criteria for Parkinson'disease, "Archives of neurology", 56, 1999, pp. 33-39.
Goetz 2005: Goetz, Christopher G. e altri, Evidence-based medical review update: pharmacological and surgical treatments of Parkinson's disease: 2001 to 2004, "Movement disorders", 20, 2005, pp. 523-539.
Gorell 1998: Gorell, Jay M. e altri, The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living, "Neurology", 50, 1998, pp. 1346-1350.
Hardy 2003: Hardy, John - Cookson, Mark R. - Singleton, Andrew, Gene and parkinsonism, "Lancet neurology", 2, 2003, pp. 221-228.
Jankovic 1990: Jankovic, Joseph e altri, Variable expression of Parkinson's disease: a base-line analysis of the DATATOP cohort. The Parkinson Study Group, "Neurology", 40, 1990, pp. 1529-1534.
Jankovic 2005: Jankovic, Joseph, Motor fluctuations and dyskinesias in Parkinson's disease: clinical manifestations, "Movement disorders", 20 (suppl. 11), 2005, pp. 11-16.
Lang 1998: Lang, Anthony E. - Lozano, Andres M., Parkinson's disease. First of two parts, "New England journal of medicine", 339, 1998, pp. 1044-1053.
Lang 1998: Lang, Anthony E. - Lozano, Andres M., Parkinson's disease. Second of two parts, "New England journal of medicine", 339, 1998, pp. 1130-1143.
Langston 1984: Langston, John M. e altri, 1-Methyl-4-phenylpyridinium ion (MPP+): identification of a metabolite of MPTP, a toxin selective to the substantia nigra, "Neuroscience letters", 48, 1984, pp. 87-92.
Nutt, Wooten 2005: Nutt, John G. - Wooten, G. Frederick, Clinical practice. Diagnosis and initial management of Parkinson's disease, "New England journal of medicine", 353, 2005, pp. 1021-1027.
Oliveri 1999: Oliveri, Robin L. e altri, Dopamine D2 receptor gene polymorphism and the risk of levodopa-induced dyskinesias in PD, "Neurology", 53, 1999, pp. 1425-1430.
Pearce 1989: Pearce, John M., Aspects of the history of Parkinson's disease, "Journal of neurology, neurosurgery, and psychiatry", 52 (suppl.), 1989, pp. 6-10.
Piccini, Whone 2004: Piccini, Paola - Whone, Alan, Functional brain imaging in the differential diagnosis of Parkinson's disease, "Lancet neurology", 3, 2004, pp. 284-290.
Takahashi, Wakabayashi 2001: Takahashi, Hiroki - Wakabayashi, Kenichi, The cellular pathology of Parkinson's disease, "Neuropathology", 21, 2001, pp. 315-322.
Tolosa 2005: Tolosa, Eduardo - Wenning, Gregor - Poewe, Werner, The diagnosis of Parkinson's disease, "Lancetneurology", 5, 2005, pp. 75-86.
Von Bohlen und Halbach 2004: von Bohlen und Halbach, Oliver - Schober, Andreas - Krieglstein, Kerstin, Genes, proteins, and neurotoxins involved in Parkinson's disease, "Progress in neurobiology", 73, 2004, pp. 151-177.