
Radicali liberi
Radicali liberi
Un atomo o un gruppo di atomi in cui è presente un elettrone non accoppiato viene chiamato radicale libero. Un biradicale è una specie chimica con due elettroni non accoppiati, mentre i poliradicali sono rari. Composti importanti della chimica inorganica, come il monossido e il diossido di azoto, sono radicali liberi. L'ossigeno, l'elemento più diffuso sulla Terra, allo stato molecolare è un biradicale. Lo studio dei radicali liberi e del loro comportamento è fondamentale per le scienze chimiche e ha avuto un grande sviluppo fin dagli inizi del Novecento specie nella chimica organica.
Nel 1900 Moses Gomberg scoprì il difenilmetile e Friedrich Adolf Paneth ottenne, nel 1929, radicali liberi metilici per decompressione termica. Negli anni Trenta, invece, William A. Waters e Donald H. Hey, spiegarono il meccanismo di molte reazioni in soluzione mediante meccanismi radicalici Morris S. Kharasch razionalizzava l'addizione anti-Markovnikov dell'acido bromidrico alle olefine e Paul J. Flory sviluppava la cinetica dei processi di polimerizzazione radicalica.
Quando i composti organici vengono portati a elevata temperatura, dell'ordine degli 800 °C, sono suscettibili di subire rotture di tipo omolitico dei loro legami covalenti, nelle quali ha luogo la scissione della coppia di elettroni che contribuiscono alla formazione del legame stesso. Ciascuno degli elettroni si unisce a uno dei due spezzoni molecolari generando così i radicali liberi. Per esempio, il cracking termico (o piroscissione) degli idrocarburi saturi è un classico processo in cui si generano radicali liberi, i quali, però, innescano un sistema complesso di reazioni dalle quali si produce una moltitudine di composti. Pertanto, per realizzare processi selettivi, coi quali si ottengono ben definiti prodotti operando per via termica, occorre agire in condizioni più blande e utilizzare, quali sorgenti, molecole nelle quali siano presenti legami con energie relativamente basse, comprese tra 20 e 40 kcal/mol, e quindi facilmente dissociabili. Le più comuni fra queste molecole sono i perossidi, in cui è presente un singolo legame fra due atomi di ossigeno, e gli azoderivati, che consentono di generare radicali liberi tra 40 e 150 °C:
[1] RO−OR → 2RO∙
[2] R−N=N−R → 2R∙+N2.
In entrambi i casi R indica un residuo idrocarburico (metile, etile, ecc.), mentre il puntino indica l'elettrone spaiato.
La fotolisi è un altro metodo generale per ottenere radicali liberi in condizioni blande. L'assorbimento da parte di una molecola di radiazioni ultraviolette eccita i suoi elettroni da uno stato fondamentale a uno stato eccitato di singoletto, che può subire molte evoluzioni, di cui alcune portano alla formazione di radicali liberi, come negli esempi che seguono:
[3] RO−Cl → RO∙ + Cl∙
[4] Br−Br → 2Br∙
[5] Ar−I → Ar∙ + I∙
[6] R3Sn−SnR3 → 2R3Sn∙
dove Ar∙ indica un residuo arilico derivante dagli idrocarburi aromatici per eliminazione di un atomo di idrogeno dal loro gruppo.
La omolisi fotochimica è più specifica di quella termica poiché alla molecola viene fornito un valore ben definito di energia. Inoltre, lo stato di singoletto può trasformarsi in uno stato eccitato di tripletto (intersystem crossing) e quindi agire direttamente come biradicale.
La rottura omolitica dei legami covalenti può essere realizzata anche mediante radiolisi, utilizzando sorgenti di raggi X, elettroni ad alta energia e il 60Co. In questi casi può avere luogo l'espulsione di un elettrone e− con formazione di un radicale cationico, che evolve ulteriormente a radicale neutro. Per esempio, l'acqua può costituire un'utile sorgente del radicale ossidrile:
[7] H2O → H2O∙+ + e−
H2O∙+ → HO∙ + H+.
Infine, sorgenti radicaliche molto versatili sono quelle che coinvolgono processi di ossidoriduzione con trasferimento di un elettrone. In tal modo, i perossidi reagiscono con vari sali metallici generando radicali all'ossigeno:
[8] RO−OR + Mn+ → RO∙ + RO− + M(n+1)+
(Mn+=Cu+, Fe2+, ecc.).
Caratterizzazione spettroscopica dei radicali liberi
Il metodo di gran lunga più utile per determinare e caratterizzare un radicale libero è la risonanza di spin elettronico o ESR (Electronic spin resonance) (o, anche EPR, Electronic paramagnetic resonance), che costituisce un metodo di sensibilità molto elevata (fino a 10−8 molare). La spettroscopia ESR si basa sul fatto che un elettrone non accoppiato presente in una molecola ha uno spin e un momento magnetico e, quindi, può esistere in due stati di spin che, in assenza di influenze esterne, sono orientati in modo indeterminato. In presenza di un campo magnetico esterno di intensità H, l'elettrone si dispone in modo parallelo o in modo indeterminato antiparallelo a esso, e la differenza di energia fra i due stati risulta espressa da:
[9] ΔE=gβH
dove g è il fattore di Landè e β il magnetone di Bohr, che esprime il momento magnetico di un elettrone atomico. La frequenza associata con il salto da un livello all'altro è data da:
[10] formula
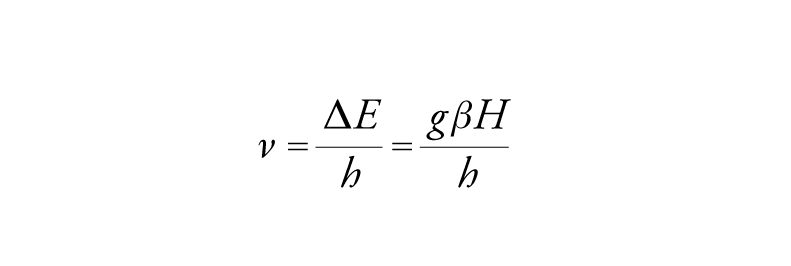
dove h è la costante di Planck.
La caratteristica più utile di uno spettro ESR ai fini della determinazione della struttura di un radicale, nota come separazione iperfine (hyperfine splitting), ha origine dall'interazione tra l'elettrone non accoppiato e i nuclei magnetici degli atomi circostanti (1H, 13C, 14N, 18O, ecc.). Per esempio, lo spettro ESR del radicale metile CH3∙ consta di 4 linee di diversa intensità, nei rapporti 1:3:3:1, dovute all'interazione dell'elettrone non accoppiato con 3 protoni magneticamente equivalenti.
Reattività dei radicali liberi
I radicali liberi possono dare origine a una grande varietà di reazioni inter- e intramolecolari, che vanno dall'addizione ai più svariati sistemi insaturi, al distacco di atomi da molecole sature (essenzialmente idrogeno o alogeni), alle sostituzioni, ossidazioni, riduzioni, dimerizzazioni, trasposizioni, frammentazioni e così via. Di particolare interesse sono i processi a catena, che possono raggrupparsi in due ampie categorie: processi radicalici a catena e processi redox.
Processi radicalici a catena
I processi radicalici a catena hanno una grande rilevanza nelle sintesi organiche, nei processi industriali e in quelli biologici. Essi hanno anche implicazioni di carattere ecologico poiché sono coinvolti in alcuni fenomeni atmosferici come, per esempio, la formazione del cosiddetto buco dell'ozono. Pertanto, è importante conoscere la velocità con cui si svolgono e questo aspetto verrà illustrato a proposito del rilevante fenomeno chimico dell'autossidazione.
Anzitutto, ricordiamo che tutti i processi a catena sono caratterizzati da tre stadi: (a) iniziazione; (b) propagazione; (c) terminazione. Il primo genera una specie radicalica in grado di innescare lo stadio successivo di propagazione. Molte delle sorgenti radicaliche menzionate in precedenza possono essere responsabili dell'iniziazione, in modo casuale o con un'opportuna programmazione.
Nella fase preliminare di un'autossidazione ha luogo l'inserimento di una molecola di ossigeno nel legame C-H di un'altra molecola:
[11] R−H+O2→RO−O−H
con formazione di un idroperossido. La reazione si svolge in condizioni di temperatura relativamente blande (da quella ambiente a non oltre i 200 °C). La possibilità che abbia luogo tale inserimento è dovuta al carattere biradicalico dell'ossigeno molecolare che lo rende particolarmente reattivo, poiché si trova in uno stato fondamentale di tripletto 3Σg− nel quale 2 elettroni non sono accoppiati ma occupano 2 diversi orbitali con spin paralleli.
L'idroperossido così ottenuto può essere stabile nelle condizioni operative oppure decomporsi in vari prodotti ossigenati (alcoli, aldeidi, chetoni, acidi carbossilici). Gli atti elementari che caratterizzano il processo globale sono mostrati nello schema seguente:
[12] formula
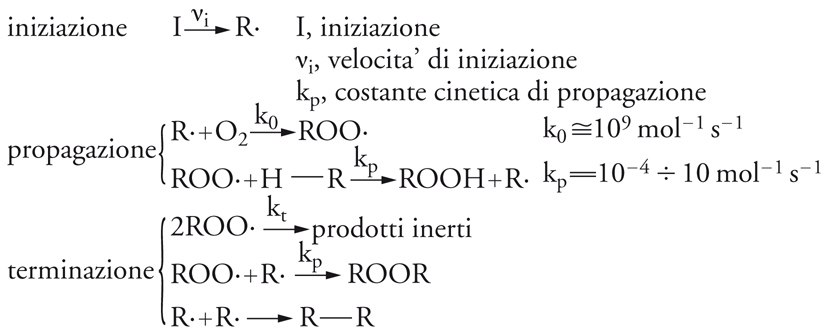
dove I indica un iniziatore, vi la velocità d'iniziazione, kp la costante cinetica di propagazione (kp=10−4÷10 mol−1s−1) e k0 una costante cinetica il cui valore è molto più elevato (k0≅109mol−1s−1).
La velocità di formazione dell'idroperossido espressa dalla derivata della sua concentrazione rispetto al tempo, compatibilmente con lo schema precedente, è data dall'espressione:
[13] formula
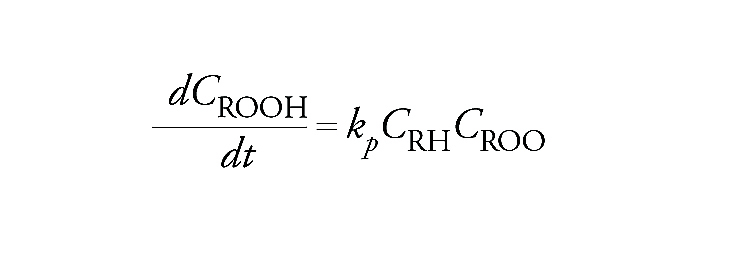
con kp costante di velocità della reazione di propagazione.
Se si ipotizza che la concentrazione del radicale ROO∙ durante la reazione si mantenga in condizioni stazionarie, poiché la sua velocità di formazione nella reazione di iniziazione eguaglia quelle di sparizione nelle reazioni di terminazione, si dimostra che l'espressione generale della velocità assume la forma seguente:
[14] formula
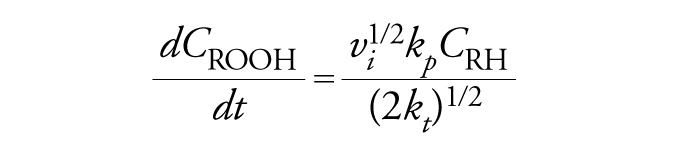
dove vi è la velocità del processo di iniziazione e kt la costante cinetica della reazione di terminazione.. Equazioni analoghe si ricavano per altri processi radicalici a catena di grande importanza, quali la polimerizzazione radicalica di monomeri vinilici (che è alla base della produzione di importanti materiali plastici), l'alogenazione radicalica, l'addizione radicalica a catena di numerose classi di composti a sistemi insaturi e così via. La caratteristica peculiare dell'autossidazione risiede nel fatto che il prodotto iniziale della reazione, l'idroperossido, può agire a sua volta da iniziatore in seguito a una decomposizione termica, fotochimica o redox.
Il processo di autossidazione ha implicazioni importanti in settori di grande rilevanza, quali la produzione di prodotti chimici di largo consumo e il deterioramento di sostanze organiche (manufatti, alimenti); esso, inoltre, è responsabile di svariati danni ai tessuti degli organismi viventi, che sono la causa di molte patologie. La prevenzione dei danni provocati dall'autossidazione dei composti organici e dei tessuti degli organismi viventi, pertanto, costituisce una problematica molto importante sia nell'ambito dell'economia industriale sia in quello medico-biologico. In questi ultimi anni tale problema ha interessato in misura crescente gli studiosi di radicali liberi che si occupano dello sviluppo di antiossidanti industriali e di quelli biologici.
Processi a catena redox
Anche i processi a catena redox hanno una notevole importanza nella chimica organica e spesso coinvolgono una catalisi metallica ossidoriduttiva, che, in linea generale, può essere indicata mediante lo schema seguente:

[15] schema
Numerose sintesi radicaliche rientrano in esso. Per esempio, due delle più classiche reazioni dei sali di diazonio, la reazione di Sandmeyer:
[16] formula
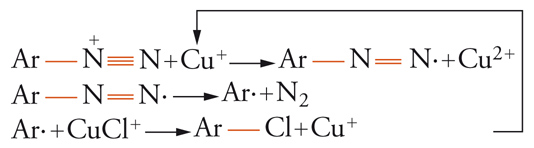
e la reazione di Meerwein:
[17] formula
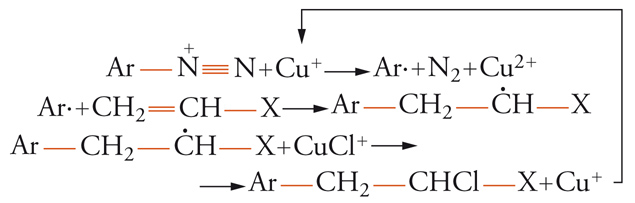
sono caratterizzate da una catena ossidoriduttiva. La stessa sostituzione radicalica aromatica è spesso caratterizzata da catalisi ossidoriduttiva.
Struttura, reattività e selettività
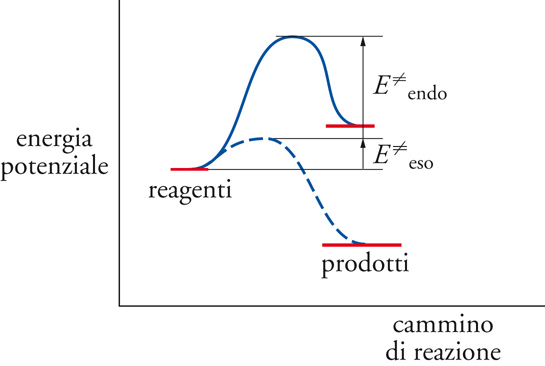
In condizioni relativamente blande di temperatura, la reattività e la selettività delle reazioni radicaliche in soluzione sono generalmente controllate dal comportamento cinetico del sistema, ovvero dalle velocità con le quali si svolgono le diverse reazioni, tranne pochi casi di reazioni reversibili il cui controllo è invece condizionato da fattori termodinamici. A temperature più elevate in fase gassosa, il controllo termodinamico diventa sempre più importante. Comunque, la variazione di entalpia dei processi elementari, che fornisce il valore dell'energia liberata o assorbita dalla reazione, condiziona, spesso in modo determinante, l'energia di attivazione e quindi la selettività globale. Infatti, una reazione esotermica ha una energia di attivazione bassa ‒ e quindi elevata velocità ‒ mentre una reazione endotermica ha un'elevata energia di attivazione e bassa velocità, come viene illustrato nella fig.1.
Un esempio classico di riscontro di tali concetti è fornito dalla clorurazione e dalla bromurazione di idrocarburi saturi:
[18] R−H + X2 → R−X + HX
(X=Cl, Br)
la cui selettività verso un particolare atomo di carbonio della catena idrocarburica è determinata dal distacco di un atomo di idrogeno del legame C-H per opera di un atomo di alogeno, che, avendo un elettrone spaiato, si comporta come un radicale:
[19] R−H + X∙ → R∙ + H−X.
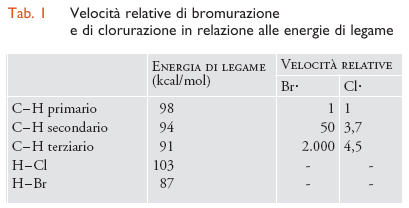
I dati riportati nella tab.1 mostrano come le energie dei legami C-H abbiano un ruolo importante nel determinare la velocità di bromurazione, che cresce al diminuire del loro valore. Le reazioni sono sempre endotermiche e l'elevata differenza di selettività tra C-H primario, secondario e terziario è sostanzialmente determinata dal distacco dell'atomo di idrogeno per azione dell'alogeno. Le clorurazioni, invece, sono sempre esotermiche e con bassa selettività. Nel caso della bromurazione, infatti, la velocità di reazione cresce di 2000 volte passando da un C-H primario a uno terziario, ma solo di 4,5 volte nel caso della clorurazione. Analogamente, l'effetto entalpico governa la selettività di distacco dell'idrogeno da parte di due radicali apparentemente simili, RO∙ e ROO∙ :
[20] R−H + RO∙ → R∙ + RO−H
[21] R−H + ROO∙ → R∙ + ROO−H.
Con i radicali alcossilici il processo di distacco dell'idrogeno è esotermico, per cui la reattività è elevata e la selettività è bassa. Viceversa, con i radicali perossidici il processo è endotermico e quindi la velocità è molto più bassa e la selettività più elevata.
Il fattore entalpico, però, non è l'unico che governa la selettività nel distacco d'idrogeno. Questo fatto si riscontra nelle reazioni regioselettive, nelle quali un isomero si forma preferenzialmente partendo da una molecola che possiede più gruppi funzionali uguali, ma distiguibili. Per esempio, nell'alogenazione dell'1-cloroeptano con N-aloammine in ambiente acido, le velocità relative di alogenazione dei vari gruppi CH2 seguono l'ordine indicato dai numeri posti sotto gli atomi di carbonio:
[22] formula
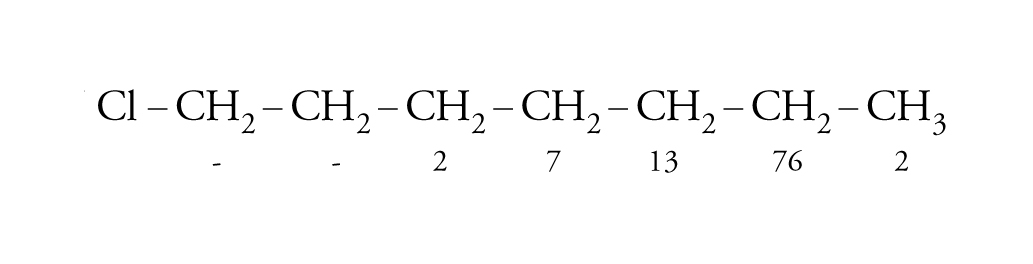
In questi casi la differenza di selettività tra il gruppo metilenico lontano dal sostituente polare e i metili è determinata dal fattore entalpico (più elevata energia dei legami C-H primari), mentre la grande differenza di reattività tra i vari gruppi metilenici è dovuta all'effetto induttivo e di campo esercitato dalla presenza dell'atomo di cloro elettronegativo, per cui reagisce più velocemente il gruppo metilenico più lontano dal sostituente polare.
Questo tipo di alogenazione, oltre a fornire un'elevata regioselettività, è caratterizzato anche dal fatto che l'introduzione di un atomo di alogeno nella catena paraffinica genera una forte disattivazione, per cui è possibile convertire tutto il substrato arrestando il processo alla monosostituzione. Questo comportamento, chiamato chemioselettività, è in netto contrasto con l'alogenazione radicalica tradizionale condotta con cloro e bromo. Quest'ultima, infatti, è soggetta a un effetto polare molto modesto per cui l'introduzione di un atomo di alogeno non modifica sostanzialmente la reattività dei legami C-H della catena paraffinica. Ne consegue che, col procedere della conversione, si formano miscele estremamente complesse di prodotti di polisostituzione in tutte le posizioni disponibili, che fanno perdere ogni interesse sintetico al processo stesso.
L'effetto polare, invece, ha un ruolo fondamentale nella sostituzione radicalica aromatica che risulta fortemente influenzata dalla polarità e dalla polarizzabilità sia del radicale che del substrato con cui reagisce. Pertanto, radicali poco polari, come quelli arilici, hanno scarso interesse applicativo nella sostituzione aromatica, in quanto caratterizzati da regio- e chemioselettività molto basse, mentre i radicali elettrofili reagiscono con elevata selettività con anelli aromatici elettron-ricchi. Il comportamento dei radicali nucleofili è del tutto opposto.
L'effetto sterico, dovuto all'ingombro spaziale dei gruppi molecolari, influenza non solo l'orientamento, ma anche la reattività nel riguardo di molti radicali, poiché si riscontra una maggior difficoltà di accedere al carbonio olefinico. Nella sostituzione aromatica, reattività e selettività sono fortemente influenzate dall'effetto sterico sia dell'anello aromatico che del radicale.
Applicazioni di alcune reazioni radicaliche
I radicali liberi, oltre a reagire per via mono- o bimolecolare tra loro, interagiscono praticamente con tutte le classi di composti organici e, pertanto, possono dare origine a innumerevoli trasformazioni che non si possono riassumere in breve spazio. Accenneremo solo ad alcune applicazioni ‒ tra le innumerevoli sviluppate in questi anni ‒ delle reazioni radicaliche a derivati olefinici, acetilenici, aromatici e paraffinici. Esse rappresentano solo qualche esempio della grande potenzialità di queste reazioni.
Reazioni di radicali liberi con composti olefinici e acetilenici
La reattività radicalica delle olefine ha rappresentato uno dei punti di partenza della chimica dei radicali liberi, e ha costituito l'area di maggiore impegno e progresso nell'ambito delle reazioni radicaliche; essa è caratterizzata, oltre che dalla polimerizzazione radicalica di monomeri vinilici, da vari processi di tipo generale.
L'addizione di un radicale può essere condotta con una grande varietà di gruppi e si può realizzare su legami sia olefinici che acetilenici:
[23] formula
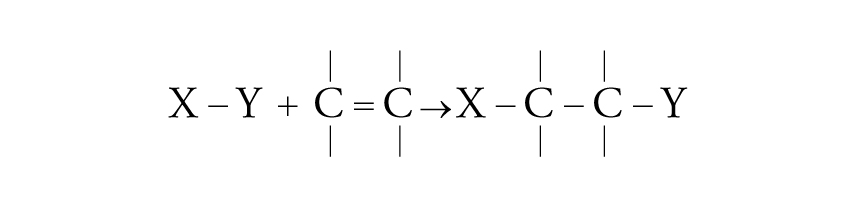
[24] formula.
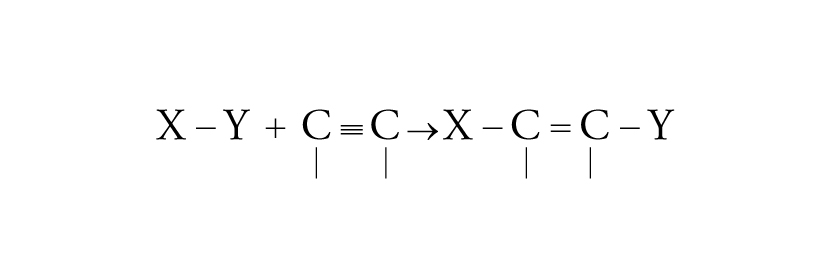
La catena radicalica può essere semplice, come per esempio nel caso di addizione di acido bromidrico o di molti altri derivati:
[25] formula
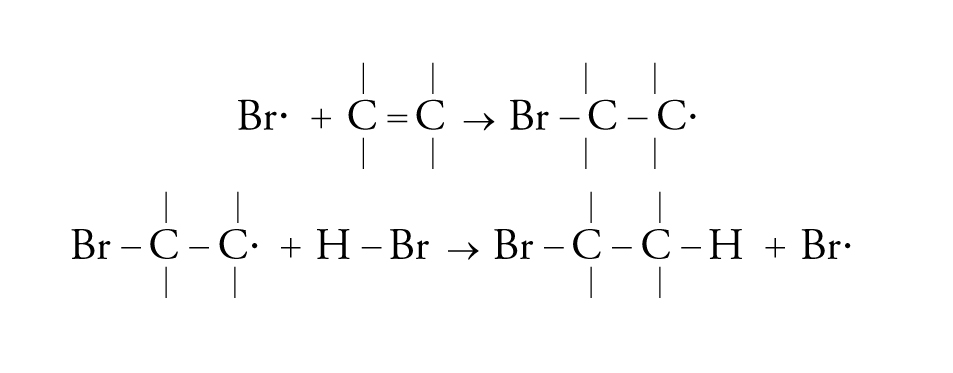
oppure può divenire più complessa attraverso una sequenza selettiva di reazioni radicaliche.
Sostituzione radicalica aromatica
Dal punto di vista sintetico, per lungo tempo la sostituzione radicalica aromatica è stata considerata un aspetto marginale della reattività aromatica; tale considerazione era basata principalmente sul fatto che gli studi si erano concentrati sulla arilazione radicalica aromatica, una reazione caratterizzata da regio- e chemioselettività molto basse. La trasposizione ingiustificata di questo comportamento a tutte le sostituzioni radicaliche ha rappresentato il principale ostacolo allo sviluppo di queste reazioni. La consapevolezza che gli effetti polari nelle sostituzioni radicaliche possono essere di entità non inferiore a quelli delle reazioni ioniche ha portato negli ultimi decenni allo sviluppo di sostituzioni radicaliche aromatiche di elevata selettività e di grande interesse sintetico. Per esempio, l'amminazione radicalica aromatica con N-cloroammine è una reazione caratterizzata da elevatissime regio- e chemioselettività, confrontabili con le più selettive sostituzioni elettrofile aromatiche. Il suo meccanismo è stato interpretato da un processo a catena di addizione ed eliminazione:
[26] formula
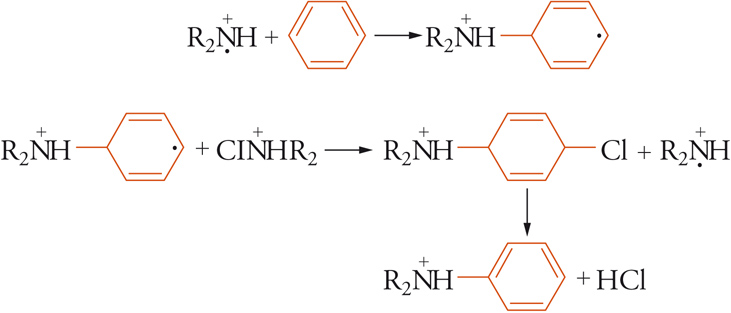
Sostituzione radicalica alifatica
Gli idrocarburi saturi sono anche chiamati paraffinea causa della loro bassa reattività (parum affinis, poco affini), che in realtà riguarda le reazioni ioniche ed è dovuta alla polarizzazione molto piccola dei legami C-H e C-C. Le paraffine, in realtà, sono molto reattive verso i radicali liberi. Infatti, una miscela di paraffina e cloro reagisce in pochi secondi a temperatura ambiente, in presenza di iniziazione radicalica chimica o fisica, mediante un processo a catena (la lunghezza cinetica della catena è di circa 106), mentre una miscela di paraffina e fluoro reagisce in modo esplosivo. La metodologia di gran lunga più versatile per la funzionalizzazione delle paraffine è pertanto quella radicalica. Una grande varietà di radicali liberi (del carbonio, dell'ossigeno, dell'azoto, atomi di alogeno, ecc.) è infatti in grado di strappare un atomo di idrogeno di legami C-H generando radicali alchilici:
[27] R−H+X∙→R∙+H−X.
Si deve comunque ricordare che la funzionalizzazione selettiva delle paraffine rimane una delle sfide di maggiore rilevanza in campo chimico a causa delle piccole differenze di energia tra i legami C-H. Si tratta di una sfida che la natura in qualche caso è riuscita a superare mediante la catalisi enzimatica, la cui emulazione mediante opportuni catalizzatori resta un importante obiettivo della ricerca.
Conclusioni
Le reazioni radicaliche hanno giocato per lungo tempo un ruolo pressoché incontrastato dell'industria chimica di base (polimerizzazione di monomeri vinilici, ossidazioni con ossigeno molecolare, clorurazione di idrocarburi, ecc.), dove l'uso di molecole semplici e la possibilità di conversioni parziali, senza i notevoli inconvenienti rappresentati dalla separazione dei prodotti, rende meno drammatici i problemi di regio- e chemioselettività. Le reazioni radicaliche, ritenute poco selettive, erano invece di scarso interesse nell'industria della chimica fine e nella sintesi di composti sofisticati o in processi biologici, dove una elevata selettività è condizione pregiudiziale per il successo. Negli ultimi decenni si è assistito a uno sviluppo davvero esplosivo nelle applicazioni delle reazioni radicaliche a sintesi selettive e nello stesso tempo è stata riconosciuta la loro grande importanza nei processi biologici e nel metabolismo dei farmaci.
La ricerca in atto lascia prevedere ulteriori, interessanti sviluppi tanto nella comprensione dei fenomeni quanto nelle applicazioni.
Bibliografia
Barton 1993: Barton, Derek H.R., Half a century of free radical chemistry, Cambridge, Cambridge University Press, 1993.
Chanon 1989: Paramagnetic organometallic species in activation/selectivity: catalysis, edited by Michel Chanon, Michel Juilliard, Jean-Claude Poite, Dordrecht-London, Kluwer, 1989.
Flory 1937: Flory, Paul J., The mechanism of vinyl polymerization, "Journal of the American Chemical Society", 59, 1937, pp. 241-253.
Giese 1986: Giese, Bernd, Radicals in organic synthesis: formation of carbon-carbon bonds, Oxford, Pergamon, 1986.
Gomberg 1900: Gomberg, Moses, On the preparation of tri-phenylchloromethane, ‟Journal of the American Chemical Society", 22, 1900, pp. 757-771.
Gomberg 1900: Gomberg, Moses,Triphenylmethyl, ein Fall von dreiwerthigem Kohlenstoff, ‟Berichte der Deutschen Chemischen Gesellschaft", 33, 1900, pp. 3150-3163.
Hey, Waters 1937: Hey, Donald H. - Waters, William A., Some organic reactions involving the occurrence of free rad-icals in solution, ‟Chemical reviews", 21, 1937, pp. 169-208.
Huyser 1969-1973: Methods in free-radical chemistry, edited by Earl S. Huyser, New York-Basel, Dekker, 1969-1973,4 v.
Huyser 1970: Huyser, Earl S., Free-radical chain reactions, New York, Wiley, 1970.
Kharasch 1937: Kharasch, Morris S. - Engelmann, Helmut - Mayo, Frank R., The peroxide effect in the addition of reagents to unsaturated compounds. XV. The addition of hydrogen bromide to 1- and 2- bromo- and chloropropenes, ‟Journal of organic chemistry", 2, 1937, pp. 288-302.
Kochi 1973: Free radicals, edited by Jay K. Kochi, New York-Chichester, Wiley, 1973, 2 v.
Minisci 1976: Minisci, Francesco, Recent aspects of homolytic aromatic substitutions, ‟Topics in current chemistry", 62, 1976, pp. 1-48.
Minisci 1989: Free radicals in synthesis and biology, edited by Francesco Minisci, Dordrecht-London, Kluwer Academic, 1989.
Nonhebel, Walton 1974: Nonhebel, Derek C. - Walton, John C., Free-radical chemistry: structure and mechanism, London-Cambridge, Cambridge University Press, 1974.
Paneth, Hofeditz 1929: Paneth, Fritz - Hofeditz, Wilhelm, Über die Darstellung von freiem Methyl, ‟Berichte der Deutschen Chemischen Gesellschaft B", 62, 1929, pp. 1335-1347.
Pryor 1966: Pryor, William Austin, Free radicals, New York, McGraw-Hill, 1966.
Viehe 1986: Substituent effects in radical chemistry, edited by Heinz G. Viehe, Zdenek Janousek, Robert Merényi, Dordrecht-Lancaster, Reidel, 1986.
Walling 1957: Walling, Cheves, Free radicals in solution, New York, Wiley, 1957.
Williams 1965-1980: Advances in free-radical chemistry, ed-ited by Gareth H. Williams, New York, Academic, 1965-1980.