
Progetto genoma
Progetto genoma
(App. V, iv, p. 290)
Negli anni Novanta vi è stato un grande sviluppo di questo programma di analisi genetica: ora si possono distinguere i risultati ottenuti con i 'piccoli genomi' - importanti soprattutto come cornice generale di conoscenza delle strutture genetiche e della modalità della loro evoluzione - da quelli relativi al genoma umano, in cui al significato di conoscenza di base si aggiunge l'importanza applicativa in biomedicina.
I progetti 'piccoli genomi'
Le prime strutture biologiche dotate d'informazione genetica la cui sequenza nucleotidica è stata analizzata erano virus. F. Sanger nel 1977 ha descritto il genoma a elica di DNA singola del fago jx174, di 5386 nucleotidi, e ha successivamente avuto bisogno di cinque anni per definire la sequenza del fago λ di 48.502 paia di basi (bp). Oggi questo risultato può essere ottenuto in un mese da qualsiasi buon laboratorio, ma allora la tecnologia era ancora in fase di sviluppo. Si deve proprio a Sanger la strategia usata tuttora e chiamata shotgun sequencing, uno "sparo di fucile" nel DNA totale mediante il trattamento con diversi enzimi di restrizione, così da ottenere frammenti tagliati in modo differente tra loro sovrapponibili per ricostruire l'intera sequenza. In seguito, sono occorsi altri tredici anni prima che fosse sequenziato il DNA totale del primo vero e proprio organismo: solo nel luglio 1995 R.D. Fleischmann e i suoi collaboratori hanno completato l'analisi dell'Haemophilus influenzae, di 1.830.240 bp.
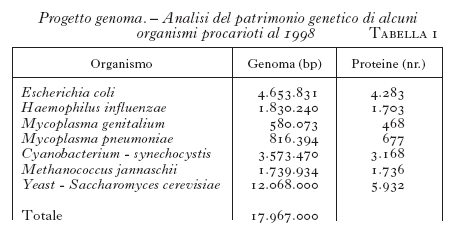
Ormai molti laboratori in tutto il mondo si sono impegnati in questa attività di sequenziamento e i risultati si sono susseguiti rapidamente. Negli USA, alla fine del 1995 il laboratorio di C.M. Fraser (Institute for Genomic Research, Rockville, Md.) ha riportato la struttura genomica del Mycoplasma genitalium, identificandovi soltanto 470 ORF (Open Reading Frame), vale a dire sequenze nucleotidiche che hanno tutte le caratteristiche per essere espresse in una proteina, e che quindi presumibilmente costituiscono un gene. Questo numero di geni rappresenta la stima più bassa identificata fino a oggi della complessità genomica necessaria alle funzioni di un organismo completo. Da allora è stata pubblicata l'analisi nucleotidica dei genomi completi di molti altri organismi procarioti (tab. 1), fra cui Escherichia coli, di gran lunga l'organismo più studiato, ospite innocuo dell'intestino di molti organismi superiori e dell'uomo, Mycoplasma pneumoniae e Helicobacter pylori, responsabili rispettivamente della tubercolosi e dell'ulcera gastroduodenale nell'uomo. Sono inoltre stati descritti i genomi di alcuni archeobatteri estremofili, quali Methanococcus jannaschii, Cyanobacterium synechocystis, Archeoglobus fulgidis, Methanobacterium thermoautotrophicum e Aquifex aeolicus, di grande importanza per lo studio dell'origine della vita sulla terra, agli albori del percorso evolutivo. Infine, è stato sequenziato interamente anche il DNA di un organismo eucariote, Saccharomyces cerevisiae.
È stato inoltre istituzionalizzato un progetto internazionale volto alla descrizione della sequenza completa di Plasmodium falciparum, protozoo della classe dei sarcodini, parassita dell'uomo in cui determina la malaria, che ancora oggi costituisce una delle cause maggiori di infermità e mortalità. La priorità data allo studio di questo organismo nasce dalla constatazione che gran parte dei ceppi di plasmodio è ora evoluta verso forme resistenti alla clorochina, farmaco tradizionalmente usato per eliminare il parassita. Solo l'analisi dell'intero genoma permetterà di acquisire sulla biologia del plasmodio tutte le informazioni necessarie per individuare altri strumenti efficaci contro la malaria. Al 4 febbraio 1998 sono state descritte di questo genoma 3945 sequenze di DNA distribuite su 14 cromosomi, di cui il cromosoma 2 è quasi completo. Data l'accelerazione crescente di queste ricerche, si calcola che nell'arco di pochi anni saranno noti nella loro interezza i genomi di almeno 100 microrganismi. La quantità di dati relativi equivarrebbero al contenuto di 1000 volumi di duecento pagine ciascuno, troppi per essere facilmente consultati e per ottenere un indice analitico complessivo e integrato. Tutte le informazioni vengono invece inserite in data base relazionali accessibili tramite Internet ai genetisti di tutto il mondo.
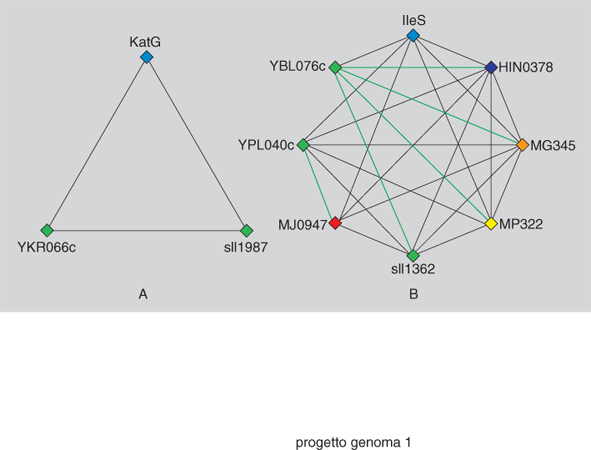
Le sequenze nucleotidiche dei diversi genomi si stanno accumulando molto più rapidamente della possibilità di razionalizzarne il significato. Il primo passo per l'analisi genetica delle nuove informazioni sulla struttura dei genomi è stato una classificazione, sulla base dell'omologia, dei geni conservati nelle diverse specie. È stato così effettuato il confronto di proteine, omologhe per struttura e possibilmente per funzione, codificate nei primi sette genomi a sequenza completamente nota e che rappresentano le cinque linee filogenetiche maggiori. Sono stati identificati 720 insiemi di gruppi di geni ortologhi; questo neologismo è la traduzione letterale dell'inglese Clusters of Orthologous Groups (COG), in cui orthologous indica, per un gene di un determinato gruppo, la corrispondenza tra una struttura e una determinata funzione. Il metodo considera identificata l'ortologia di un gene se vi è corrispondenza fra almeno tre linee filogenetiche diverse, assumendo così una comune origine evolutiva di quel gene (fig. 1).
L'omologia di funzione identificata all'interno dei gruppi di geni ortologhi permette di estendere quella funzione a nuovi membri del gruppo di cui è nota la struttura nucleotidica. Inoltre, questa relazione fra geni permette di formulare in modo diretto una serie di previsioni sulla funzionalità di genomi solo parzialmente caratterizzati. In questo senso i COG delineano una cornice di analisi funzionale ed evolutiva dei genomi che si va progressivamente delineando. In concreto, quindi, quale interesse culturale e scientifico possono avere queste collezioni immense di dati sulla struttura molecolare dei genomi?
In prima istanza, i p. g. possono aiutare a studiare la complessità dell'informazione genetica, soprattutto negli eucarioti, in cui, come riferito sopra, una larga parte del genoma sembra priva di funzione genica. Per es., il genoma dei lieviti contiene almeno 6000 possibili geni (possibili, perché definiti come ORF) e di questi solo dal 10 al 15% potrebbero essere essenziali per le funzioni vitali (molte sono sequenze geniche ripetute e quindi ridondanti), mentre per la gran parte hanno funzione ignota, perché codificano proteine non equiparabili ad alcuna proteina descritta in altri organismi.
Un secondo scopo riguarda proprio queste ORF a espressione ignota, definite orfane. Molti laboratori stanno cercando di identificarne la funzione producendo mutazioni inattivanti e verificando poi il relativo fenotipo mutato. Questo procedimento ricade sotto una nuova prassi d'indagine definita reverse genetics, proprio perché, al contrario di quella classica, in cui si procede da un fenotipo mutato all'identificazione della proteina responsabile, fino allo studio molecolare del gene, nella genetica 'a rovescio' si parte invece dalla struttura molecolare di un gene per descrivere una sequenza amminoacidica ancora ignota da esso codificata e per identificarne il relativo fenotipo.
Un terzo aspetto è la possibilità di disporre del repertorio completo dei geni di un organismo per reperire facilmente le informazioni sulla struttura di proteine identificate soltanto per una parte limitata della loro sequenza amminoacidica. Ciò è di particolare utilità per lo studio di meccanismi biologici complessi, dove più catene polipeptidiche svolgono una funzione integrata.

Un quarto aspetto è la concretizzazione a livello molecolare degli eventi evolutivi identificati dai paleontologi e amplificati dagli studi di genetica delle popolazioni. Il confronto fra le variazioni amminoacidiche di proteine con funzione analoga nella scala evolutiva e quelle nucleotidiche nei geni corrispondenti permette di stabilire i tempi dell'orologio evolutivo (fig. 2), l'origine comune e le divergenze delle specie e di interpretare la funzione di geni non noti in una specie, per analogia con quelli a funzione nota di un'altra specie. Recentemente sono state ottenute le sequenze di 658 geni a localizzazione nucleare, che sono riconoscibili e confrontabili in 207 specie di Vertebrati che rappresentano pietre miliari del percorso evolutivo. Lo studio delle differenze tra le sequenze fossili nei genomi di specie diverse ha rappresentato un metodo, denominato orologio molecolare dell'evoluzione, che permette di determinare la 'nascita' della specie e la sua provenienza evolutiva.
La datazione dei fossili è necessariamente utilizzata e serve a riconoscere gli eventi più distanti fra loro, mentre l'orologio molecolare è usato per determinare la scala temporale degli eventi più ravvicinati. La corrispondenza fra i due metodi è però risultata poco precisa in molti eventi, perché una semplice valutazione statistica ammette per la scala temporale errori fino al 10% se i geni considerati sono dieci, fino al 5% se sono cinquanta, fino al 3% se sono cento. Il numero di geni disponibile attualmente (658 geni in 270 specie diverse) porta la precisione della valutazione vicina al 99%.
I nuovi dati hanno confermato precedenti determinazioni, quali quelle di divergenza delle scimmie antropomorfe, cioè di 8,2 milioni di anni per l'orango, 6,7 per il gorilla e 5,5 per lo scimpanzé. Al contrario, eventi molto importanti, quali la radiazione dei primi mammiferi e l'insorgenza di roditori hanno subito importanti modifiche nella scala temporale. I mammiferi sarebbero sorti prima di 100 milioni di anni fa (rispetto agli 80 milioni già stimati), cioè assai anterioremente all'estinzione dei dinosauri, contraddicendo l'ipotesi che l'esplosione evolutiva dei mammiferi fosse potuta avvenire in un pianeta ormai privo di tali pericolosi rettili. L'origine dei roditori è stata fissata a 110 milioni di anni fa, cioè, in modo inaspettato, molto più precocemente della radiazione topo-ratto, avvenuta 40 milioni di anni or sono.
In conclusione, la sequenza completa del genoma di un organismo è come un affascinante dizionario di una lingua sconosciuta al lettore. Da analogie con lingue note, dalle radici comuni e dalle desinenze differenti fra i vocaboli, dalle articolazioni delle parole in successione nelle frasi, sarà possibile determinare progressivamente la struttura di quella lingua.
Il progetto 'genoma umano'
A mano a mano che le sequenze di geni umani conosciuti si sono accumulate nei data base, nei laboratori di genetica umana in tutto il mondo ha preso corpo un progetto immenso, ma affascinante: clonare e sequenziare l'intero genoma umano, più di cinque miliardi di nucleotidi, che è costituito per circa il 3% dall'informazione necessaria per tutti i prodotti genici (circa 50.000÷100.000 geni) e per il 97% da sequenze senza apparente significato genotipico. A partire dal 1989 questo progetto è stato sviluppato da numerosi laboratori, tra loro coordinati, che si sono ripartiti i frammenti di genoma o cromosomi per le analisi. Il progetto si muove tramite una strategia complessa, in cui sono identificabili procedure diverse, ma integrate fra loro.
a) Seguire il criterio della curiosità dello scienziato, tipico della ricerca di base: partendo sia da fenotipi mutati o normali sia da mappe geniche molto particolareggiate, procedere all'identificazione di tratti di DNA che codificano la proteina responsabile di quel fenotipo e quindi alla clonazione e all'analisi nucleotidica del gene, incluse le regioni di controllo della sua espressione.
b) Clonare tutto il genoma umano suddiviso in più di 52.000 grossi frammenti usando come vettori gli YAC (Yeast Artificial Chromosomes: v. biologia molecolare, in App. V e in questa Appendice), cromosomi di lievito modificati in modo da accogliere e amplificare tratti singoli di DNA, di cui è noto il cromosoma umano di origine.
c) Isolare RNA messaggeri (che rappresentano i geni trascritti e codificano catene polipeptidiche), retrotrascriverli in DNA complementare (cDNA) e clonarli in genoteche di cDNA. I cloni di trascritti genici sono stati distribuiti a laboratori di tutto il mondo dove sono usati, tramite la PCR (Polymerase Chain Reaction), per produrre oltre 16.000 marcatori genici individuali o STS (Sequence Tagged Sites), la cui localizzazione cromosomica viene identificata mediante gli YAC e controllata mediante ibridazione in situ.
d) Identificare marcatori molecolari a distanza regolare (circa uno ogni 100.000) che servano da pietre miliari di riferimento lungo il tragitto lineare di ogni cromosoma, dal telomero del braccio corto al centromero, e da questo al telomero del braccio lungo, possibilmente con un riferimento puntuale al bandeggio cromosomico, cioè alla successione lineare di marcatori citogenetici lungo il cromosoma definito morfologicamente. Nel marzo 1997 un gruppo di ricercatori della Washington University di Saint Louis (Mo.) ha terminato la descrizione della mappa molecolare del cromosoma X, lungo 164 milioni di paia di basi, identificando 2100 marcatori molecolari distanti circa 75.000 bp l'uno dall'altro, con questa analogia geografica: se il cromosoma X fosse la strada da Saint Louis a San Francisco, gli intervalli fra i marcatori, come per le pietre miliari, sarebbero distanti proprio un miglio.
e) Procedere all'analisi della sequenza nucleotidica di ciascun tratto di circa 100.000 bp fra un marcatore e l'altro. Nel corso di questo procedimento analitico, lo studio dei singoli tratti, dei quali è nota con precisione la localizzazione cromosomica, può portare all'analisi di sequenze nucleotidiche già precedentemente descritte nella loro sequenza, come indicato al punto a. In questo caso è possibile confermare la posizione del locus relativo rispetto agli altri loci di quel cromosoma. Frequentemente questa procedura porta all'analisi di sequenze ignote, spesso ripetute a tandem un numero variabile di volte (VNTR, Variable Number of Tandem Repeats) o, con minore frequenza, permette l'identificazione di ORF, probabili geni egualmente ignoti, dai quali si deduce la sequenza amminoacidica per indagarne, in analogia a catene polipeptidiche note, la possibile funzione.
f) Analizzare una grande collezione di EST (Expressed Sequence Tags), ottenuta raccogliendo la sequenza di un numero molto grande di cDNA prodotti dalla trascrizione inversa di RNA messaggeri estratti da tessuti umani diversi, in cui ogni messaggero è identificato per una sua breve sequenza terminale. Al maggio 1998 risultavano depositate oltre 1.026.012 diverse sequenze umane, ovviamente in parte sovrapposizioni di uno stesso gene, poiché il numero complessivo di geni umani viene stimato, come è stato già detto, fra 50.000 e 100.000. Queste sequenze sarebbero inutili se non fossero identificate per almeno due parametri: la collocazione cromosomica e il significato funzionale. Il primo parametro viene affrontato attraverso la capacità delle sequenze EST di formare ibridi molecolari sia con cromosomi umani (identificati in situ per fluorescenza, FISH, Fluorescence in Situ Hybridization) sia con cromosomi artificiali di lievito (YAC) portatori di lunghe sequenze umane di cui è nota la localizzazione cromosomica. Il secondo parametro viene studiato utilizzando l'osservazione che le regioni funzionalmente significative dei genomi sono altamente conservate anche in specie evolutivamente distanti, per cui un'importante strategia di identificazione di geni sequenziati si basa sul confronto fra sequenze umane, di cui si ignora la funzione, con sequenze omologhe a funzione nota provenienti da specie diverse anche evolutivamente molto lontane dall'uomo, come il lievito e la drosofila. Di quest'ultimo organismo, che ha avuto sempre una particolare attenzione dai genetisti, sono note molte mutazioni e attraverso il progetto FlyBase sono stati catalogati più di 10.000 geni e 30.000 alleli. Un'analisi computerizzata che confrontasse sequenze di drosofila responsabili di mutazioni fenotipicamente caratterizzate ed EST umane ha permesso di identificare la funzione di geni umani che, per alcuni alleli mutati, danno luogo a patologie molto simili a quelle della drosofila.
Con questo metodo e con il confronto diretto con geni umani già identificati è stato possibile fino a tutto dicembre 1997 identificare 91 geni umani responsabili di patologie ereditarie e 94 geni umani noti come oncogeni o soppressori di crescita tumorale.
A giugno 1998 la situazione del p. g. umano (HUGO, Human Genome Organization) risulta la seguente: sono state inserite informazioni per 9342 oggetti genetici, di cui 6253 sono loci genici controllati e mappati, 499 fenotipi relativi a genotipi ancora non caratterizzati e 2590 altre sequenze. Circa 1400 geni sono stati identificati, ma non ancora assegnati ai cromosomi. L'assegnazione dei geni identificati ai loci sui cromosomi procede al ritmo di circa 600 geni per anno.
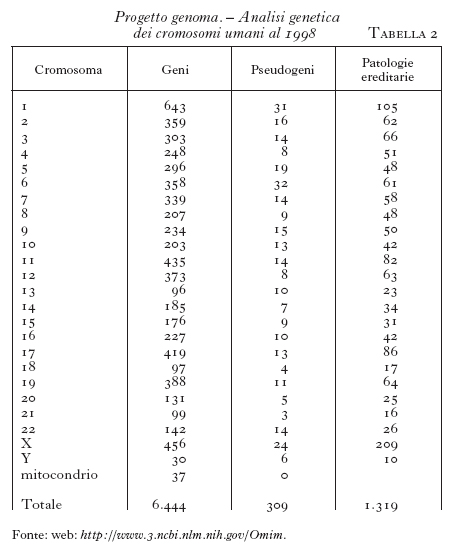
Nel maggio 1998 la porzione del genoma umano analizzata nella sequenza nucleotidica è stata di poco inferiore al 3% (tab. 2). I geni di cui sono state identificate sequenza nucleotidica e localizzazione cromosomica sono 6444 e, di questi, 1319 si riferiscono ad alleli mutati che sono responsabili di malattie ereditarie. Per quest'ultimo aspetto nell'aprile 1996 si è reso disponibile su Internet un nuovo sito informatico dedicato esplicitamente all'analisi delle mutazioni della linea germinale a carico di geni nucleari responsabili di malattie ereditarie nell'uomo, lo HGMD (Human Gene Mutation Database), che oggi contiene più di 13.000 sequenze di alleli mutati riferiti a 636 loci, con una crescita di informazione di almeno 2000 nuove sequenze per anno.
bibliografia
M.D. Adams, J.M. Kelley, J.D. Gocayne, Complementary DNA sequencing: expressed sequence tags and human genome project, in Science, 1991, 252, pp. 1651-56.
A.S. Wilcox, A.S. Khan, J.A. Hopkins et al., Use of 3' untranslated sequences of human cDNAs for rapid chromosome assignment and conversion to STSs: implications for an expression map of the genome, in Nucleic acids research, 1991, 19, pp. 1837-43.
R.D. Little, G. Pilia, S. Johnson et al., Yeast artificial chromosomes spanning 8 megabases and 10-15 centimorgans of human cytogenetic band Xq26, in Proceedings of the National Academy of Sciences of USA, 1992, 89, pp. 177-81.
M.S. Boguski, T.M. Lowe, C.M. Tolstoshev, dbEST database for 'expressed sequence tags', in Nature genetics, 1993, 4, pp. 332-33.
V.A. McKusick, J.S. Amberger, The morbid anatomy of the human genome: chromosomal location of mutations causing disease, in Journal of medical genetics, 1993, 30, pp. 1-26.
C.M. Fraser, J.D. Gocayne, O. White et al., The minimal gene complement of Mycoplasma genitalium, in Science, 1995, 270, pp. 397-403.
S. Banfi, G. Borsani, E. Rossi et al., Identification and mapping of human cDNAs homologous to Drosophila mutant genes through EST database searching, in Nature genetics, 1996, 13, pp. 167-74.
R.L. Tatusov, E.V. Koonin, D.J. Lipman, A genomic perspective on protein families, in Science, 1997, 278, pp. 631-37.
D.N. Cooper, E.V. Ball, M. Krawczak, The human gene mutation database, in Nucleic acids research, 1998, 26, pp. 285-87.
S. Kumer, B. Hedges, A molecular timescale for vertebrate evolution, in Nature, 1998, 392, pp. 917-20.
S.I. Letovsky, R.W. Cottingham, C.J. Porter et al., GDB: the human genome database, in Nucleic acids research, 1998, 26, pp. 94-99.
Sviluppi recenti
Malgrado l'impegno di università e istituzioni governative di molti paesi sul progetto, ad aprile del 2000, una società privata statunitense, la Celera Genomics di Washington fondata da C. Venter, sembra abbia completato la mappatura del genoma umano, dopo aver completato quella del genoma di Drosophila. *