
Radicali liberi
RADICALI LIBERI
Biologia e patologia di John M. C. Gutteridge
Sommario: 1. Introduzione. 2. Cenni di storia e di chimica dell'ossigeno: a) l'ossigeno e i suoi intermedi di riduzione; b) ossigeno singoletto; c) radicale superossido; d) perossido di idrogeno; e) radicale idroperossile e radicale ossidrile. 3. Origine biologica dei radicali liberi: a) formazione del radicale ossidrile in sistemi che generano superossidi e in altri sistemi biologici; b) la natura sito-specifica della reazione di Fenton; c) alternative al radicale ossidrile. 4. Metabolismo dei metalli di transizione: a) ferro; b) provenienza del ferro reattivo in vivo; c) rame. 5. La perossidazione lipidica, una reazione radicalica a catena: a) iniziazione mediata da complessi del ferro; b) iniziazione mediata da ioni ferrosi; c) stimolazione mediata da complessi del ferro; d) tipi di perossidazione lipidica; e) conseguenze della perossidazione lipidica; f) importanza della perossidazione lipidica nel danno indotto da radicali; g) perossidazione delle lipoproteine e suo ruolo nell'aterosclerosi. 6. Introduzione agli antiossidanti: a) antiossidanti cellulari; b) antiossidanti di membrana; c) antiossidanti extracellulari; d) equilibrio proossidanti/antiossidanti e stress ossidativo. 7. Stress ossidativo e patologie umane: alcuni esempi: a) polmone da shock o sindrome da sofferenza respiratoria negli adulti; b) artrite reumatoide; c) patologie del sistema nervoso. □ Bibliografia.
1. Introduzione
L'ossigeno viene usato dall'organismo per ossidare (bruciare) i substrati ricchi di carbonio e idrogeno (gli alimenti) e ricavarne l'energia indispensabile ai processi vitali. Nei diversi processi ossidativi la molecola dell'ossigeno, caratterizzata da una struttura chimica particolare, viene ridotta ad acqua mediante l'acquisto di quattro elettroni, uno per volta, formando immancabilmente dei radicali liberi come intermedi di reazione. I radicali liberi vengono costantemente prodotti nell'organismo durante il metabolismo dell'ossigeno ed eliminati, per evitarne i danni, da antiossidanti altamente specializzati. Quando la produzione di radicali liberi dell'ossigeno supera le capacità di controllo da parte dell'organismo, si sviluppa uno stato di stress ossidativo che può causare un danno tissutale; questo, a sua volta, può incrementare la formazione di radicali liberi amplificando così il danno.
Gli intermedi ridotti del metabolismo dell'ossigeno, compresi i radicali liberi, possono svolgere anche funzioni utili all'organismo: l'esempio più calzante è rappresentato dalla produzione di superossido e perossido di idrogeno da parte dei fagociti attivati durante la ‛combustione respiratoria' o respirazione cellulare (processo completamente diverso dalla respirazione a livello polmonare). Composti inorganici dell'ossigeno come il superossido, l'ossido nitrico e il perossido di idrogeno svolgono funzioni importanti come molecole ‛segnale' o ‛messaggeri'. Attualmente la nostra capacità di inibire, ritardare o riparare il danno tissutale causato dai radicali dell'ossigeno dipende dalla natura sito-specifica del danno e dai sistemi di trasporto delle molecole dei farmaci destinati a interagire con tali radicali. Entrambi i fattori vengono ulteriormente complicati dalla estrema rapidità delle reazioni di formazione dei radicali liberi.
2. Cenni di storia e di chimica dell'ossigeno
L'elemento ossigeno, presente nell'aria sotto forma di molecola (O2) nota come diossigeno od ossigeno molecolare, è stato per la prima volta isolato e caratterizzato, tra il 1770 e il 1775, dagli scienziati europei K. W. Scheele, J. Priestley e A.-L. Lavoisier, indipendentemente. Il diossigeno (d'ora in poi indicato come ossigeno) è apparso in quantità significative sulla superficie terrestre 2,5 × 109 anni fa, ed esistono testimonianze geologiche che fanno risalire la sua comparsa all'attività fotosintetica di alcuni microrganismi come le alghe blu-verdi. Queste ultime, scindendo l'acqua per soddisfare il proprio fabbisogno di idrogeno, rilasciarono tonnellate di ossigeno nell'atmosfera, dando luogo al caso più rilevante di ‛inquinamento ambientale' mai verificatosi sul nostro pianeta. L'aumento lento e costante della concentrazione atmosferica dell'ossigeno fu accompagnato dalla formazione della fascia di ozono nella stratosfera che, unitamente all'ossigeno, ha agito da filtro nei confronti dei raggi solari ultravioletti, i quali in precedenza raggiungevano liberamente la superficie della Terra (v. tab. I). Salendo dalla superficie terrestre verso gli strati più alti dell'atmosfera, le specie relativamente più pesanti, come O2, N2, H2O, sono sostituite da specie più leggere - atomi e ioni, come H•, H+, •OH, ed elettroni - che prevalgono al di sopra degli 800 km. La Terra rappresenta quindi l'unico centro di ossidazione in un universo completamente riducente, nel quale sono presenti in prevalenza idrogeno (H) ed elio (He).

L'aria secca è oggi costituita per circa il 21% (in volume) di ossigeno, elemento che risulta così secondo per abbondanza solo all'azoto (78%); tuttavia, la percentuale di ossigeno nell'aria è trascurabile se confrontata con quella presente nelle molecole di acqua degli oceani, dei laghi e dei fiumi (88,8%) e con quella presente nelle riserve minerarie della crosta terrestre, dove è di gran lunga l'elemento più abbondante (46,6%). Quando l'atmosfera terrestre è passata da uno stato altamente riducente allo stato attuale ricco di ossigeno, l'esistenza di forme di vita anaerobie fu limitata a luoghi dove non vi era ossigeno, mentre l'evoluzione di antiossidanti specializzati nella protezione dagli effetti tossici dell'ossigeno fu necessaria per la nascita di forme di vita aerobie, nelle quali l'ossigeno è indispensabile per lo svolgersi dei processi metabolici.
In condizioni normali, l'ossigeno è un gas stabile, inodore, insapore e incolore, scarsamente solubile in acqua (3 volumi di gas si sciolgono in 100 volumi di acqua). La sua limitata solubilità è indispensabile per la vita acquatica ed essenziale per le normali funzioni respiratorie dell'uomo. L'aria disciolta nell'acqua contiene una percentuale di ossigeno (34%) maggiore di quella dell'aria secca (21%), poiché la solubilità in acqua dell'azoto è di solo 2 volumi per 100. L'ossigeno è, inoltre, considerevolmente più solubile nei solventi organici che in acqua: per esempio, nel cloroformio (CHCl3), un solvente dei grassi, a 10 °C possono essere disciolti fino a 219,5 ml di ossigeno per litro a 1 atmosfera, mentre alle stesse condizioni nell'acqua possono essere sciolti solo 38,2 ml di ossigeno. Queste differenze di solubilità sono importanti, se si considera la quantità di ossigeno disponibile per le reazioni chimiche nei compartimenti delimitati da membrane biologiche, che sono costituite in prevalenza da lipidi.
Quando l'ossigeno viene ridotto, per aggiunta di un elettrone per volta (v. equazioni 1-5), si formano composti pericolosi per la cellula: due radicali liberi (HO2•, •OH) e il perossido di idrogeno (H2O2). A pH fisiologico (7,4) il radicale idroperossido (HO2•), con un pKa di 4,8 (il valore del pH a cui sono presenti, alla stessa concentrazione, sia l'acido, HO2•, che la base, O−2•), si dissocia e forma il radicale anionico superossido (O−2•):
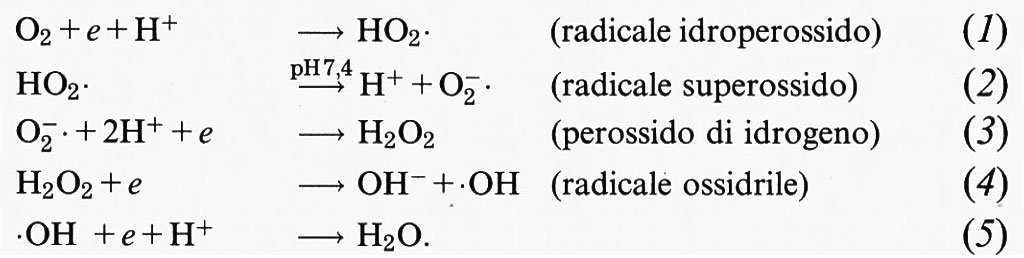
Non è necessario indicare l'elettrone spaiato (punto a destra) sul radicale anionico superossido (O−2•), poiché questo può essere considerato meno ‛radicale' dell'ossigeno molecolare, che possiede due elettroni spaiati, ma non è mai indicato come O2••. A questo punto, prima di continuare la discussione, è necessario chiarire che cosa sia un radicale libero.
‟Un radicale libero può essere definito come una specie chimica, contenente uno o più elettroni spaiati, capace di esistere in modo autonomo". Questa è una definizione biologica generica (v. Halliwell e Gutteridge, 19892), poiché non specifica esattamente dove è posizionato l'elettrone spaiato; la adottiamo, comunque, perché ci permette di classificare come radicali liberi la maggior parte degli ioni dei metalli di transizione e quindi di capire meglio la stretta correlazione tra ossigeno e ioni metallici reattivi.
Le molecole di interesse biologico e chimico differiscono nella loro tendenza ad acquistare o cedere elettroni; si definisce ‛potenziale redox' il voltaggio necessario per contrastare tale tendenza in soluzione acquosa, in condizioni standard.
Osservazioni sperimentali hanno dimostrato che, nonostante l'elevata affinità per gli elettroni, l'ossigeno mostra una reattività inferiore a quella prevista; le ragioni di tale comportamento sono imputabili alla sua particolare struttura elettronica: solo quando quest'ultima viene forzata l'ossigeno può esprimere la sua reattività ‛effettiva'.
a) L'ossigeno e i suoi intermedi di riduzione
L'ossigeno molecolare (O2) allo stato diatomico fondamentale è esso stesso un radicale, con due elettroni spaiati, localizzati ciascuno in un orbitale di antilegame π* e aventi lo stesso numero quantico di spin, o spins paralleli (per una spiegazione più dettagliata, v. Halliwell e Gutteridge, 19892). Pertanto, quando l'O2 ossida un atomo o una molecola, i due elettroni che acquista da tali specie riducenti devono necessariamente avere spins paralleli, così da riempire gli spazi vuoti negli orbitali π*. Al contrario, la maggior parte delle molecole biologiche è costituita da specie non radicaliche, caratterizzate dalla presenza di legami covalenti, formati da due elettroni a spins antiparalleli che occupano lo stesso orbitale molecolare. La reazione dell'ossigeno con le biomolecole è, dunque, soggetta a restrizione di spin: tale restrizione imposta dal numero quantico di spin risulta molto vantaggiosa alla vita in un ambiente ricco di ossigeno, poiché ritarda la reazione dell'ossigeno molecolare con le molecole biologiche. La sopravvivenza dipende comunque dalla costante riparazione del danno ossidativo, sia mediante una protezione antiossidante specifica (enzimatica), sia attraverso molecole scavenger (letteralmente ‛spazzino': molecola che inattiva un radicale, formando una specie meno tossica per la cellula) che proteggono i siti chiave e limitano l'estensione del danno (v. cap. 6). La restrizione di spin dell'ossigeno può tuttavia essere vinta dai metalli di transizione presenti nel sito attivo della maggior parte degli enzimi ad attività ossidasica e ossigenasica, grazie alla loro capacità di acquistare o donare singoli elettroni.
b) Ossigeno singoletto
La reattività dell'O2 può essere aumentata spostando uno degli elettroni spaiati, in modo da attenuare la restrizione di spin. Tale processo richiede energia e genera gli stati di singoletto dell'ossigeno. Lo stato di O2 singoletto 1Δg - uno tra i più importanti nei sistemi biologici - non possiede elettroni spaiati e così non viene classificato come radicale. L'O2 singoletto 1Σ +g, di solito, decade allo stadio 1Δg senza aver tempo di reagire. L'eccitazione dell'O2 agli stati di singoletto può essere raggiunta quando vari pigmenti vengono illuminati in sua presenza: il pigmento, infatti, assorbe la luce, entra in uno stato di eccitazione più alto e trasferisce energia alla molecola dell'O2, formando così l'O2 singoletto. Tale fenomeno può quindi avvenire in qualsiasi sistema pigmentato esposto alla luce, come nel caso del cristallino (v. Zigler e Goosey, 1981) e dei cloroplasti esposti alla luce (v. Halliwell e Gutteridge, Oxygen toxicity..., 1984).
Talune malattie possono indurre un eccessivo accumulo di O2 singoletto, come accade per esempio nelle porfirie, alterazioni generalmente congenite del metabolismo delle porfirine. Queste ultime sono spesso escrete nelle urine e accumulate nella pelle: in tal caso l'esposizione alla luce causa l'insorgenza di eruzioni cutanee sgradevoli, cicatrici e pruriti. La gravità del danno dipende dalla struttura delle porfirine accumulate e dunque è differente nelle diverse forme di porfiria. Inoltre, è stato osservato che alcune porfirine vengono accumulate in cellule neoplastiche. Questo fenomeno può essere sfruttato per il rilevamento e la terapia di tumori: infatti, somministrando un derivato porfirinico noto come HPD (Haemato-Porphyrin Derivative), possono essere individuati i tessuti tumorali, in quanto questi trattengono prodotti fluorescenti. Dato che la successiva irradiazione con luce della lunghezza d'onda assorbita dall'HPD può danneggiare il tumore, tali reazioni possono trovare applicazione in chemioterapia, specialmente nella cura del cancro della pelle e del polmone, in quanto sia i radicali ossidrile che l'ossigeno singoletto possono danneggiare le cellule tumorali. Un'altra applicazione delle reazioni di fotosensibilizzazione in medicina consiste nell'uso degli psoraleni nel trattamento di alcune malattie della pelle, come, ad esempio, la psoriasi (v. Pathak e Joshi, 1984). Il trattamento, definito terapia PUVA (psoralene-ultravioletto), consiste nell'applicazione combinata di luce ultravioletta, nell'intervallo di lunghezze d'onda compreso tra 320 e 400 nm (UVA), e uno psoralene. Gli psoraleni sono una classe di composti prodotti dalle piante che agiscono come potenti fotosensori della produzione di O2 singoletto. È importante sottolineare che anche alcuni farmaci (per es. le tetracicline e il benoxaprofene, un farmaco antinfiammatorio non steroideo) e alcuni composti costituenti di cosmetici possono danneggiare la pelle per fotosensibilizzazione.
È stato spesso sostenuto che l'O2 singoletto si formerebbe nella reazione di dismutazione del radicale O−2 e durante la respirazione cellulare dei fagociti, ma per nessuno dei due processi si possiedono prove conclusive; generalmente, l'unica prova della formazione di O2 singoletto in un sistema è rappresentata dalla produzione di luce da parte del sistema stesso, oppure dall'inibizione di tale produzione, in presenza di scavengers dell'O2 singoletto, quali ad esempio il DABCO (diazabiciclo-ottano o trietilendiammina), il difenilisobenzofurano, l'istidina o gli azotidrati.
Tuttavia, non esistono scavengers specifici dell'O2 singoletto: tutti reagiscono con il radicale ossidrile, spesso con una costante di velocità maggiore rispetto alle reazioni con l'O2 singoletto. Una parte di essi reagisce anche con almeno un radicale organico del perossido (v. Packer e altri, 1981) e con l'acido ipocloroso prodotto dall'azione della mieloperossidasi nei neutrofili attivati (v. Harrison e altri, 1978). Se, aggiungendo alte concentrazioni delle molecole scavenger prima menzionate a un sistema, nessuna di esse inibisce la reazione studiata, allora si può concludere che l'O2 singoletto non è richiesto da tale reazione; non è però valida la considerazione inversa. È da notare, tuttavia, che i prodotti delle reazioni del colesterolo (v. Kulig e Smith, 1973) e del triptofano (v. Singh e altri, 1981) con l'O2 singoletto sono diversi da quelli ottenuti dalle reazioni di queste molecole con il radicale ossidrile, cosicché l'isolamento e la caratterizzazione di tali prodotti potrebbero fornire prove migliori della formazione di O2 singoletto.
Le molecole dell'ossigeno singoletto, quando decadono allo stato fondamentale, emettono luce nella banda dell'infrarosso (1.270 nm), ma si può avere emissione di luce anche a 634 e 702 nm, a causa di un fenomeno di ‛emissione dimolare', che coinvolge la cooperazione di due molecole di O2 singoletto (v. Lengfelder e altri, 1983).
Un metodo utile per seguire la respirazione cellulare consiste nel misurare l'emissione di luce da parte di fagociti attivati in presenza di molecole stimolatrici, come il luminolo o la lucigenina. Tale misurazione, però, non può esser considerata una dimostrazione della produzione di O2 singoletto, poiché lo spettro di emissione della luce non ha alcuna analogia con quello atteso per l'O2 singoletto. Si può dimostrare che una miscela di mieloperossidasi, H2O2 e Cl- produce O2 singoletto, ma solamente in condizioni che difficilmente si realizzano in vivo (v. Kanofsky e altri, 1984). In ogni caso, la reattività dell'acido ipocloroso prodotto dal sistema della mieloperossidasi è sufficiente per giustificare la citotossicità del sistema, anche in assenza di O2 singoletto. La mieloperossidasi svolge solo un ruolo secondario nell'eliminazione dei Batteri da parte dei neutrofili umani; infatti una carenza congenita di tale enzima porta raramente all'insorgenza di problemi di natura clinica. Tuttavia l'HOCl prodotto dalla mieloperossidasi potrebbe inattivare l'α1-antitripsina nei siti di infiammazione, contribuendo in tal modo al danno proteolitico (v. Weiss, 1986).
La chemiluminescenza è un saggio utile per valutare lo ‛stress ossidativo' in organelli isolati, cellule intere e organi perfusi (v. Cadenas e altri, 1981). Una frazione della luce emessa potrebbe derivare dall'O2 singoletto, ma il resto probabilmente proviene da altre fonti, come, ad esempio, dalle reazioni di Fenton (v. Andersen e Harvath, 1979), dalle reazioni catalizzate dalla perossidasi (v. Cilento, 1982) e dall'interazione delle porfirine con H2O2 (v. Slawinski e altri, 1981).
c) Radicale superossido
La riduzione dell'ossigeno a opera di un solo elettrone produce il radicale superossido, O−2, che si forma in quasi tutte le cellule aerobie (v. Fridovich, 1975, 1978 e 1983; v. Halliwell e Gutteridge, 19892): in gran parte esso si forma per cessione di elettroni all'O2 da parte dei vari componenti delle catene di trasporto degli elettroni nella cellula, quali quelle dei mitocondri, dei cloroplasti e del reticolo endoplasmatico. L'entità della cessione di elettroni, e quindi il tasso di produzione di O−2, aumenta con l'aumentare della concentrazione di O2. È stato inoltre dimostrato che O−2 viene prodotto durante la respirazione dei fagociti (neutrofili, monociti, eosinofili e macrofagi). La Km per l'O2 del complesso della NADPH-ossidasi, che produce l'O−2 nei neutrofili, ha un valore compreso tra i livelli di concentrazione dell'O2 nei liquidi corporei, così che un'elevata concentrazione di O2 può incrementare la produzione di O−2 nei fagociti attivati. La produzione di superossido da parte dei fagociti assume un ruolo chiave nella eliminazione di diversi ceppi batterici; se il superossido non viene prodotto, come nel caso della malattia congenita nota come granulomatosi cronica, molti ceppi batterici non vengono completamente eliminati e ciò dà origine a infezioni multiple (v. Tauber e altri, 1983). Molti altri ceppi batterici vengono, comunque, completamente eliminati dai fagociti nella malattia cronica granulomatosa e quindi devono esistere altri meccanismi antibatterici altrettanto efficaci.
Nei solventi organici l'O−2 è una specie estremamente reattiva, per esempio può rimuovere lo ione Cl- da idrocarburi clorurati non reattivi come il CCl4. Al contrario, in soluzione acquosa, l'O−2 è poco reattivo e agisce principalmente da agente riducente, come nel caso del ferro del citocromo c o del nitro-blu di tetrazolio, o come un ossidante debole di molecole come l'adrenalina e l'acido ascorbico. L'O−2 può, inoltre, esser soggetto alla seguente reazione di dismutazione, che, nel suo complesso, può essere scritta nella maniera seguente:
2O−2 + 2H+ → H2O2 + O2, (6)
benché a pH fisiologico la reazione si componga di due stadi:
O−2 + H+ → HO2• (7)
HO2• + O−2 + H+ → H2O2 + O2. (8)
La costante complessiva di dismutazione a pH 7 è circa 5 × 105 mol-1s-1 (v. Bielski e Allen, 1977) e ogni reazione in soluzione acquosa cui sia soggetto O−2 competerà con questa reazione di dismutazione.
Anche se l'O−2 in soluzione acquosa ha una bassa reattività, è stato osservato che i sistemi chimici, enzimatici e fagocitari che generano O−2 in soluzione acquosa provocano un considerevole danno biologico (v. Halliwell e Gutteridge, The importance..., 1985). Una ulteriore prova dell'opportunità di rimuovere l'O−2 in vivo deriva dai molti dati che mostrano come gli enzimi appartenenti alla famiglia delle superossido-dismutasi (SOD), tra i principali antiossidanti presenti nei sistemi biologici (v. Fridovich, 1975), si sono evoluti in modo da assumere una carica di superficie tale da facilitare l'utilizzo di O−2 come substrato (v. Getzoff e altri, 1983) e sono in grado di far aumentare la velocità della reazione (6) di circa 4 ordini di grandezza. Si può, quindi, concludere che per la cellula la rimozione di O−2 è più importante della sua conversione in H2O2, sebbene nelle cellule umane le SOD agiscano in presenza di enzimi che decompongono l'H2O2, come la catalasi e la glutatione-perossidasi (v. cap. 6; v. Chance e altri, 1979; v. Halliwell e Gutteridge, The importance..., 1985; v. Gutteridge e Halliwell, 1988).
Spesso si ritiene che l'inibizione di una reazione per aggiunta di SOD implichi necessariamente che questa reazione, per procedere, richieda O−2, essendo quest'ultimo un substrato catalitico specifico delle SOD. Prima di giungere a tale conclusione si devono tuttavia mettere a punto anche degli esperimenti di controllo, usando la proteina denaturata al calore o l'apoenzima. Inoltre, è necessario considerare con cautela anche le inibizioni mediate dalle SOD in sistemi contenenti chinoni; molti semichinoni, infatti, reagiscono reversibilmente con l'O2:
semichinone +O2 ⇌ O−2 + chinone. (9)
L'aggiunta di SOD accelera il consumo di semichinone attraverso l'eliminazione di O−2: la reazione, che è in realtà indotta dal semichinone, potrebbe essere erroneamente attribuita all'O−2, in conseguenza del fatto che c'è una inibizione dovuta all'aggiunta di SOD (v. Winterbourn, Hydroxyl..., 1981). Interpretazioni erronee possono anche insorgere quando si forma O−2 per ossidazione di molecole come l'adrenalina, il diidrossifumarato o l'idrossidopammina, ossidazione che produce altro O−2 che ossiderà ulteriormente tali molecole.
d) Perossido di idrogeno
L'H2O2 potrebbe essere normalmente prodotto nelle cellule sia indirettamente, in un qualsiasi sistema che produce O−2 per dismutazione non enzimatica o catalizzata dalle SOD (6), sia direttamente da alcune ossidasi, come la glicolato- e l'urato-ossidasi (v. Chance e altri, 1979). Infatti, se si ipotizza che l'O−2 sia prodotto in vivo nelle cellule umane e che in seguito venga eliminato dalle SOD, dobbiamo supporre che si produca anche H2O2. In realtà, la produzione di H2O2, probabilmente soprattutto a partire da O−2, è stata osservata in molti batteri di diverse specie, nei fagociti, negli spermatozoi, nei mitocondri, nei microsomi e nei cloroplasti (v. Chance e altri, 1979). Concentrazioni micromolari di H2O2 sono state rilevate nel cristallino (v. Bhuyan e Bhuyan, 1977) e inoltre sono stati individuati vapori di H2O2 nell'aria espirata dall'uomo (v. Williams e altri, 1983); nel plasma umano se ne riscontrano livelli discreti e, in concentrazioni quasi micromolari, l'H2O2 è tra i costituenti della maggior parte delle riserve naturali d'acqua (v. Zafiriou, 1987).
L'H2O2 non ha elettroni spaiati e non è un radicale; allo stato puro possiede una reattività limitata, ma può attraversare le membrane biologiche, mentre le specie O−2 cariche possono farlo solo lentamente, a meno che non attraversino un ‛canale anionico' come quello presente nella membrana eritrocitaria. Se le cellule vengono esposte a sistemi che generano O−2 e il danno ossidativo viene evitato mediante l'aggiunta di catalasi ma non di SOD, ciò non significa necessariamente che tale danno derivi direttamente da H2O2: l'O−2 formatosi all'esterno della cellula non può facilmente attraversarne la membrana, al contrario dell'H2O2 che all'interno della cellula può dare origine a diverse specie radicaliche reattive. Analogamente, l'H2O2 generato all'interno della cellula può essere decomposto per aggiunta di catalasi all'esterno della cellula stessa, perché tale enzima turba l'equilibrio di diffusione dell'H2O2 attraverso la membrana e ne facilita la fuoriuscita; al contrario, la SOD aggiunta esternamente non può eliminare l'O−2 generato all'interno. In questo tipo di esperimenti, si dovranno comunque effettuare dei controlli con la catalasi denaturata al calore. Tuttavia, i dati sulla tossicità dell'H2O2 per cellule e organismi sono contrastanti: alcune cellule batteriche e animali vengono uccise anche da concentrazioni micromolari di H2O2, mentre altri batteri e alghe fotosintetiche ne producono e ne rilasciano grandi quantità (v. Halliwell, 1981). Tale variabilità è dovuta all'attività degli enzimi che decompongono l'H2O2 e probabilmente alla percentuale di H2O2 convertito in radicali maggiormente reattivi.
È stato osservato che frazioni microsomiali isolate da diversi tessuti animali producono livelli elevati di H2O2 e di O−2, se incubati in presenza di NADPH; tali frazioni vengono spesso usate per studiare il ciclo di ossidoriduzione di alcuni farmaci e la perossidazione dei lipidi. Nonostante il loro nome, i microsomi non sono degli organelli, ma degli artefatti del frazionamento cellulare: infatti, nelle cellule sottoposte a omogeneizzazione la membrana plasmatica e il reticolo endoplasmatico vengono frammentati e la frazione microsomiale, ottenuta per centrifugazione ad alta velocità, risulta formata da un insieme eterogeneo di vescicole derivanti da questi due sistemi di membrane. L'O−2 e l'H2O2 prodotti nei microsomi derivano dal sistema NADPH-citocromo P450 reduttasi-citocromo P450; se si aumenta la quantità del P450 e della sua reduttasi, pretrattando gli animali con fenobarbitale, verrà incrementata la produzione di H2O2 nei microsomi del fegato successivamente isolati dagli animali (v. Hildebrandt e Roots, 1975). Nel fegato perfuso di ratto, però, il valore basale di glutatione ossidato (GSSG) liberato, considerato come indice della produzione di H2O2, è minore di quello atteso in base ai valori osservati nei microsomi in vitro (v. Oshino e altri, 1975) e non aumenta pretrattando gli animali con fenobarbitale; se ne deduce che la formazione di H2O2 e, di conseguenza, di O−2, nel reticolo endoplasmatico in vivo non è così rapida come ci si aspetterebbe dagli esperimenti condotti con i microsomi. Forse durante la frammentazione e la formazione delle vescicole di membrana che avviene rompendo le cellule per formare i microsomi, la disposizione dei componenti del sistema P450 all'interno della membrana viene alterata, cosicché gli elettroni raggiungono più facilmente l'ossigeno; tale osservazione deve essere tenuta presente negli esperimenti condotti con frazioni microsomiali isolate. Inoltre, la concentrazione di ossigeno in prossimità del reticolo endoplasmatico in vivo deve sicuramente essere inferiore a quella misurata nei microsomi incubati in vitro.
Quando si studiano i microsomi, che apparentemente producono O−2 e H2O2 a livelli abnormi, si deve sempre ricordare che si tratta di artefatti che hanno origine dal frazionamento subcellulare.
e) Radicale idroperossile e radicale ossidrile
È improbabile che il danno causato dai sistemi che generano O−2 sia dovuto direttamente alla tossicità dell'O−2 e dell'H2O2, data la reattività relativamente bassa di queste due molecole in soluzione acquosa; bisogna dunque indagare su quale sia la causa del danno molecolare in tali sistemi.
Radicale idroperossile. - La protonazione dell'O−2 dà origine al radicale dell'idroperossido HO2• (v. eq. 7). Il pKa dell'HO2• è 4,7-4,8 (v. Bielski e Allen, 1977) e pertanto, se si presuppone che il pH fisiologico sia di circa 7,4, solo lo 0,25% dell'O−2 generato sarà presente sotto forma di HO2•. In prossimità delle membrane, però, il pH può essere considerevolmente più basso, favorendo così la formazione di maggiori quantità di HO2•; per esempio, in corrispondenza di macrofagi attivati il pH può raggiungere valori inferiori a 5 e pertanto una quantità considerevole dell'O−2 da essi prodotto potrà essere protonato, dando luogo ad HO2•.
Non è ancora stato chiaramente dimostrato se l'HO2• svolga un ruolo citotossico in qualche sistema biologico, ma è possibile fare due considerazioni: in primo luogo, essendo meno polare dell'O−2, dovrebbe essere in grado di attraversare le membrane biologiche con la stessa facilità dell'H2O2; in secondo luogo, è molto più reattivo dell'O−2, e infatti, a differenza dell'O−2, può reagire direttamente con gli acidi grassi, ed è stato dimostrato che gli acidi linolenico, linoleico e arachidonico vengono convertiti in perossidi da HO2• (v. Bielsky e altri, 1983).
Radicale ossidrile. - Il radicale ossidrile •OH viene prodotto esponendo l'acqua a radiazioni ionizzanti ad alta energia; per questo motivo le sue proprietà sono ben note ai radiochimici. L'•OH possiede un'elevata reattività (v. Anbar e Neta, 1967), tanto che ogni radicale ossidrile che si forma in vivo reagisce nel suo sito di formazione o in sua prossimità (entro pochi ångström). Il radicale •OH prodotto in prossimità del DNA può danneggiarlo, modificandone le basi o provocando la rottura dei filamenti, mentre prodotto in prossimità di un enzima presente in eccesso nella cellula (come la lattato-deidrogenasi) potrebbe non avere conseguenze biologiche. La reazione dell'•OH con una biomolecola produce un altro radicale, generalmente meno reattivo, il quale a sua volta può causare dei danni, diffondendo dal proprio sito di formazione e interagendo poi con specifiche biomolecole. Per esempio, l'interazione del radicale •OH con l'acido urico protegge l'enzima lattatodeidrogenasi dall'inattivazione da parte dell'•OH stesso, ma facilita l'inattivazione dell'alcoldeidrogenasi, poiché, essendo i radicali derivati dall'urato complessivamente meno reattivi, ne sopravvive un numero maggiore, che può raggiungere i siti sensibili dell'alcoldeidrogenasi e reagirvi. Forse il miglior esempio dell'importanza dei radicali secondari è rappresentato dalla capacità dell'•OH di iniziare la perossidazione dei lipidi, per sottrazione di un atomo di idrogeno, con conseguente formazione di un carbonio reattivo e di radicali perossido (v. cap. 5).
La maggior parte dell'•OH formatosi in vivo, con l'eccezione di quello prodotto per eccessiva esposizione alle radiazioni ionizzanti, deriva dalla rottura metallo-dipendente della molecola di H2O2, secondo l'equazione generica:
Mn+ + H2O2 → M(n+1)+ + •OH + OH-, (10)
in cui Mn+ è lo ione di un metallo, che può essere titanio (III), rame (I), ferro (II) (v. Walling, 1982) o cobalto (II) (v. Gutteridge e Bannister, 1986). Anche alcuni complessi del vanadio, del rame e del nichel possono generare •OH da H2O2 (v. Quinlan e altri, 1992; v. Sunderman, 1987). Tutte queste reazioni possono essere importanti, se consideriamo gli effetti tossicologici dell'avvelenamento da metalli, ma probabilmente solo le reazioni dipendenti da ferro (II) e rame (I) avvengono in vivo in condizioni normali. Grande attenzione è stata data alla decomposizione dell'H2O2 ferro-dipendente, detta ‛reazione di Fenton' (v. Walling, 1982):
Fe2+ + H2O2 → Fe3+ + •OH + OH-. (11)
Tracce di Fe3+ possono reagire successivamente con H2O2, anche se questa reazione procede lentamente a pH fisiologico:
Fe3+ + H2O2 → Fe2+ + O−2 + 2H+, (12)
e possono verificarsi altre reazioni, per esempio:
•OH + H2O2 → H2O + H+ + O−2 (13)
O−2 + Fe3+ → Fe2+ + O2 (14)
•OH + Fe2+ → Fe3+ + OH-. (15)
Sebbene sia stato spesso detto che i radicali •OH provengono quasi esclusivamente da una miscela di Fe(II) e H2O2, le equazioni precedenti dimostrano che anche le miscele Fe(III)-H2O2 possono produrli, per quanto più lentamente, per combinazione delle reazioni (11), (12) e (14) (v. Walling, 1982). La SOD, rimuovendo O−2 (v. eqq. 6 e 14), può inibire in determinate circostanze la produzione di •OH in un sistema Fe(III)-H2O2.
L'ipotesi che la reazione di Fenton avvenga in vivo (v. Halliwell e Gutteridge, Oxygen toxicity...,1984) è stata contestata (v. Winterbourn, Hydroxyl..., 1981) sulla base delle seguenti obiezioni: a) la costante di velocità della reazione (11) è troppo bassa per avere un significato biologico; b) la specie prodotta nel sistema di Fenton non è il radicale •OH; c) non ci sono ioni metallici catalizzatori in vivo.
La prima obiezione è valida se, in termini chimici, la costante della reazione (11) è bassa, sicuramente inferiore a 102 mol-1 s-1. Bisogna però fare una considerazione: è improbabile che, nella maggior parte dei casi, le concentrazioni di H2O2 e del catalizzatore ferro (II) in vivo raggiungano livelli superiori a qualche micromole, anche se ciò potrebbe verificarsi per H2O2 (v. Chance e altri, 1979), ma comunque, per concentrazioni di entrambi i reagenti pari a 1 µmol/l, la velocità di produzione (v) dell'•OH sarebbe pari a:
v = k [Fe2+] [H2O2] = k [10-6] [10-6] = 7,6 × 10-11, (16)
considerando il valore della costante di secondo ordine k pari a 76 mol-1 s-1. Visto che il valore medio per una cellula del parenchima epatico è un po' maggiore di 10-12 litri, il numero (N) di radicali •OH generati per secondo sarebbe:
N=7,6 × 10-11 × 10-12 × 6,023 × 1023 ≅ 46. (17)
Quindi, approssimativamente, anche con questa stima per difetto, potrebbero essere generati circa 46 radicali •OH in ogni cellula per secondo. L'alta reattività dell'•OH potrebbe, anche a tale concentrazione, avere enormi conseguenze biologiche nel sito di formazione. Naturalmente, con il procedere della reazione sia H2O2 che Fe(II) verranno consumati e il tasso di produzione di •OH diminuirà, a meno che i reagenti non siano continuamente ripristinati. Occorre poi puntualizzare che la costante di reazione del Cu (I) con l'H2O2:
Cu+ + H2O2 → Cu2+ + •OH + OH-, (18)
è di 4,7 × 103 mol-1 s-1 ed è quindi molto più grande di quella del Fe(II).
3. Origine biologica dei radicali liberi
a) Formazione del radicale ossidrile in sistemi che generano superossidi e in altri sistemi biologici
La formazione del radicale •OH in una vasta gamma di sistemi che generano O−2, compresi i fagociti attivati e le miscele ipoxantina/xantinossidasi, è stata dimostrata utilizzando diverse tecniche, tra cui l'idrossilazione aromatica, la risonanza di spin elettronica (ESR, Electron Spin Resonance), detta anche risonanza paramagnetica elettronica (EPR, Electron Paramagnetic Resonance), associata allo spin trapping (v. radicali liberi: Chimica, vol. XI), l'ossidazione del metionale, l'attacco sul triptofano, l'ossidazione del dimetilsolfossido, la decarbossilazione dell'acido benzoico marcato con 14C al carbossile, l'attacco sul benzoato per produrre materiale che dà colorazione con l'acido tiobarbiturico o la produzione di prodotti fluorescenti, come il 4-idrossi- e il 3-idrossibenzoato, e la degradazione del desossiribosio per dare prodotti che generano la reazione cromatica con l'acido tiobarbiturico (v. Halliwell e Gutteridge, 19892). In termini chimici, i metodi più efficaci per evidenziare la formazione del radicale •OH sono probabilmente l'ESR, l'idrossilazione aromatica e l'analisi di prodotti specifici, derivanti da diverse biomolecole. Dopo l'idrossilazione aromatica, i prodotti idrossilati derivati dall'attacco dell'•OH sugli anelli benzenici possono essere separati mediante tecniche cromatografiche (HPLC o GLC), quantificati e distinti dai prodotti formati da sistemi enzimatici di idrossilazione. La prova decisiva della formazione del radicale •OH richiede diversi esperimenti di controllo (v. Finkelstein e altri, 1981). La corretta interpretazione dei risultati ottenuti in questo tipo di esperimenti comporta, però, alcune osservazioni.
1. L'inibizione provocata dall'aggiunta di una singola molecola scavenger del radicale ossidrile a una reazione biologica non prova nulla, specialmente se lo scavenger è la tiourea, che reagisce con H2O2, HOCl e i radicali alcossido e può anche chelare gli ioni metallici necessari alla produzione di •OH; anche l'etanolo, diversamente dal mannitolo e dal formiato, reagisce con i radicali alcossido. Bisognerebbe provare con diverse molecole scavenger e confrontare il grado di inibizione che producono con le costanti, note, delle reazioni degli scavengers con •OH.
2. Le molecole scavenger e quelle utilizzate per determinare la presenza di •OH dovrebbero avere delle cinetiche di competizione, ossia dovrebbero competere per le stesse specie.
3. La reazione del radicale •OH con una molecola scavenger produce un radicale secondario, che potrebbe esso stesso risultare dannoso in determinati sistemi: per esempio, i radicali del formiato e dell'etanolo possono attaccare la sieroalbumina e il radicale azide (formato dalla reazione dell'•OH con l'anione azotidrato, N−3) attacca il triptofano e la tirosina.
4. Il radicale •OH viene spesso prodotto nei sistemi biologici dalla reazione dell'H2O2 con ioni metallici legati a siti specifici: quando l'•OH reagisce con le molecole con cui si lega, diviene inaccessibile alle molecole scavenger dell'•OH aggiunte. Per esempio, il danno causato dall'H2O2 al DNA delle cellule dei Mammiferi può essere dovuto alla reazione dell'H2O2 stesso con ioni metallici legati al DNA. Questo concetto di ‛sito-specificità' sarà discusso in seguito (v. sotto, § b). La formazione del radicale •OH nei sistemi che generano O−2 è generalmente inibita dall'aggiunta di catalasi o di SOD. A tal proposito è possibile fare due considerazioni: innanzitutto, l'inibizione da parte della catalasi non è sorprendente, dal momento che l'•OH proviene dalla scissione metallo-dipendente dell'H2O2 (v. eq 11); in secondo luogo, l'azione inibitoria delle SOD, se accompagnata dai controlli appropriati discussi in precedenza, mostra chiaramente che anche l'O−2 è coinvolto nella produzione di •OH. Una semplice spiegazione del suo ruolo potrebbe essere che l'O−2 riduce gli ioni metallici ossidati, promuovendo in tal modo la reazione di Fenton. Quindi, prendendo il ferro come esempio, si può scrivere il seguente ciclo di reazioni:
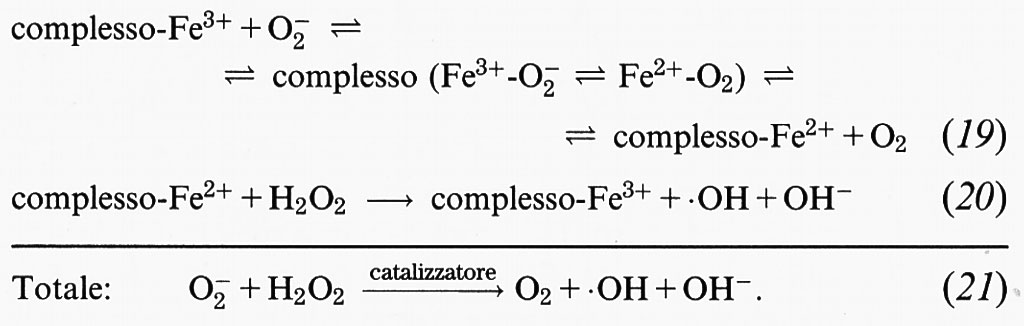
La somma delle reazioni (19) e (20) è chiamata spesso ‛reazione di Haber-Weiss catalizzata da metalli' o ‛reazione di Fenton superossido-guidata'. Tale interpretazione del ruolo dell'O−2, cioè di semplice agente riducente, è probabilmente ingenua, ma spiega diverse osservazioni sperimentali. Tuttavia solleva anche il problema del perché il radicale O−2 sia così importante, dal momento che altri agenti riducenti sono presenti in vivo. Rowley e Halliwell (v. i contributi del 1982) hanno studiato l'effetto dell'aggiunta di altri agenti riducenti a sistemi ferro-dipendenti che generano il radicale •OH da O2- e da H2O2, concludendo che è improbabile che l'NADH, l'NADPH o i composti tiolici come il glutatione (GSH) o la cisteina siano in grado di impedire la formazione in vivo dei radicali •OH dipendente dall'O−2. Anzi, sia l'NAD(P)H che i composti tiolici, in determinate condizioni, possono interagire con gli ioni metallici e con l'H2O2, facendo aumentare la formazione del radicale •OH dipendente dall'O−2.
Un altro importante agente riducente biologico è l'acido ascorbico, che può sostituire l'O−2 come riducente in molti dei sistemi in vitro impiegati per dimostrare la formazione O−2-dipendente dei radicali •OH in presenza di sali di ferro o di rame. Quando l'agente riducente è l'ascorbato, le SOD non impediscono la produzione di •OH, che tuttavia è ancora inibita dalla catalasi; se l'O−2 e l'ascorbato sono entrambi disponibili, il contributo relativo di ciascuno dipende dalle rispettive concentrazioni. L'ascorbato è normalmente presente nei liquidi extracellulari umani a concentrazioni tali da poter rimpiazzare solo parzialmente l'O−2 nella riduzione del Fe(III), ma, essendo esso rapidamente ossidato durante le reazioni con l'O−2 e l'•OH, la produzione di •OH diventa, alla fine, completamente O−2-dipendente (v. Rowley e Halliwell, 1983). Al contrario, le elevate concentrazioni (millimolari) dell'ascorbato presenti in alcuni tessuti dei Mammiferi, come l'occhio, il midollo spinale o gli pneumociti, rendono improbabile che l'O−2 competa come agente riducente, a meno che l'ascorbato e l'O2- non siano localizzati in differenti compartimenti subcellulari o che grandi quantità di O−2 siano prodotte in un sito localizzato; in effetti, a così alte concentrazioni, la capacità dell'ascorbato di eliminare direttamente il radicale •OH diventa significativa. Bisogna aggiungere che l'acido ascorbico nei siti di infiammazione può essere rapidamente consumato dalla reazione con l'ossidante HOCl prodotto dalla mieloperossidasi (v. Halliwell e altri, 1987).
b) La natura sito-specifica della reazione di Fenton
L'irradiazione ad alta energia di una soluzione acquosa produce una notevole quantità di radicali ossidrile:
H2O → •OH + H• + Eaq-; (22)
tali radicali possono reagire con molecole scavenger aggiunte e la velocità di reazione dipende esclusivamente dalle concentrazioni e dalle costanti di secondo ordine delle reazioni di tali molecole con l'•OH. Le cinetiche di competizione vengono anzi usate proprio per determinare le costanti di reazione delle molecole scavenger, mediante la tecnica della radiolisi a impulsi. Con tale approccio, le cinetiche di solito sono semplici, dal momento che i radicali •OH vengono prodotti in soluzione e possono reagire direttamente e in maniera competitiva con le molecole scavenger aggiunte. Comunque, quando questi scavengers con costanti di reazione note vengono aggiunti durante la reazione di Fenton, non sempre impediscono il danno provocato dal radicale •OH alla molecola usata come rivelatore (una sostanza chimica che, aggiunta alla reazione, sarà danneggiata dai radicali •OH, formando un composto rilevabile facilmente e, possibilmente, in modo specifico), come ci si aspetterebbe dalle loro costanti di reazione (v. Gutteridge, 1984).
Questa evidente anomalia può essere spiegata facilmente: dal momento che gli ioni ferro non possono esistere liberi in soluzione, essi devono o essere legati a una molecola biologica, oppure precipitare sotto forma di idrossidi e ossidrossidi ferrici polimerizzati. Quindi, il ferro coinvolto nella reazione di Fenton deve esser legato a un componente della miscela di reazione in cui ha luogo la reazione stessa. Nell'ipotesi più semplice, si può supporre che un carboidrato rivelatore, come il desossiribosio, possa essere un sito di legame per il ferro, cosa che è stata poi dimostrata (v. Aruoma e altri, 1987; v. Gutteridge, Ferrous-salt..., 1987). Il legame del ferro con il rivelatore molecolare rende la reazione di Fenton sito-specifica e solo quelle molecole scavenger capaci di interagire con questo sito e di interferire con la formazione dell'•OH agiranno da inibitori. Secondo un'ipotesi alternativa, il fatto che solo alcuni ben definiti scavengers dell'•OH siano in grado di inibire la reazione di Fenton dipenderebbe dalla relativa affinità di legame con il ferro del rivelatore molecolare e dello scavenger esaminati (v. Gutteridge, 1984, e Ferrous salt..., 1987). Prove della differente affinità di legame col ferro delle diverse molecole scavenger dell'•OH derivano da studi condotti con l'EDTA, un efficace agente chelante del ferro: quando l'EDTA è presente nella reazione di Fenton, la maggior parte degli scavengers dell'•OH ha un tasso di inibizione molto vicino a quello determinato con esperimenti di radiolisi a impulsi. Questo dato sperimentale può essere spiegato considerando che l'EDTA lega tutto il ferro rimuovendolo dal rivelatore molecolare e dagli scavengers dell'•OH; in questo modo alcuni dei radicali •OH liberati dal sistema ferro-EDTA reagiranno con l'EDTA stesso, mentre molti altri, a causa della struttura ‛aperta' del complesso ferro-EDTA, verranno liberati in soluzione e reagiranno con gli scavengers addizionati, come avviene durante l'irradiazione ad alta energia. Questo fenomeno è stato definito ‛effetto radiomimetico' dell'EDTA sugli scavengers del radicale •OH (v. Gutteridge, Ferrous-salt...,1987).
Le implicazioni biologiche di questo meccanismo sono rilevanti, dal momento che sarà estremamente difficile bloccare il danno causato dai radicali •OH nei sistemi biologici con la semplice aggiunta stechiometrica di scavengers dei radicali stessi. Naturalmente, gli agenti chelanti del ferro con costanti di stabilità e cinetiche che permettono loro di rimuovere il ferro dai siti di attacco dell'•OH sono comunque degli importanti antiossidanti (v. cap. 6).
c) Alternative al radicale ossidrile
Che la specie reattiva prodotta al termine della reazione di Fenton sia l'•OH è, oggi, convinzione comune, anche se in un primo tempo tale identificazione è stata oggetto di controversie. Per esempio, nel 1932 Bray e Gorin ipotizzarono che il prodotto finale fosse il radicale ferrile, in cui il ferro ha numero di ossidazione 4 (v. Bray e Gorin, 1932; v. Walling, 1982):
Fe2+ + H2O2 → FeOH3+ (oppure FeO2+) + OH-. (23)
Il radicale ferrile è probabilmente la specie reattiva presente nei siti attivi dei composti I e II della perossidasi di rafano e nel citocromo P450 e quindi deve essere notevolmente reattivo; non sorprende, perciò, che tale controversia si sia estesa alla reazione di Haber-Weiss, nella quale l'•OH viene prodotto dalla reazione di Fenton. In ogni caso, se la specie reattiva prodotta dai sistemi che generano O−2 non è l'•OH, essa deve comunque effettuare spin trap per produrre il segnale •OH corretto, deve idrossilare composti aromatici per formare specifici prodotti idrossilati, reagire con gli scavengers dell'•OH in maniera paragonabile a quella dell'•OH stesso e richiedere sia O−2 che H2O2 per la sua formazione. Che i radicali ferrile agiscano in tal modo non è stato ancora dimostrato; essi sono stati chiaramente identificati solo in una struttura ad anello eminico. Il sistema del citocromo P450, naturalmente, forma substrati idrossilati, ma l'insieme dei prodotti è diverso da quello dovuto all'attacco dell'•OH.
Un'altra specie con caratteristiche simili a quelle del radicale ossidrile è l'[O]x. L'ossidazione dell'acido arachidonico a prostaglandina PGG2 è catalizzata da un enzima noto come ciclossigenasi; lo stesso enzima riduce poi la PGG2 per dare PGG2H2 e allo stesso tempo genera una specie eme-associata nota come [O]x. Quest'ultimo inattiva la ciclossigenasi, a meno che non sia eliminato da uno scavenger, ed è anche in grado di ossidare il metionale a etene, di convertire il fenolo in prodotti polimerici, di ossidare l'adrenalina o il difenilisobenzofurano e di attaccare il gallato di propile. L'[O]x possiede perciò molte proprietà simili a quelle del radicale •OH, sebbene sia noto che non è un radicale.
Il radicale perferrile, FeO+ o Fe3+-O−2, che si forma come intermedio durante la riduzione del complesso-Fe3+ mediata dall'O−2 (v. cap. 3, § a), è un'altra specie spesso chiamata in causa, pur essendo dotata di una bassa capacità ossidativa. Il perferrile contribuisce in misura modesta alla struttura di risonanza che si ottiene dal legame dell'O2 con il Fe(II) negli anelli dell'eme della mioglobina e dell'emoglobina (Fe2+-O2 ⇌ Fe3+-O−2).
Abbiamo già visto che l'elevata reattività dell'•OH con quasi tutte le molecole presenti nelle cellule viventi fa sì che esso non si allontani molto dal proprio sito di formazione. È quindi probabile che la tossicità dell'O−2 e dell'H2O2 per le cellule dipenda dalla disponibilità e dal sito degli ioni metallici catalizzatori della formazione del radicale •OH. Se, per esempio, i sali di ferro sono legati al DNA o ai lipidi di membrana, la formazione di H2O2 e O−2 può, rispettivamente, frammentare il DNA o dare inizio alla perossidazione dei lipidi: il sito di attacco dei radicali •OH dipenderà in maniera determinante dal sito di legame dello ione metallico e tale danno ‛sito-specifico' viene raramente prevenuto dall'azione degli scavengers dell'•OH.
4. Metabolismo dei metalli di transizione
a) Ferro
Sebbene la presenza del ferro nel sangue non sia stata scoperta fino al XVIII secolo, si hanno notizie del trattamento di malattie umane con ferro sin dal 2735 a. C. in Cina e dal 1550 a. C. nel bacino del Mediterraneo. Testi del 1681 descrivono un liquore composto da limatura di ferro, zucchero e vino, usato con successo dal medico inglese Thomas Sydenham per curare una malattia successivamente attribuita a deficienza di ferro. Intorno al 1832 si osservò che i pazienti anemici avevano una concentrazione di ferro nel sangue inferiore al normale, per cui si cominciò a trattare la malattia prescrivendo l'assunzione per via orale di sali di ferro. Questi rimedi impiegati nel trattamento dell'anemia sono ancora ampiamente diffusi in tutto il mondo, con o senza prescrizioni mediche. Inoltre, in alcuni paesi, vari alimenti e bevande sono arricchiti con ferro o con sali di ferro; è stato stimato che in alcuni casi da essi derivi circa il 10-15% del ferro totale assunto con la dieta. È con una certa preoccupazione che si osserva come nei paesi in via di sviluppo vi sia un eccessivo arricchimento in ferro degli alimenti, specialmente considerando che in alcuni soggetti di sesso maschile vi è una correlazione tra riserve di ferro dell'organismo, cancro e malattie cardiache (v. cap. 5, § g); ciò è particolarmente rilevante vista l'alta incidenza (1 su 300) del gene per l'emocromatosi idiopatica.
Un uomo adulto normale possiede circa 4,5 g di ferro, ne assume in media 1 mg al giorno con la dieta e, se il bilancio del ferro è in equilibrio, ne espelle circa la stessa quantità. Dal momento che il ricambio del ferro nel plasma è di circa 35 mg al giorno, devono esistere dei meccanismi estremamente efficaci di conservazione del ferro: disturbi minimi nel suo metabolismo provocano in breve tempo una deficienza o un eccesso di ferro nell'organismo. È stato stimato che più di 500 milioni di persone nel mondo soffrono di carenze di ferro, mentre diversi milioni ne posseggono in eccesso. Non esistono comunque meccanismi fisiologici specifici per l'eliminazione del ferro, che avviene attraverso il ricambio delle cellule epiteliali dell'intestino, l'escrezione di feci e urine, la sudorazione e, nelle donne, il flusso mestruale.
Circa i due terzi del ferro presente nell'organismo sono reperibili nell'emoglobina; quantità minori si trovano nella mioglobina, in diversi enzimi e nella transferrina, la proteina di trasporto del ferro. La ferritina e l'emosiderina sono le molecole organiche deputate al suo deposito; la ferritina è una proteina globulare, il cui nucleo centrale può contenere fino a 4.500 ioni ferro per molecola proteica, nella quale il ferro entra come Fe(II), viene poi ossidato dalla proteina a Fe(III) e depositato al suo interno; analogamente, il ferro può essere rimosso dalla ferritina come Fe(II), dopo essere stato ridotto. Nei lisosomi la ferritina può essere trasformata in un prodotto insolubile, noto come emosiderina; attualmente si ritiene che tale cambiamento sia dovuto a un attacco proteolitico, sebbene ciò non sia stato ancora chiaramente dimostrato. O'Connell e i suoi collaboratori hanno formulato un'ipotesi suggestiva, secondo la quale nella trasformazione della ferritina in emosiderina potrebbero essere coinvolte reazioni di radicali liberi (v. O'Connell e altri, Haemosiderin-like..., 1986).
La mioglobina e l'emoglobina contenute nei tessuti animali rappresentano la principale fonte di ferro eminico per i non vegetariani; tuttavia, la maggior parte del ferro assunto con la dieta è ferro non eminico, presente come Fe(III). L'eme è assorbito come tale e il ferro ne viene rimosso all'interno delle cellule della mucosa intestinale, mentre altre forme di ferro, per poter essere assorbite, devono essere solubilizzate e ridotte allo stato di Fe(II); l'acido cloridrico presente nello stomaco ne permette la solubilizzazione e la vitamina C (o acido ascorbico, un agente riducente) assunta con la dieta ne facilita l'assorbimento. I siti più attivi di assorbimento del ferro sono il duodeno e la parte superiore del digiuno, ma non tutto il ferro assorbito entra in circolo: una parte viene infatti immagazzinata come ferritina all'interno delle cellule della mucosa intestinale e viene alla fine eliminata per desquamazione delle stesse durante il normale processo di ricambio.
Il ferro assorbito dall'intestino che viene trasferito in circolo penetra nel plasma legato alla proteina transferrina, la quale agisce da molecola trasportatrice. La transferrina è una glicoproteina con due siti di legame distinti ai quali, a pH fisiologico, il Fe(III) si lega in modo estremamente stabile. Un legame di questo tipo richiede in ciascun sito la presenza di un anione, di solito l'HCO−3. Nell'uomo, in condizioni normali, solo il 25% della transferrina presente nel sangue è legato al ferro, così che la quantità di sali di ferro liberi nel plasma sanguigno è virtualmente nulla, come risulta da numerosi dati sperimentali (v. Gutteridge e altri, 1981). Una proteina simile alla transferrina, prodotta dai fagociti e nota come lattoferrina, è stata trovata in diversi liquidi corporei e nel latte; anch'essa è in grado di legare due grammoioni Fe(III) per mole di proteina.
Il ferro legato alla transferrina deve entrare nelle diverse cellule dell'organismo per essere utilizzato nella sintesi degli enzimi e delle proteine contenenti ferro. Attualmente si ritiene che la maggior parte delle cellule che necessitano di ferro assuma la transferrina per endocitosi mediata da recettori, trasferendola quindi in un vacuolo citoplasmatico; il contenuto del vacuolo viene poi acidificato, in modo da facilitare il rilascio di ferro dalla transferrina, e il ferro viene probabilmente chelato da vari costituenti cellulari, come il citrato o esteri fosforici come l'ATP e il GTP.
La transferrina priva di ferro (apotransferrina) viene poi espulsa dalla cellula, mentre un piccolo compartimento di ‛ferro non legato a proteine' (v. Jacobs, 1977) può essere usato per la sintesi delle proteine contenenti ferro e del DNA. Sebbene la quantità e la natura chimica di tale compartimento di ‛ferro non legato a proteine' contenuto nelle cellule e negli organelli siano ancora poco definite, è stato osservato che i mitocondri, per esempio, possono contenere piccole quantità di sali di ferro chelati nella matrice e che sono in grado di assumerli rapidamente per incorporare il ferro in proteine eminiche e non.
Esaminiamo ora i diversi potenziali catalizzatori contenenti ferro ai quali potrebbe esser dovuta la formazione del radicale •OH.
Lattoferrina e transferrina. - In letteratura esistono dati contrastanti riguardo alla capacità della transferrina e della lattoferrina di agire da promotori della formazione del radicale •OH (v. Gutteridge e Halliwell, 1989). Una possibile spiegazione di tali discrepanze è data dal fatto che, se queste proteine vengono studiate in condizioni di saturazione del ferro, questo può talvolta essere legato a siti non specifici sulla molecola proteica, da cui può distaccarsi durante l'esperimento per poi divenire l'effettivo catalizzatore della formazione del radicale •OH. Sono stati condotti molti esperimenti nel tentativo di verificare questa ipotesi: si è concluso che la transferrina e la lattoferrina saturate con ferro non accelerano la produzione di •OH, a meno che non siano presenti nella miscela di reazione degli agenti chelanti, in special modo quando le proteine sono legate al ferro in maniera non corretta o quando i sistemi utilizzati per tali esperimenti rendono necessari bassi valori di pH. Come è stato già detto, la transferrina presente nel plasma umano è solo parzialmente legata al ferro, cioè solo alcune molecole trasportano uno ione Fe(III) in ciascun sito di legame. Analogamente, la lattoferrina liberata dai fagociti contiene poco ferro e sembra anzi che il suo rilascio costituisca un meccanismo antibatterico, in quanto lega il ferro necessario per la sopravvivenza dei Batteri (v. Bullen e altri, 1978). Secondo queste osservazioni sembra quindi decisamente poco probabile che la transferrina e la lattoferrina siano tra i principali catalizzatori della produzione del radicale •OH in vivo.
In circa il 40% dei pazienti affetti da artrite, il liquido sinoviale dell'articolazione del ginocchio mostra la presenza di ferro, rilevabile mediante il saggio con bleomicina (v. Gutteridge e altri, 1981; v. Gutteridge, Bleomycin-detectable..., 1987): a pH acido il ferro viene ceduto alla bleomicina da un componente proteico del liquido sinoviale. In questi pazienti le concentrazioni di transferrina, lattoferrina e ceruloplasmina sono inferiori a quelle dei pazienti in cui non vi è rilascio di ferro alla bleomicina, perché l'attività di queste proteine, che sono degli importanti antiossidanti extracellulari, è estremamente bassa nei liquidi che rilasciano ferro. La tendenza di alcuni liquidi a rilasciare ferro a bassi valori di pH, come avviene nel microambiente intorno ai macrofagi, unitamente alla diminuita capacità di difesa antiossidante contro i danni dovuti ai radicali dell'ossigeno indotti dal ferro, può spiegare le correlazioni tra la gravità di alcune malattie, il contenuto di perossidi nei lipidi e la presenza di ferro chelabile dalla bleomicina.
Ferritina ed emosiderina. - La ferritina è spesso considerata una innocua forma di immagazzinamento del ferro, nonostante stimoli la formazione di radicali •OH da O−2 e da H2O2 (v. Bannister e altri, 1984). Il superossido sembra mobilizzare una piccola quantità di ferro dalla ferritina, inducendo così la produzione catalizzata dal ferro del radicale •OH. Questo ferro è probabilmente in una forma labile, in quanto legato alla ferritina in un sito diverso dal suo nucleo proteico. O'Connell e i suoi collaboratori hanno dimostrato che anche il ferro dell'emosiderina può partecipare alla formazione di •OH (v. O'Connell e altri, Haemosiderin-like..., 1986), sebbene l'emosiderina sia meno attiva della ferritina, vista la maggiore difficoltà con cui il ferro ne viene rimosso (v. Gutteridge e Hou, 1986).
Emoglobina. - Sono state formulate diverse ipotesi sul coinvolgimento dell'emoglobina o della metemoglobina nella stimolazione della reazione di Fenton, anche se tali ipotesi sono state basate su assunzioni errate circa i saggi effettuati e la specificità degli scavengers (v. Halliwell e Gutteridge, 19892); è comunque certo che miscele di ossiemoglobina, metemoglobina e H2O2 possono ossidare un certo numero di specie diverse. L'azione ossidante del complesso metemoglobina-H2O2 potrebbe essere dovuta a specie costituite da ferro associato a eme, simili a quelle presenti nei composti I e II della perossidasi di rafano. Pertanto, non esistono ancora prove certe del coinvolgimento dell'emoglobina nella formazione del radicale •OH in vivo. Comunque, il perossido di idrogeno può indurre il rilascio di ioni ferro dall'emoglobina attaccando l'anello dell'eme, e studi recenti hanno evidenziato che il ferro rilasciato dalla metemoglobina per azione del perossido di idrogeno o di perossidi organici può catalizzare la formazione di radicali dell'ossigeno; una volta libero, il ferro può essere complessato dall'apotransferrina, dalla bleomicina o dalla desferrioxammina (v. Gutteridge, 1986).
Chelati semplici del ferro. - Com'è stato già detto, all'interno delle cellule è presente un modesto insieme transiente di chelati del ferro a basso peso molecolare (v. Jacobs, 1977). L'esatta natura chimica di questo insieme di molecole non è chiara, ma probabilmente esso è formato da ioni ferro legati a esteri fosforici come l'ATP, l'ADP o il GTP, ad acidi organici e forse all'estremità polare dei lipidi di membrana o al DNA. Tutti questi composti del ferro sono in grado di scindere H2O2 per formare •OH. Sembra infatti che l'effetto letale dell'H2O2 sui fibroblasti sia dovuto agli ioni metallici intracellulari, probabilmente legati al DNA o vicini a esso (v. Schraufstatter e altri, 1986).
Gutteridge e i suoi collaboratori hanno messo a punto un test per misurare la disponibilità del ferro contenuto in complessi catalitici nei liquidi corporei umani (v. Gutteridge e altri, 1981; v. Gutteridge e Hou, 1986). Tale metodo si basa sull'osservazione che la bleomicina, un antibiotico glicopeptidico antitumorale, degrada il DNA solo in presenza di sali del ferro, di ossigeno e di un adatto agente riducente; il legame della bleomicina con il DNA rende questa reazione sito-specifica e minima è l'interferenza di antiossidanti biologici. Il saggio può quindi essere condotto direttamente in tutti i liquidi biologici per rivelare il ferro chelabile dalla molecola di bleomicina, ma la sua affidabilità dipende dalla totale rimozione del ferro dall'acqua, dai reagenti e dalla bleomicina stessa. Il ferro misurato con tale metodo fornisce una buona stima di quello disponibile nel campione biologico e può essere saggiato fino a concentrazioni di 0,5 µmol/l. L'applicazione del test della bleomicina mostra che il contenuto in ferro disponibile del siero e del plasma umano è praticamente nullo, eccetto in alcuni casi di sideremia elevata; al contrario, quantità significative di ferro sono state rilevate nel sudore degli atleti, nel liquido cerebrospinale normale e nel liquido sinoviale del ginocchio in molti pazienti affetti da artrite reumatoide, in pazienti leucemici sotto chemioterapia, durante interventi chirurgici in circolazione extracorporea e in pazienti con lesioni polmonari acute (v. Gutteridge, 1986). L'esatta struttura molecolare del ferro chelabile dalla bleomicina non è chiara: potrebbe trattarsi di ferro legato a semplici chelanti, quali esteri fosforici o acidi organici, oppure debolmente legato a proteine, come l'albumina, o al DNA, o rilasciato, in condizioni di basso pH, da proteine che normalmente possiedono un'elevata affinità di legame per il ferro.
b) Provenienza del ferro reattivo in vivo
L'ipotesi che l'•OH fosse coinvolto nella tossicità dell'H2O2 e dell'O−2 aprì un acceso dibattito sulla disponibilità di ioni metallici in vivo (v. Winterbourn, Hydroxyl..., 1981). Dopo circa quindici anni, il quadro è divenuto più chiaro: il test della bleomicina, appena descritto, fu introdotto nel 1981 in un primo tentativo di rilevare e misurare il ferro disponibile nei liquidi biologici. L'applicazione del test al siero di pazienti con elevata sideremia ha confermato i dati ottenuti da Hershko e altri (v., 1978) sulla presenza di ferro complessato sotto forma di composti a basso peso molecolare. Altri liquidi corporei, come il liquido cerebrospinale, il sudore e il liquido sinoviale, contengono ferro complessabile che può essere rilevato con il test della bleomicina. La complessità della reazione con la bleomicina, unita alla sua sensibilità e flessibilità entro una vasta gamma di valori di pH, può essere di valido aiuto per capire in che modo il ferro si renda disponibile per le reazioni radicaliche in vivo (v. Gutteridge e Hou, 1986). In condizioni normali, le proprietà chelanti della molecola di bleomicina non sono da sole sufficienti a rimuovere il ferro, strettamente associato alle proteine eminiche o non eminiche come la ferritina, la transferrina, l'emoglobina e la mioglobina. Come abbiamo già detto, sia il perossido d'idrogeno che gli idroperossidi organici possono indurre il rilascio di ferro dall'eme, rendendolo complessabile da chelanti quali la desferrioxammina, il dipiridile e la bleomicina (v. Gutteridge, 1986) e quindi disponibile per le reazioni radicaliche. Sembra certo, quindi, che anche lo stress ossidativo fornisca il ferro necessario per completare la dannosa sequenza di eventi che termina con la reazione di Fenton in vivo, rendendo ancor più importante l'azione antiossidante della superossidodismutasi, della catalasi e della glutationeperossidasi contro il danno cellulare. Comunque, il sequestro degli ioni metallici, o altrimenti il tentativo di impedire il loro coinvolgimento in reazioni radicaliche, costituisce un'importante difesa antiossidante extracellulare (v. cap. 6).
c) Rame
In un uomo adulto normale sono presenti mediamente 80 mg di rame, assorbito con la dieta a livello dello stomaco o della parte superiore dell'intestino tenue, probabilmente complessato con amminoacidi (come l'istidina) o con piccoli peptidi. Il trasporto verso il fegato è facilitato dal legame con gli amminoacidi e con l'albumina, la quale possiede un sito di legame a elevata affinità per il rame (eccetto che nel cane); nel fegato il rame si lega alla ceruloplasmina, la proteina del plasma umano con maggiore contenuto di questo elemento. Questa proteina ha un peso molecolare di circa 132.000, con 6 o 7 ioni rame per molecola, 6 dei quali sono legati fortemente e possono venire rilasciati solo a bassi valori di pH in presenza di agenti riducenti. La ceruloplasmina può, tuttavia, fornire rame alle cellule, che lo incorporano in altre proteine contenenti rame, come la superossidodismutasi e la citocromossidasi. Il ruolo svolto dalla ceruloplasmina come ‛donatore di rame' viene spesso indicato come funzione di ‛trasporto del rame', anche se, in realtà, nella ceruloplasmina il legame e il trasporto del rame non sono così specifici come avviene invece per il ferro nella transferrina. Secondo quanto riportato in letteratura, circa il 5% del rame plasmatico è legato all'albumina o ad amminoacidi come l'istidina, mentre il restante 95% è legato alla ceruloplasmina. Purtroppo la ceruloplasmina viene degradata durante la conservazione del siero a 4 °C, ed è stato ipotizzato che parte del ‛rame serico non legato alla ceruloplasmina' nell'uomo possa essere un artefatto conseguente alla degradazione della ceruloplasmina durante il trattamento dei campioni (v. Gutteridge e altri, 1985).
In vitro, la ceruloplasmina catalizza l'ossidazione di una vasta gamma di substrati costituiti da poliammine o polifenoli ma, con la possibile eccezione di alcune bioammine, queste ossidazioni non hanno alcun significato biologico noto. È stato ipotizzato che il ruolo biologico della ceruloplasmina sia quello di una ‛ferrossidasi' (v. Osaki e altri, 1966), in quanto essa catalizza l'ossidazione del Fe(II) a Fe(III), facilitando così il legame del ferro con la transferrina. Diversamente dalle ossidazioni non enzimatiche del Fe(II) in presenza di O2, l'attività ferrossidasica della ceruloplasmina (a volte chiamata ferrossidasi I) non produce radicali O−2 e •OH (v. Gutteridge e Stocks, 1981). Considerando la sua capacità di inibire le reazioni radicaliche Fe(II)-dipendenti, la ceruloplasmina è stata considerata un importante agente antiossidante extracellulare (v. Stocks e altri, 1974; v. cap. 6). Al contrario, gli ioni rame legati all'albumina e agli amminoacidi possono interagire con l'O−2 e l'H2O2 per formare radicali •OH; sembra però che tale specie reattiva attacchi il ligando al quale è unito il rame e non venga lasciata ‛libera' in soluzione. Il legame del rame con il DNA, con virus o con enzimi (v. Samuni e altri, 1981) può causare danno sito-specifico dovuto alla formazione di radicali dell'ossigeno, il che rappresenta un'ulteriore dimostrazione che la tossicità dell'O−2 e dell'H2O2 in vivo dipende dalla localizzazione degli ioni metallici che catalizzano la formazione del radicale •OH. In questo contesto la capacità dell'albumina di legare ioni rame può rappresentare un importante meccanismo biologico di protezione: la presenza di concentrazioni molto elevate di albumina nel sangue, infatti, può evitare che il rame si leghi a siti sensibili, come, per esempio, la membrana cellulare. Pertanto, se nel plasma vengono prodotti O−2 e H2O2, per esempio da fagociti attivati, il radicale •OH può formarsi nel sito di legame per il Cu2+ dell'albumina, ma il danno prodotto avrà effetti biologici irrilevanti, vista l'abbondanza di albumina nel sangue.
L'agente chelante 1,10-fenantrolina degrada il DNA in presenza di ioni rame, ossigeno e agenti riducenti appropriati, per esempio il mercaptoetanolo; il risultato della degradazione consiste nel rilascio dal DNA di sostanze in grado di reagire con l'acido tiobarbiturico. Questa reazione è alla base di una tecnica per rilevare e misurare la presenza del rame chelabile disponibile presente nei liquidi biologici (v. Gutteridge, 1984). Il rame, legato alla fenantrolina e aggiunto ai liquidi biologici, è ridotto dal mercaptoetanolo e il danno al DNA che ne deriva risulta essere proporzionale alla quantità di rame presente. Il ‛saggio della fenantrolina' può rivelare il rame legato ai siti ad alta affinità dell'albumina e all'istidina, ma non quello legato ai sei siti di legame ad alta affinità della ceruloplasmina. Al contrario, lo ione rame legato debolmente alla ceruloplasmina può agire come proossidante e causare ossidazione delle lipoproteine in vivo (v. Ehrenwald e altri, 1994). Quando il test è stato applicato ai liquidi extracellulari umani, si è trovato rame chelabile nel liquido cerebrospinale, nel sudore e nel siero di pazienti con gravi danni epatici (v. Halliwell e Gutteridge, 19892).
5. La perossidazione lipidica, una reazione radicalica a catena
In tossicologia e in patologia gli effetti dannosi delle reazioni indotte dai radicali liberi sono spesso misurati mediante la stima del grado di perossidazione lipidica. Una vasta gamma di tecniche è oggi a disposizione per quantificare questo processo, ma nessuna di esse è applicabile in tutti i casi.
Il primo evento di una sequenza di perossidazioni a catena in una membrana o in un acido grasso polinsaturo consiste nell'attacco di una qualsiasi specie con reattività sufficiente a staccare un atomo di idrogeno (H) da un gruppo metilenico (−CH2−). Dal momento che l'atomo di idrogeno possiede un solo elettrone, tale evento produrrà un elettrone spaiato sul carbonio, −C•H−. In un acido grasso la presenza di un doppio legame rende più debole il legame C−H del carbonio a esso adiacente e di conseguenza più facile la rimozione dell'atomo di H; per questa ragione le catene laterali degli acidi grassi polinsaturi presenti nei lipidi delle membrane sono particolarmente sensibili alla perossidazione.
Il centro radicalico del carbonio subisce un riarrangiamento molecolare per formare un diene coniugato che si combina con l'ossigeno a formare un radicale perossido, capace di staccare un atomo di idrogeno da un altro acido grasso, dando così inizio a una reazione a catena che continuerà fino all'esaurimento del substrato, a meno che non intervenga un antiossidante (per es. la vitamina E) in grado di bloccare la catena di reazioni. Pertanto la perossidazione lipidica, come tutte le reazioni a catena, si compone di tre stadi: iniziazione, propagazione e termine. Tra i ricercatori più autorevoli in questo campo esiste un discreto accordo sui risultati sperimentali ottenuti, mentre permane una certa confusione riguardo alla terminologia descrittiva usata, soprattutto in relazione alla parola ‛iniziazione'. Secondo l'autore di questo articolo il termine ‛iniziazione' dovrebbe essere utilizzato solo per descrivere l'inizio della prima catena di reazioni (cioè la rimozione dell'atomo di idrogeno), mentre la decomposizione dei perossidi, che dà origine a una nuova reazione a catena, dovrebbe essere descritta con il termine ‛stimolazione' o ‛accelerazione' o ‛perossidazione lipidica'. In ultima analisi, potrà causare la perossidazione qualsiasi specie che possieda sufficiente energia da staccare un atomo di idrogeno da un lipide, come il radicale •OH, i radicali alcossido (RO•), i radicali perossido (ROO•) e in qualche modo l'HO2•, ma non l'H2O2 o l'O−2 (v. Halliwell e Gutteridge, 19892). Al termine della reazione a catena si accumula una miscela di perossidi lipidici e perossidi ciclici; in effetti, tutti i campioni di acidi grassi poliinsaturi in commercio contengono quantità variabili di perossidi contaminanti.
a) Iniziazione mediata da complessi del ferro
A temperatura fisiologica, i perossidi lipidici sono molecole piuttosto stabili, ma la loro decomposizione è catalizzata da metalli di transizione e da complessi metallici (v. cap. 4, § a). Tutti i complessi del ferro presenti in vivo che partecipano alla reazione di Fenton sono in grado di promuovere anche la decomposizione dei perossidi, come pure, del resto, la metemoglobina e i citocromi. Sia la ferritina che l'emosiderina sono catalizzatori molto efficienti, mentre in alcune circostanze lo è meno la catalasi, e ciò rende poco indicato il suo impiego come ‛sonda' per stabilire il ruolo dell'H2O2 nei sistemi di perossidazione lipidica (v. Gutteridge e altri, 1983). Al contrario, il ferro correttamente legato ai due siti specifici presenti nella transferrina o nella lattoferrina non sembra stimolare la decomposizione dei perossidi.
I complessi metallici ridotti (per es., Fe[II] o Cu[I]) reagiscono con i perossidi lipidici (LOOH) per dare radicali alcalossido:
LOOH+Mn+ → LO• + M(n+1)+ + OH-, (24)
mentre i complessi metallici ossidati (per es., Fe[III] o Cu[II]) reagiscono più lentamente per formare radicali perossido e alcalossido, i quali stimolano la reazione a catena di perossidazione lipidica staccando ulteriori atomi di idrogeno.
La maggior parte delle presunte iniziazioni di perossidazione lipidica che si osservano quando si aggiungono chelati di metalli a un sistema lipidico in vitro è in realtà imputabile alla decomposizione di perossidi lipidici preformati da parte di complessi metallici e alla rimozione di un atomo di idrogeno da parte di radicali LO• e LOO• risultanti. Alcuni ioni metallici con numero di ossidazione fisso possono influenzare il grado di perossidazione lipidica: ad esempio, in appropriate condizioni gli ioni Ca2+, Al3+ e Pb2+ sono in grado di accelerare la perossidazione stimolata da sali del ferro. Questo effetto potrebbe essere dovuto a cambiamenti nella struttura e nell'organizzazione della membrana (v. Quinlan e altri, 1992).
Alcuni autori hanno ipotizzato che durante le complesse reazioni di degradazione della perossidazione lipidica si formi O2 singoletto e che esso contribuisca alla reazione a catena causando ulteriori iniziazioni.
b) Iniziazione mediata da ioni ferrosi
Come abbiamo già detto, gli ioni ferro sono essi stessi radicali liberi (v. Halliwell e Gutteridge, 19892) e lo ione ferroso può essere coinvolto nelle reazioni di trasferimento di elettroni all'ossigeno molecolare:
Fe2+ + O2 ⇌ Fe2+−O2 ⇌ Fe3+−O−2 ⇌ Fe3++O−2. (25)
Una qualsiasi formazione di radicale superossido in presenza di ioni ferro può provocare la formazione di radicali ossidrile attraverso la reazione di Fenton. Studi con radiazioni ad alta energia hanno dimostrato che il radicale •OH prodotto in soluzione può iniziare la perossidazione dei lipidi (LH) per rimozione di idrogeno da acidi grassi purificati (v. Bielski e altri, 1983):
LH + •OH → L• + H2O. (26)
Non c'è dunque alcuna ragione per cui la reazione di Fenton dipendente da superossidi non possa dare inizio alla perossidazione lipidica in questo modo. Nonostante queste considerazioni, attenti studi basati sull'osservazione di sistemi in cui si svolgono reazioni di Fenton dipendenti da superossidi (che generano l'H2O2 e riducono il Fe3+ a Fe2+) hanno inequivocabilmente mancato di mostrare un coinvolgimento significativo dei radicali ossidrile nei sistemi di perossidazione dei microsomi o dei liposomi, come dimostrato dall'azione degli scavengers (v. Gutteridge, 1982). Tuttavia, in questi sistemi si formano radicali •OH che possono essere rilevati tramite spin trapping o misurazione della degradazione del desossiribosio, un'anomalia, questa, che deve ancora trovare una spiegazione.
c) Stimolazione mediata da complessi del ferro
Il fattore più importante da considerare negli studi sulle reazioni di iniziazione è la purezza della preparazione lipidica. Come abbiamo già sottolineato, tutti i campioni commerciali e biologici di acidi grassi insaturi contengono da poche tracce a grosse quantità di materiale già perossidato. L'aggiunta di un complesso del ferro a tali preparazioni stimolerà, pertanto, la perossidazione mediante reazioni di decomposizione dei perossidi, che generano radicali alcossido (LO•) e perossido (LO2•):
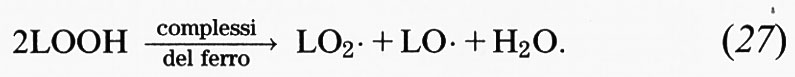
Quando sono coinvolti ioni ferrosi, la costante di velocità di questa reazione, che è di 1,5 × 103 mol-1s-1, è superiore a quella della reazione di Fenton (76 mol-1s-1) in cui gli ioni ferrosi reagiscono con l'H2O2. Ne consegue che i complessi del ferro stimolano preferenzialmente la perossidazione mediante reazioni di decomposizione dei lipidi, vista la generale abbondanza di idroperossidi.
Sali di ferro o di rame attaccati alle membrane biologiche possono causare, in presenza di H2O2, la formazione sito-specifica di radicali •OH, con conseguente perossidazione. La maggior parte delle membrane biologiche è formata per il 50% da proteine; nelle membrane tilacoidali del cloroplasto e nella membrana mitocondriale interna, tale valore può superare l'80%; pertanto, le reazioni di propagazione della perossidazione lipidica in una membrana biologica in vivo raggiungono facilmente le proteine, danneggiandole. Talvolta questi danni possono essere biologicamente più rilevanti di quelli subiti dai lipidi di membrana, così che le cellule hanno sviluppato meccanismi di riconoscimento e distruzione delle proteine modificate da processi ossidativi.
d) Tipi di perossidazione lipidica.
Perossidazione lipidica non enzimatica. - Come abbiamo visto, un qualsiasi radicale libero (R•) che possieda energia sufficiente a rimuovere un atomo di idrogeno da un gruppo metilenico di un acido grasso insaturo (LH) può iniziare una reazione a catena nella maggior parte dei lipidi. La reazione a catena di perossidazione lipidica da parte dei radicali liberi si propagherà fintanto che due radicali liberi non si annulleranno a vicenda, terminando la catena:
LO2• + LO2• → LOOL + O2 (28)
LO2• + L• → LOOL. (29)
Durante una perossidazione lipidica non enzimatica, i radicali perossido hanno un'emivita sufficiente per poter raggiungere altre molecole di acido grasso, prima di essere intercettati e inattivati da una serie di diversi antiossidanti chimici. I perossidi lipidici (LOOH) prodotti durante le reazioni a catena sono costituiti da una complessa miscela di isomeri.
Perossidazione enzimatica. - Si può parlare di perossidazione enzimatica solo quando si formano prodotti di perossidazione lipidica nel sito attivo di un enzima. Gli idroperossidi e gli endoperossidi prodotti in tal modo sono stereospecifici e svolgono importanti funzioni biologiche: gli enzimi ciclossigenasi e lipossigenasi rispondono pienamente a questa definizione. Probabilmente i radicali liberi sono importanti intermedi di reazione, ma sono localizzati nei siti attivi delle proteine. Durante la formazione di endoperossidi catalizzata dalla ciclossigenasi si forma un potente ossidante (Ox), che può essere inattivato da alcuni antiossidanti.
Ossidoriduzioni enzimatiche cicliche del ferro durante la perossidazione non enzimatica. - Da molto tempo è nota l'importanza del ferro nelle reazioni di perossidazione lipidica, per tutte le ragioni prima esposte. Le ossidoriduzioni cicliche del ferro stimolano notevolmente la perossidazione lipidica ed enzimi come la riduttasi del citocromo P450, in presenza di NADPH, possono ridurre i complessi del ferro così come fanno i radicali O−2 prodotti da miscele di xantinossidasi con i suoi substrati. In tal modo l'enzima svolge una funzione non diversa da quella dell'acido ascorbico e la sua stessa natura viene alterata. La reazione a catena dei radicali liberi è uguale a quella descritta per la perossidazione non enzimatica e non vi è formazione di prodotti stereospecifici.
e) Conseguenze della perossidazione lipidica
L'estesa perossidazione dei lipidi delle membrane biologiche causa perdita di fluidità, diminuzione del potenziale di membrana, aumento della permeabilità agli ioni H+ e ad altri ioni e rottura finale, con conseguente rilascio del contenuto della cellula e degli organelli. Alcuni prodotti terminali della frammentazione dei perossidi sono anche citotossici (v. Esterbauer e altri, 1991). Non tutti i processi di ossidazione lipidica sono dannosi, anzi alcuni prodotti di perossidazione possono svolgere ruoli utili nella cascata dell'acido arachidonico e nella ‛risposta alle lesioni' dei tessuti vegetali (v. Halliwell e Gutteridge, 19892), perché la produzione di perossidi lipidici e la loro frammentazione in composti carbonilici può aiutare la pianta a difendersi da batteri o da spore fungine penetrati nel tessuto danneggiato (v. Galliard, 1978).
f) Importanza della perossidazione lipidica nel danno indotto da radicali
Una vasta gamma di tecniche è stata utilizzata per dimostrare che la perossidazione lipidica aumenta in diversi stati patologici (v. Gutteridge, 1993) e in tessuti avvelenati da varie tossine; alla base di molti di questi studi c'è l'implicita assunzione che le malattie e le tossine provochino un aumento della perossidazione lipidica, la quale sarebbe quindi responsabile della tossicità. Tuttavia, diversi anni fa, è stato dimostrato che nei tessuti danneggiati le reazioni di perossidazione lipidica avvengono più velocemente che nei tessuti sani: per esempio, i perossidi lipidici si accumulano in omogenati di cervello molto più velocemente di quanto non avvenga in cervelli isolati integri (v. Stocks e altri, 1974). L'aumento della facilità di perossidazione nei tessuti danneggiati è imputabile a una serie di fattori diversi, tra cui l'inattivazione di alcuni antiossidanti, la fuoriuscita degli antiossidanti cellulari, il rilascio di ioni metallici (specialmente ferro e rame) dai rispettivi siti di deposito e dalle metalloproteine, la cui idrolisi è mediata dagli enzimi rilasciati dai lisosomi danneggiati. Pertanto la sequenza di avvenimenti ‛malattia o tossina → danno o morte cellulare → aumento della perossidazione lipidica', può spiegare molti dati sperimentali sull'aumento della perossidazione lipidica in patologia e in tossicologia (v. Halliwell e Gutteridge, Oxygen..., 1984). Per poter stabilire la causalità della perossidazione bisognerebbe però dimostrare che essa precede o accompagna il danno cellulare e che la sua prevenzione mediante antiossidanti può preservare le cellule dall'insorgere di tali danni. La misura della perossidazione lipidica può, in tal senso, essere un eccellente indicatore del danno tessutale.
g) Perossidazione delle lipoproteine e suo ruolo nell'aterosclerosi
Nei macrofagi in coltura incubati con elevate concentrazioni di lipoproteine a bassa densità (LDL) native non si accumulano esteri del colesterolo, nonostante l'assorbimento delle LDL da parte di tali cellule. I lavori pionieristici di Brown, Goldstein e colleghi (v. Goldstein e altri, 1979) hanno dimostrato che le LDL, quando sono chimicamente modificate, vengono riconosciute da un recettore specifico, chiamato scavenger o recettore per le acetil-LDL, che si trova sulla superficie dei macrofagi e da questi ultimi vengono attivamente inglobate. L'attività di questo recettore non è regolata negativamente dalla concentrazione locale di colesterolo ed è per questo motivo che in macrofagi trattati con LDL ossidate si accumulano grandi quantità di colesterolo. Questa osservazione ha fornito il primo legame a livello molecolare tra LDL, colesterolo, formazione delle cellule schiumose e sviluppo dell'aterosclerosi, e ha indotto a formulare l'ipotesi che simili meccanismi possano agire anche in vivo (v. Steinberg e altri, 1989).
Quando le LDL vengono isolate da plasma umano normale e sottoposte a stress ossidativi mediante l'aggiunta di quantità micromolari di sali di rame, esiste una fase di latenza prima che inizi una perossidazione lipidica misurabile: questo periodo di latenza è stato assunto come indice del ‛potenziale antiossidante' delle molecole di LDL. Quando gli antiossidanti delle LDL - come la vitamina E, il β-carotene, il licopene e il fitofluene - sono completamente esauriti, ha termine la fase di latenza (che ha la durata di circa un'ora), la perossidazione aumenta per propagazione (un'altra ora) e poi continua per diverse ore, mentre la reazione di decomposizione produce aldeidi (v. Esterbauer e altri, 1991), responsabili della citotossicità e della cattura delle LDL da parte dei macrofagi. Tutte le molecole di vitamina E presenti nelle LDL vengono consumate nei primi 10 minuti della fase di latenza, seguite dagli altri antiossidanti liposolubili nei successivi 40-50 minuti. La vitamina E è di gran lunga l'antiossidante più abbondante contenuto nelle LDL, essendo presente in ragione di circa 6 molecole per particella di LDL; gli altri antiossidanti liposolubili ammontano, invece, a meno di una molecola per particella di LDL.
Abbiamo già ricordato la stretta associazione tra elevati livelli di colesterolo plasmatico, colesterolo trasportato dalle LDL e malattie cardiache: questi fattori, unitamente ai classici fattori di rischio come l'elevata pressione sanguigna e il fumo, aumentano il pericolo di malattie cardiache del 50% al massimo; altri fattori di rischio sono probabilmente legati al tipo di alimentazione, poiché in gruppi che adottano una dieta mediterranea o vegetariana la morbilità e la mortalità legate alle malattie cardiache sono risultate minori (v. Gey e altri, 1991). Per provare l'ipotesi della ‛dieta antiossidante', Gey e i suoi collaboratori hanno condotto studi epidemiologici su 16 popolazioni europee, analizzando le relazioni tra livelli di vitamine antiossidanti presenti nel plasma e tasso di mortalità dovuta a malattie cardiache (v. Gey e altri, 1987; v. Gey e Puska, 1989). In tutte le popolazioni studiate, i valori del colesterolo e della pressione sanguigna erano moderatamente associati alla mortalità dovuta a malattie cardiache, ma tale correlazione era comunque inferiore a quella relativa ai livelli plasmatici di vitamina E. Così il tasso di mortalità è stato prevedibile al 62% valutando la vitamina E a contenuto lipidico standardizzato, al 79% valutando vitamina E e colesterolo, all'83% dopo l'inclusione della valutazione di retinolo a contenuto lipidico standardizzato e all'87% valutando tutti i parametri sopra indicati più la pressione sanguigna.
I risultati ottenuti sono in accordo con il ben noto gradiente nord-sud di distribuzione delle malattie cardiache in Europa e sembrano avvalorare il ruolo degli antiossidanti nella protezione delle LDL dall'ossidazione. È noto che nell'Europa settentrionale vengono assunte con la dieta minori quantità di verdura e frutta fresche rispetto a quanto avviene nei paesi mediterranei; questo può spiegare le differenze significative tra i livelli plasmatici di antiossidanti misurati nelle popolazioni del nord e del sud.
Benché queste scoperte siano interessanti, è importante sottolineare che si tratta di studi epidemiologici, che mostrano le correlazioni con le malattie cardiache ma non le cause; è quindi possibile che le vitamine antiossidanti siano semplicemente dei marcatori di altri fattori chiave presenti nella dieta. Infatti, l'autore di questo articolo ha avanzato l'ipotesi che bassi livelli di vitamina E siano dovuti alla perdita causata da sostanze proossidanti piuttosto che a una sua minore assunzione. I proossidanti, come il ferro, sono capaci di distruggere la vitamina E ed è stato ipotizzato che sia lo stato del ferro sia quello del rame siano fattori di rischio per le malattie cardiache (v. Sullivan, 1989; v. Reunanen e altri, 1992).
6. Introduzione agli antiossidanti
Alcuni autori ritengono probabile che il prezzo che paghiamo durante la nostra vita per la sopravvivenza quotidiana sia l'accumulo di danni molecolari che in seguito saranno la causa di malattie mortali (v. Ames e Shigenaga, 1993).
Il termine ‛antiossidante' è molto usato nella letteratura scientifica, ma di rado è messo chiaramente in relazione con sostanze chimiche in grado di interrompere le reazioni a catena dei radicali, come la vitamina E (α-tocoferolo) e la vitamina C (acido ascorbico). Secondo una definizione più ampia si considera antiossidante ‟una qualsiasi sostanza che, presente in concentrazione più bassa di quella del substrato ossidabile, rallenta in modo rilevante o inibisce l'ossidazione del substrato stesso" (v. Halliwell e Gutteridge, 19892).
È possibile descrivere il comportamento degli antiossidanti considerando la perossidazione lipidica che avviene nelle membrane cellulari o in substrati ricchi di lipidi. Essi possono agire a diversi livelli di una sequenza di ossidazioni, secondo i seguenti meccanismi: 1) sottraendo ossigeno o diminuendo localmente la sua concentrazione; 2) sottraendo ioni metallici catalitici; 3) sottraendo importanti specie reattive dell'ossigeno (ROS), come i superossidi e il perossido d'idrogeno; 4) inattivando radicali liberi in grado di promuovere la fase d'iniziazione, come i radicali idrossido, alcossido e perossido; 5) interrompendo una catena di reazioni già iniziata; 6) schermando o inattivando l'ossigeno singoletto.
Gli antiossidanti che inibiscono l'ossidazione lipidica (perossidazione) secondo i meccanismi elencati nei punti 1, 2, 4 e 6 sono detti antiossidanti ‛preventivi'; quelli che operano secondo il meccanismo 3 sono anch'essi preventivi, ma, trattandosi di enzimi (per es. catalasi, superossidodismutasi e glutationeperossidasi), non vengono consumati nel corso della reazione; gli antiossidanti che interrompono la catena (5), quelli che schermano l'ossigeno singoletto (6) e quelli che sottraggono ioni metallici (2) vengono consumati mentre svolgono la loro funzione protettiva. Molti antiossidanti possiedono meccanismi d'azione multipli, come per esempio il gallato di propile, un composto fenolico di sintesi parzialmente solubile in acqua ma discretamente solubile nei lipidi, che può agire sia interrompendo la catena di reazioni, sia inattivando il radicale •OH, sia legando il ferro.
a) Antiossidanti cellulari
Le cellule possiedono formidabili sistemi di difesa contro il danno ossidativo, alcuni dei quali non sono immediatamente riconoscibili come antiossidanti. La protezione di tipo antiossidante può avvenire a vari livelli: 1) impedendo la formazione di radicali; 2) intercettando i radicali quando si sono formati; 3) riparando i danni ossidativi causati dai radicali; 4) facilitando l'eliminazione delle molecole danneggiate; 5) evitando di riparare le molecole eccessivamente danneggiate per limitare il rischio di mutazioni.
L'ossigeno viene metabolizzato all'interno della cellula ed è qui che si trovano antiossidanti in grado di interagire velocemente e specificamente (per via enzimatica) con intermedi ridotti dell'ossigeno (v. Gutteridge e Halliwell, 1988; v. tab. II). Enzimi come la superossidodismutasi (SOD) fanno avvenire la dismutazione del superossido in perossido d'idrogeno e ossigeno in modo molto più rapido di quanto avverrebbe se tale reazione non venisse catalizzata (v. Fridovich, 1975, 1978 e 1983):
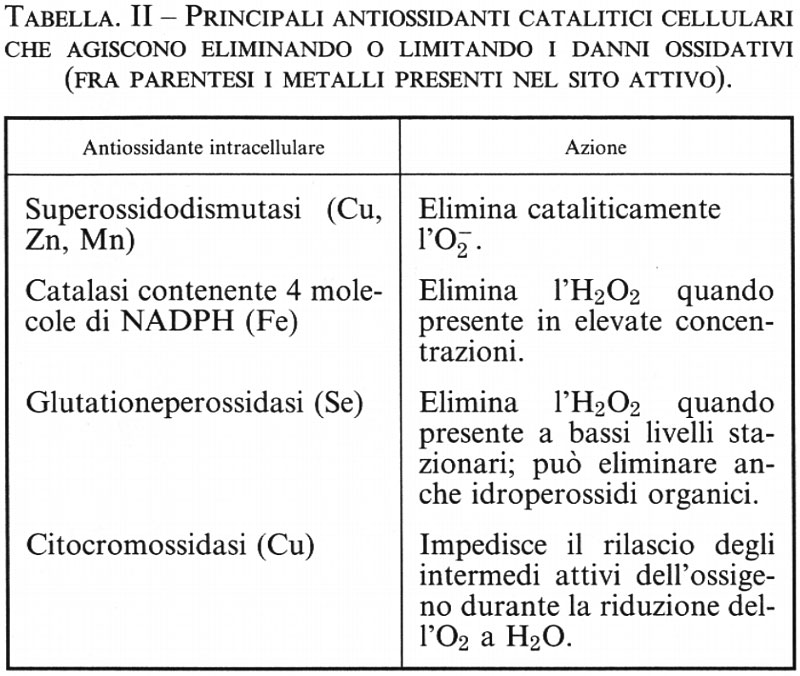
2O−2 + 2H+ → H2O2 + O2. (30)
Il perossido d'idrogeno, prodotto dalla reazione di dismutazione, può essere inattivato da due differenti enzimi, la catalasi (v. eq. 31) e la glutationeperossidasi (GSHPx), un enzima contenente selenio che richiede glutatione (GSH; v. eq. 32):
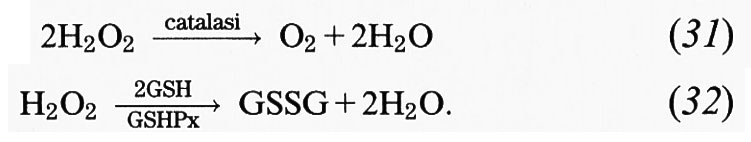
Durante il normale metabolismo questi enzimi eliminano gli intermedi di riduzione dell'ossigeno tossici che si formano all'interno della cellula: ciò consente l'accumulo di una piccola quantità di ferro sotto forma di composti a basso peso molecolare, necessaria per la sintesi del DNA e la formazione delle proteine contenenti ferro (v. Jacobs, 1977).
L'inibizione della formazione di radicali all'interno della cellula è il meccanismo più ovvio che deve essersi evoluto per limitare la tossicità dell'ossigeno. Ne è un esempio la citocromossidasi - l'ossidasi terminale della catena di trasporto degli elettroni nei mitocondri - la quale, mentre agisce cataliticamente, non rilascia intermedi reattivi dell'ossigeno dal suo sito attivo (v. Chance e altri, 1979).
b) Antiossidanti di membrana
All'interno degi strati lipidici idrofobici delle membrane si possono formare vari tipi di radicali lipofili a partire da quelli presenti nel citoplasma, che vengono poi inattivati da diversi antiossidanti (v. tab. III). La vitamina E (α-tocoferolo), liposolubile, ha scarso potere antiossidante al di fuori del doppio strato lipidico della membrana, ma è molto efficace quando vi è incorporata (v. Gutteridge, 1978). È difficile stabilire quale sia l'antiossidante biologicamente più importante, in quanto questo dipende dal tipo di proossidante che ha provocato la formazione di radicali liberi; per questo motivo la vitamina E è considerata, a seconda delle condizioni di reazione, l'antiossidante più efficace (v. Burton e altri, 1982) o meno efficace (v. Stocks e altri, 1974).

L'organizzazione della componente lipidica della membrana è un elemento importante per la sua stabilità e la sua protezione; una corretta organizzazione strutturale implica che fosfolipidi e colesterolo siano presenti in determinate proporzioni e che i diversi tipi di fosfolipidi siano legati agli appropriati acidi grassi (v. Gutteridge e Halliwell, 1988).
c) Antiossidanti extracellulari
I liquidi corporei extracellulari non contengono gli enzimi che abbiamo descritto come antiossidanti intracellulari. Tuttavia, la superossidodismutasi e la glutationeperossidasi sono state recentemente identificate anche come distinte proteine glicosilate extracellulari (v. Maddipati e Marnett, 1987). Consentendo alle specie reattive dell'ossigeno, come il superossido e il perossido d'idrogeno, una breve sopravvivenza all'interno dei liquidi extracellulari, l'organismo può utilizzare queste molecole, unitamente ad altre come l'ossido d'azoto (NO), in qualità di molecole segnale, messaggere o d'innesco (v. Saran e Bors, 1989), purché il superossido e il perossido d'idrogeno non incontrino all'esterno della cellula ferro e rame reattivi e i meccanismi antiossidanti extracellulari mantengano questi ioni metallici in una forma reattiva scarsa o nulla (v. tab. IV).
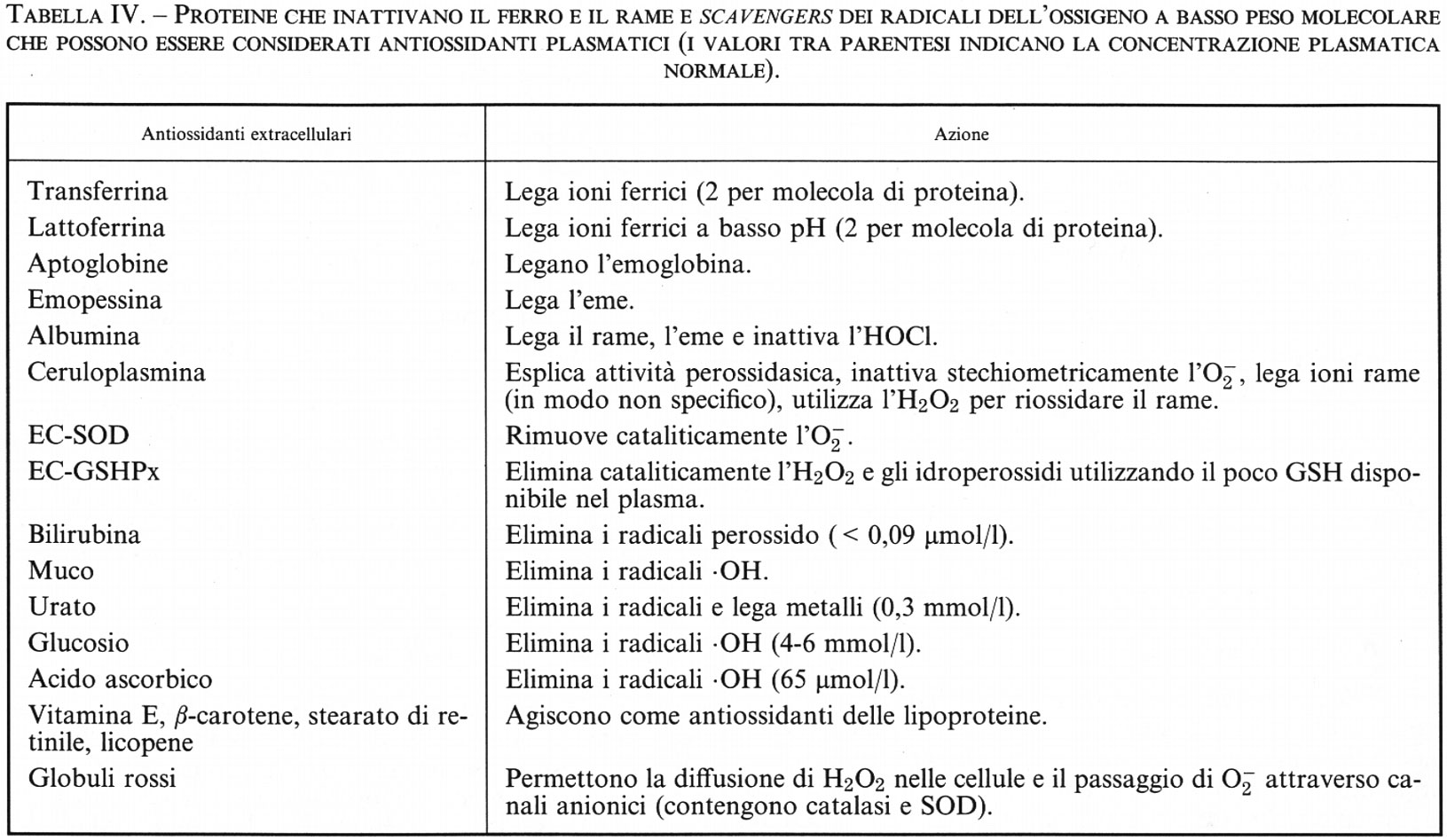
Come abbiamo già detto, solo un terzo delle molecole di transferrina, proteina trasportatrice del ferro, è legato a questo metallo; e ciò fa sì che la concentrazione plasmatica del ferro ‛libero' sia praticamente nulla. Il ferro legato alla transferrina non partecipa a reazioni radicaliche e la residua capacità di legare il ferro che ha questa proteina le conferisce forti proprietà antiossidanti nei confronti delle reazioni radicaliche stimolate dal ferro (v. Gutteridge e altri, 1981). Considerazioni analoghe possono essere fatte per la lattoferrina che, come la transferrina, può legare due moli di ferro per ogni mole di proteina, mantenendole legate anche quando il pH diminuisce fino a 4,0. Come abbiamo visto in precedenza, l'emoglobina, la mioglobina e i composti eminici possono accelerare la perossidazione lipidica, ma il plasma contiene anche proteine, come le aptoglobine e l'emopessina, che legano e conservano, rispettivamente, l'emoglobina e il ferro eminico e allo stesso tempo diminuiscono sensibilmente la loro capacità di accelerare la perossidazione lipidica (v. Gutteridge e Smith, 1988).
La ceruloplasmina, tipica per la sua intensa colorazione azzurro-blu, è la principale proteina plasmatica contenente rame, ma la sua funzione biologica, a parte la nota capacità di reagire durante la fase acuta, rimane un enigma. Il suo coinvolgimento nel metabolismo del ferro come ferrossidasi (catalizzando l'ossidazione degli ioni ferrosi a ferrici, così da facilitare il loro legame alla transferrina) non è stato ancora pienamente accertato (v. Gutteridge e Stocks, 1981). Comunque, Gutteridge e i suoi collaboratori hanno evidenziato che l'attività ferrossidasica della ceruloplasmina fornisce un notevole contributo alla protezione antiossidante extracellulare contro le reazioni radicaliche indotte dagli ioni ferro (v. Gutteridge e Stocks, 1981), rimuovendo rapidamente il ferro in soluzione e contemporaneamente riducendo l'ossigeno ad acqua; il trasferimento di 4 elettroni al sito attivo dell'enzima avviene senza rilascio di forme reattive dell'ossigeno. È stato dimostrato che la ceruloplasmina reagisce stechiometricamente con il superossido e con il perossido d'idrogeno (v. Calabrese e Carbonaro, 1986), pur non possedendo attività catalitiche del tipo della SOD e della perossidasi, come è stato spesso asserito.
Il plasma contiene una gran quantità di molecole a basso peso molecolare con attività redox, a molte delle quali sono state attribuite funzioni antiossidanti primarie. Così, studiando la formazione ferro-dipendente di radicali perossido da iniziatori azotati, sono stati isolati importanti fattori antiossidanti, come la vitamina E, il β-carotene (provitamina A), l'acido urico, la bilirubina e l'acido ascorbico (v. Frei e altri, 1988): molti di questi antiossidanti sono vitamine e il loro ruolo è oggetto di vivaci discussioni tra i nutrizionisti.
d) Equilibrio proossidanti/antiossidanti e stress ossidativo
Quando l'equilibrio tra la produzione di specie derivanti dall'ossigeno - come l'O−2, l'acido ipocloroso (HOCl) e l'ossigeno singoletto - e le difese antiossidanti è alterato, le cellule vanno incontro a stress ossidativo. Questa condizione può fornire il ferro necessario perché la reazione di Fenton proceda fino a indurre danni molecolari (v. Gutteridge, 1986), ma, come già accennato, l'organismo mette in atto diverse strategie per combattere tale stress. Parallelamente a questi sistemi di difesa immediati esiste una protezione regolata geneticamente, che comprende l'induzione della sintesi di proteine da shock termico e da stress ossidativo (v. Storz e altri, 1990).
7. Stress ossidativo e patologie umane: alcuni esempi
L'aumento della formazione di specie reattive dell'ossigeno (ROS) è stato osservato in più di un centinaio di malattie umane (v. Gutteridge, 1993), ma in nessuna di esse è mai stato dimostrato che questo processo possa svolgere un ruolo patologico centrale. Prenderemo in esame alcune patologie nelle quali i danni indotti da ROS sono ritenuti rilevanti.
a) Polmone da shock o sindrome da sofferenza respiratoria negli adulti
Con l'espressione ‛polmone da shock' si designa una forma acuta di lesione polmonare, che si ritiene colpisca negli Stati Uniti fino a 150.000 pazienti ogni anno. Nella descrizione originale della malattia (v. Ashbaugh e altri, 1967), dodici pazienti avevano manifestato sintomi clinici simili a quelli della sindrome di sofferenza respiratoria infantile, entro 96 ore da altri traumi o disturbi associati (per es., sepsi, pancreatite, bypass chirurgici, aspirazione dei contenuti gastrici o traumi violenti), così che venne denominata ARDS (Adult Respiratory Distress Syndrome). Il tasso di mortalità dovuto all'ARDS è superiore al 50% e non sembra diminuire nonostante i progressi compiuti nel trattamento dei pazienti.
Anatomicamente, la fase acuta del polmone da shock (entro i primi 5 giorni) è caratterizzata da un forte danno transmurale con distruzione dell'epitelio alveolare e dell'endotelio del microcircolo, con edema alveolare e interstiziale e aumento del numero dei neutrofili nel polmone. La fase subacuta (fra il 5° e il 10° giorno) è caratterizzata da edema interstiziale persistente, con meno neutrofili ma più cellule mononucleate. Col progredire dei processi di riparazione alveolare si verifica una sequenza coordinata di replicazione e migrazione cellulare che, se coronata da successo, ricostituisce l'interfaccia aria-polmone, quella polmone-sangue e l'interstizio. Tali processi comprendono la proliferazione di cellule alveolari di tipo II (a sostituire le cellule di tipo I precedentemente danneggiate), la proliferazione dei fibroblasti, la deposizione di tessuto connettivo e la crescita di nuovi vasi sanguigni. I pazienti che sopravvivono a questo stadio possono recuperare la maggior parte della funzione polmonare; se, invece, a questo stadio il processo di recupero non ha inizio, si sviluppa una fase cronica (fra la 1a e la 2a settimana), caratterizzata da risposte fibro-proliferative apparentemente incontrollate, occlusione dei vasi polmonari e successivo deterioramento della funzione di scambio di gas, che può rimanere alterata anche se in seguito il paziente migliora.
È stato più volte ipotizzato che il polmone da shock conseguente a sepsi sia causato da lipopolisaccaridi batterici in grado d'indurre l'espressione sequenziale di diversi tipi di citochine (per es., α-TNFa, IL-1, IL-2, IL-8) e l'attivazione delle cascate del complemento e della coagulazione, che hanno importanti effetti di attivazione e di innesco su diversi sistemi cellulari, tra cui i fagociti circolanti e quelli normalmente presenti nel polmone. Questi fenomeni sono accompagnati dal sequestro dei neutrofili nel polmone, dove rilasciano O−2, H2O2, HOCl, eicosanoidi, proteasi e altri agenti dannosi; tuttavia, il polmone da shock può svilupparsi anche in pazienti neutropenici: i neutrofili, infatti, non sono sempre essenziali, poiché non va dimenticato che anche i macrofagi alveolari possono essere stimolati e attivati con conseguente rilascio di proteasi, di specie reattive che derivano dall'ossigeno e di altre specie potenzialmente tossiche.
Prove a sostegno dell'aumento dello stress ossidativo in pazienti affetti da ARDS sono il reperto di mieloperossidasi e α1-antitripsina ossidata nei liquidi di lavaggio broncoalveolare, l'espulsione dell'eccesso di H2O2 nell'aria espirata, l'aumento del danno alle proteine e la perdita con i liquidi corporei di acido ascorbico e α-tocoferolo. Un aumento del tasso di perossidazione lipidica è stato evidenziato anche nel plasma. Nel polmone da shock, lo stress ossidativo, inevitabile complicazione del trattamento della malattia, viene fortemente potenziato dalla necessità di aiutare la respirazione fornendo al paziente O2 in elevate concentrazioni.
Molte delle attuali attività di ricerca sono indirizzate verso possibili strategie atte a prevenire o migliorare le fasi iniziali dei processi di attivazione, come l'infusione di anticorpi contro i lipopolisaccaridi batterici, l'α-TNF (una citochina che probabilmente svolge un ruolo fondamentale nell'ARDS conseguente a sepsi) e le molecole glicoproteiche che aderiscono all'endotelio vascolare e ai leucociti circolanti, le quali svolgono un ruolo nel sequestro dei neutrofili attivati nel polmone. Comunque, data la molteplicità degli stimoli iniziali e dei susseguenti meccanismi di danno tessutale, è poco probabile che i soli antiossidanti possano risultare efficaci nel trattamento dell'ARDS.
b) Artrite reumatoide
Il pionieristico lavoro di J. M. McCord (v., 1974) ha focalizzato l'attenzione sul possibile ruolo svolto dai radicali liberi e da altre specie derivanti dall'ossigeno nella malattia reumatoide umana. L'autore ha preso in esame la diminuzione della viscosità del liquido sinoviale nelle articolazioni dei pazienti affetti da artrite reumatoide e ha riprodotto in vitro un'analoga diminuzione esponendo il liquido sinoviale o lo ialuronato (il glicosamminoglicano responsabile della maggior parte della viscosità del liquido sinoviale) a un sistema che genera superossido. Un successivo lavoro (v. Halliwell, 1978) ha dimostrato che la depolimerizzazione in vitro dello ialuronato in sistemi in cui si formano ROS non è imputabile a O−2 o a H2O2, ma alla formazione ferrodipendente di •OH. I radicali ossidrile provocano la frammentazione casuale dello ialuronato in oligosaccaridi. Sia il ferro che il rame possono catalizzare la formazione di •OH, ma non è stato chiaramente individuato rame ‛catalitico' nel liquido sinoviale appena aspirato dall'articolazione del ginocchio di pazienti affetti da artrite reumatoide, mentre in circa il 40% dei casi è presente del ferro rilevabile con il saggio della bleomicina (v. Gutteridge, Bleomycin-detectable..., 1987). Nelle articolazioni infiammate il ferro potrebbe provenire da cellule lisate o derivare dalla degradazione dell'emoglobina (rilasciata dagli eritrociti, liberati in conseguenza di microemorragie traumatiche nell'articolazione) esposta a elevate concentrazioni di H2O2 (v. Gutteridge, 1986). Un'altra potenziale fonte di ferro nel liquido sinoviale è costituita dalla ferritina (v. Biemond e altri, 1986); è da notare che il livello del ferro nei pazienti affetti da artrite reumatoide influenza i sintomi della malattia (v. Blake e altri, 1981). Con la spettroscopia NMR sono stati rilevati danni provocati dall'attacco dei radicali liberi allo ialuronato del liquido sinoviale (v. Grootveld e altri, 1991), benché in corso di artrite reumatoide lo ialuronato possa essere secreto in forme anormali a basso peso molecolare.
Per quanto riguarda la provenienza dell'O−2 e dell'H2O2 presenti nelle articolazioni reumatoidi infiammate, una fonte è rappresentata dai neutrofili attivati, che rilasciano O−2, H2O2, elastasi, HOCl ed eicosanoidi. La produzione di O−2 e H2O2 da parte dei neutrofili può essere influenzata dalla tensione dell'ossigeno nel liquido sinoviale e quindi può essere limitata nelle articolazioni in ipossia. Il panno sinoviale che ricopre la cartilagine contiene molte cellule simili ai macrofagi, che presumibilmente secernono O−2, H2O2, eicosanoidi, IL-1 e NO•. È stato suggerito che nell'artrite reumatoide le articolazioni infiammate siano soggette a ripetuti cicli di ipossia e riperfusione che, attraverso svariati meccanismi, inclusa l'attività della xantinossidasi, possono produrre radicali liberi (v. Allen e altri, 1987). Inoltre, le specie reattive dell'ossigeno potrebbero accelerare il riassorbimento osseo da parte degli osteoclasti (v. Garrett e altri, 1990).
Esistono pochi dubbi sul fatto che nell'artrite reumatoide le articolazioni infiammate siano soggette a danno ossidativo; non è ancora chiara l'importanza del ruolo svolto dal danno ossidativo in questa patologia. Alcune indicazioni preliminari (v. Flohe, 1988) avevano suggerito che le SOD potessero essere di aiuto nel trattamento dell'artrite, ma questa ipotesi non è stata convalidata (v. Greenwald, 1991).
c) Patologie del sistema nervoso
Rispetto agli altri tessuti, il sistema nervoso centrale è particolarmente sensibile ai danni ossidativi (v. Evans, 1993), dovuti all'elevato tasso di attività metaboliche ossidative, all'alta concentrazione di acidi grassi polinsaturi, ai bassi livelli di catalasi, glutationeperossidasi e, probabilmente, superossidodismutasi, alle ossidazioni e alle autossidazioni neurochimiche e alla presenza di zone a elevato contenuto di ferro e di ascorbato.
Trauma cerebrale, ischemia e riossigenazione. - Il danneggiamento del sistema nervoso centrale costituisce uno dei maggiori problemi clinici, poiché genera spesso lesioni più estese di quanto non avvenga in altri tessuti sottosposti a traumi analoghi. Le reazioni dei radicali liberi risultano implicate nel danneggiamento del SNC (v. Halliwell e Gutteridge, The importance..., 1985), specialmente in tessuti ischemici riossigenati. Ciò suggerisce che la xantinossidasi contribuisca all'aumentata formazione di ossidanti, anche se il suo ruolo nelle lesioni del cervello umano riossigenato rimane oscuro e la rilevanza dei danni causati dai radicali liberi è strettamente legata soprattutto alla durata della mancanza di ossigeno e all'estensione della zona interessata. Gutteridge e collaboratori (v., 1991), analizzando periodi di ischemia seguiti da riossigenazione in cervello di gerbillo in vivo, non hanno osservato alcun significativo aumento di composti del ferro a basso peso molecolare rispetto ai controlli. Tuttavia, l'incubazione in vitro di cervelli di ratto in condizioni aerobie, a differenti valori di pH, ha mostrato un aumento di tali composti non appena il pH scende al valore di 5,0, mentre in condizioni di anossia, agli stessi valori di pH, il rilascio di ferro aumentava notevolmente (v. Bralet e altri, 1992).
I traumi del sistema nervoso centrale inducono una rapida amplificazione del danno tessutale che, probabilmente, implica reazioni ferro-dipendenti di proteine contenenti ferro eminico e non eminico. Esiste, quindi, un considerevole interesse clinico verso la possibilità di utilizzare gli antiossidanti e gli agenti chelanti per arrestare l'espansione del danno tessutale dal sito della lesione iniziale.
Morbo di Parkinson. - In questa patologia si osserva una selettiva e progressiva distruzione dei neuroni dopamminergici della via nigro-striatale. G. Cohen (v., 1988) suggerì che l'aumento compensatorio dei neuroni dopamminergici potesse indurre un aumento della formazione di H2O2 da parte della monoamminossidasi. Si è anche ipotizzato che le reazioni dei radicali liberi ferro-dipendenti contribuiscano all'insorgenza dei danni alla substantia nigra osservati in pazienti affetti da morbo di Parkinson. A sostegno di tale ipotesi esistono diverse osservazioni effettuate in cervelli parkinsoniani: Dexter e altri (v., 1991) hanno rilevato l'aumento del ferro nella substantia nigra e la diminuzione dei livelli tessutali di ferritina, mentre Riederer e altri (v., 1989) hanno riscontrato l'aumento del ferro nella substantia nigra, l'aumento della ferritina e la diminuzione del glutatione ridotto (GSH). Inoltre, la scoperta che il gene per la CuZn-SOD è preferenzialmente espresso nella substantia nigra rafforza l'ipotesi che questa regione possa essere particolarmente suscettibile allo stress ossidativo (v. Ceballos e altri, 1990).
Il liquido cerebrospinale di pazienti parkinsoniani mostra valori normali del contenuto totale di ferro, ma Pall e altri (v., 1987) hanno osservato in alcuni pazienti un aumento dei valori del rame chelabile. Quando un sale di ferro viene iniettato nella substantia nigra del ratto, esso provoca un comportamento ‛di tipo parkinsoniano' (v. Ben-Shachar e Youdim, 1991), in accordo con l'ipotesi che le reazioni ossidative indotte dal ferro svolgano un qualche ruolo nel morbo di Parkinson. Tuttavia, i dati disponibili sulla natura chimica e la reattività del ferro presente nel cervello umano sono insufficienti, e il danno ossidativo osservato nel morbo di Parkinson potrebbe essere solo una semplice conseguenza della lesione dei tessuti.
Morbo di Alzheimer. - Tuttora oggetto di controversie è l'ipotesi di un legame tra esposizione ambientale all'alluminio e sviluppo del morbo di Alzheimer, basata sulla scoperta dell'aumento degli alluminosilicati nel core delle placche senili e nei neuroni contenenti neurofibrille addensate in aggregati (v. Perl e Brody, 1980). Esperimenti condotti su animali hanno mostrato che gli ioni alluminio sono neurotossici e possono entrare nel cervello utilizzando i recettori della transferrina (v. Roskams e Connor, 1990); è stato inoltre osservato che la deposizione di ferro risulta aumentata all'interno o in prossimità delle placche e negli aggregati neurofibrillari (v. Youdim, 1988). I sali di alluminio possono stimolare i fagociti a produrre specie reattive dell'ossigeno (v. Evans e altri, 1989), aumentando così le potenzialità del danno ossidativo, oltre che agire sinergicamente con il ferro (v. Gutteridge e altri, 1985) e favorirne probabilmente il rilascio. Il plasma di pazienti affetti da morbo di Alzheimer presenta bassi livelli di diversi antiossidanti (v. Zaman e altri, 1992) e fibroblasti della cute di tali pazienti, tenuti in coltura, sono più sensibili ai danni ossidativi (v. Piersanti e altri, 1992), in accordo con l'alterazione dei sistemi di difesa. Anche in questo caso non è chiaro se queste osservazioni sono una conseguenza della malattia o se contribuiscono sostanzialmente al suo sviluppo.
Morbo di Batten. - Le lipofuscinosi ceroidi neuronali (LCN) sono un gruppo di malattie neurodegenerative ereditarie recessive, caratterizzate da accumulo lisosomiale, ad alta incidenza nei paesi nordici. Nei vari tipi di LCN si osserva un accumulo diffuso di ceroidi e lipofuscine in tutto il corpo che, in passato, ha condotto a formulare l'ipotesi semplicistica che la perossidazione lipidica fosse l'evento chiave per lo sviluppo della malattia. Recenti studi, che utilizzano un modello ovino di LCN, hanno dimostrato che il pigmento accumulato è essenzialmente costituito da una singola proteina di peso molecolare pari a circa 3.500, che corrisponde alla subunità c intatta e non modificata dell'ATP-sintasi.
Il liquido cerebrospinale di pazienti affetti da LCN mostra livelli aumentati di ioni ferro a stabilità dipendente dal pH, che possono complessarsi con la bleomicina (v. Heiskala e altri, 1988). Le concentrazioni di ferro e rame debolmente legati aumentano nei pazienti affetti da LCN in funzione dell'età, ma ciò potrebbe semplicemente riflettere l'aumento della distruzione tessutale che accompagna una progressiva degenerazione (v. Heiskala e altri, 1988).
A-β-lipoproteinemia. - I pazienti affetti da a-β-lipoproteinemia sono portatori di un raro errore congenito del metabolismo lipidico che causa l'incapacità di sintetizzare apolipoproteina β, facendo sì che i grassi ingeriti con la dieta vengano assorbiti, ma non trasportati fuori dalle cellule della mucosa intestinale. Questi pazienti hanno livelli plasmatici di vitamina E trascurabili e in alcuni casi mostrano neuropatie, degenerazione della retina ed eritrociti di forma anormale (acantociti). Studi recenti hanno dimostrato che sia le neuropatie che le retinopatie possono essere prevenute somministrando ai pazienti massicce dosi di vitamina E per via orale (v. Runge e altri, 1986), e ciò conferma l'ipotesi che la vitamina E sia un importante antiossidante del cervello (v. Goss-Sampson e Muller, 1987).
Benché oggi esistano prove considerevoli che mettono in relazione gli ioni dei metalli di transizione e i radicali liberi dell'ossigeno con importanti malattie degenerative del cervello, non esiste ancora nessuna prova definitiva a sostegno del ruolo svolto dalle specie reattive dell'ossigeno nello sviluppo di queste patologie, così che queste possano essere trattate con successo con terapie antiossidanti.
BIBLIOGRAFIA
Allen, R. E., Outhwaite, J., Morris, C. J., Blake, D. R., Xanthine oxido-reductase is present in human synovium, in ‟Annals of the rheumatic diseases", 1987, XLVI, pp. 843-845.
Ames, B. N., Shigenaga, M. K., Oxidants are a major contributor to cancer and ageing, in DNA and free radicals (a cura di B. Halliwell e O. I. Aruoma), London 1993, pp. 1-15.
Anbar, M., Neta, P., A compilation of specific bimolecular rate constants for the reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals with inorganic and organic compounds in aqueous solution, in ‟International journal of applied radiation and isotopes", 1967, XVIII, pp. 495-523.
Andersen, B. R., Harvath, L. A., Light generation with Fenton's reagent. Its relationship to granulocyte chemiluminescence, in ‟Biochimica et biophysica acta", 1979, DLXXXIV, pp. 164-173.
Aruoma, O. I., Grootveld, M., Halliwell, B., The role of iron in ascorbate-dependent deoxyribose degradation. Evidence consistent with site-specific hydroxyl radical generation caused by iron ions bound to the deoxyribose molecule, in ‟Journal of inorganic biochemistry", 1987, XXIX, pp. 289-299.
Ashbaugh, D. G., Bigelow, D. B., Petty, T. L., Levine, B. E., Acute respiratory distress in adults, in ‟The lancet", 1967, n. 2, pp. 19-23.
Bannister, J. V., Bannister, W. K., Thornalley, P. J., The effect of ferritin iron loading on hydroxyl radical production, in ‟Life chemistry reports", 1984, II, suppl., pp. 64-72.
Ben-Shachar, D., Youdim, M. B. H., Intranigral iron injection induces behavioural and biochemical ‛parkinsonism' in rats, in ‟Journal of neurochemistry", 1991, LVII, pp. 2133-2135.
Bhuyan, K. C., Bhuyan, D. K., Regulation of hydrogen peroxide in eye humours. Effect of 3-amino-1H-1,2,4-triazole on catalase and glutathione peroxidase of rabbit eye, in ‟Biochimica et biophysica acta", 1977, CDXCVII, pp. 641-651.
Bielski, B. H. J., Allen, A. O., Mechanism of the disproportionation of superoxide radicals, in ‟Journal of physical chemistry", 1977, CCLVIII, pp. 4759-4761.
Bielski, B. H. J., Arudi, R. L., Sutherland, M. W., A study of the reactivity of HO2/O_2 with unsaturated fatty acids, in ‟Journal of biological chemistry", 1983, CCLVIII, pp. 4759-4761.
Biemond, P., Swaak, A. J. G., Eijk, H. G. van, Koster, J. F., Intraarticular ferritin-bound iron in rheumatoid arthritis. A factor that increases oxygen free radical-induced tissue destruction, in ‟Arthritis and rheumatism", 1986, XXIX, pp. 1187-1193.
Blake, D. R., Hall, N. D., Bacon, P. A. e altri, The importance of iron in rheumatoid disease, in ‟The lancet", 1981, n. 2, pp. 1142-1144.
Bralet, J., Schreiber, L., Bouvier, C., Effect of acidosis and anoxia on iron delocalization from brain homogenates, in ‟Biochemical pharmacology", 1992, XLIII, pp. 979-983.
Bray, W. G., Gorin, M., Ferryion, a compound of tetravalent iron, in ‟Journal of the American Chemical Society", 1932, LIV, pp. 2124-2125.
Bullen, J. J., Rogers, H. J., Griffiths, M., Role of iron in bacterial infection, in ‟Current topics in microbiology and immunology", 1978, LXXX, pp. 1-35.
Burton, G. W., Joyce, A., Ingold, K. V., First proof that vitamin E is major lipid-soluble, chain-breaking antioxidant in human blood plasma, in ‟The lancet", 1982, n. 2, pp. 327-328.
Cadenas, E., Wefers, H., Sies, H., Low-level chemiluminescence of isolated hepatocytes, in ‟European journal of biochemistry", 1981, CXIX, pp. 531-536.
Calabrese, L., Carbonaro, M., An e.p.r. study of the non-equivalence of the copper sites of caeruloplasmin, in ‟Biochemical journal", 1986, CCXXXVIII, pp. 291-295.
Ceballos, I., Lafon, M., Javoy-Agid, F. e altri, Superoxide dismutase and Parkinson's disease, in ‟The lancet", 1990, CCCXXXV, pp. 1035-1036.
Chance, B., Sies, H., Boveris, A., Hydro-peroxide metabolism in mammalian organs, in ‟Physiological review", 1979, LIX, pp. 527-605.
Cilento, G., Electronic excitation in dark biological processes, in Chemical and biological generation of excited states (a cura di W. Adam e G. Cilento), New York 1982, pp. 277-307.
Cohen, G., Oxygen radicals and Parkinson's disease, in Oxygen radicals and tissue injury (a cura di B. Halliwell), Bethesda, Md., 1988, pp. 130-135.
Davies, K. J., Protein damage and degradation by oxygen radicals. I: general aspects, in ‟Journal of biological chemistry", 1987, CCLXII, pp. 9895-9901.
Dexter, D. T., Carayon, A., Javoy-Agid, F. e altri, Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia, in ‟Brain", 1991, CXIV, pp. 1953-1975.
Ehrenwald, E., Chisolm, G. M., Fox, P. L., Intact human caeruloplasmin oxidatively modifies low density lipoprotein, in ‟Journal of clinical investigation", 1994, XCIII, pp. 1493-1501.
Esterbauer, H., Schaur, R. J., Zollner, H., Chemistry and biochemistry of 4-hydroxynonenal malonaldehyde and related aldehydes, in ‟Free radical biology and medicine", 1991, XI, pp. 82-128.
Evans, P. H., Free radicals in brain metabolism and pathology, in ‟British medical bulletin", 1993, IL, pp. 577-587.
Evans, P. H., Klinowski, J., Yano, E., Urano, N., Alzheimer's disease: a pathogenic role for aluminosilicate-induced phagocytic free radicals, in ‟Free radical research communications", 1989, VI, pp. 317-321.
Finkelstein, E., Rosen, G. M., Rauckman, L., Production of hydroxyl radical by decomposition of superoxide spin-trapped adducts, in ‟Molecular pharmacology", 1981, XXI, pp. 262-275.
Flohe, L., Superoxide dismutase for therapeutic use: clinical experience, dead ends and hopes, in ‟Molecular and cellular biochemistry", 1988, LXXXIV, pp. 123-131.
Frei, B., Stocker, R., Ames, B. N., Antioxidant defenses and lipid peroxidation in human blood plasma, in ‟Proceedings of the National Academy of Sciences", 1988, LXXXV, pp. 9748-9752.
Fridovich, I., Superoxide dismutases, in ‟Annual review of biochemistry", 1975, XLIV, pp. 147-159.
Fridovich, I., The biology of oxygen radicals, in ‟Science", 1978, CCI, pp. 875-880.
Fridovich, I., Superoxide radical: an endogenous toxicant, in ‟Annual review of pharmacology and toxicology", 1983, XXIII, pp. 239-257.
Galliard, T., Lipolytic and lipoxygenase enzymes in plants and their action in wounded tissue, in Biochemistry of wounded plant tissues (a cura di G. Kahl), Berlin 1978, pp. 156-201.
Garrett, I. R., Boyce, B. F., Oreffo, R. O. e altri, Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo, in ‟Journal of clinical investigation", 1990, LXXXV, pp. 632-639.
Getzoff, E. D., Tainer, J. A., Weiner, P. K., Kollman, P. A., Richardson, J. S., Richardson, J. C., Electrostatic recognition between superoxide and copper, zinc superoxide dismutase, in ‟Nature", 1983, CCCVI, pp. 287-290.
Gey, K. F., Puska, P., Plasma vitamin E and A inversely correlated to mortality from ischemic heart disease in cross-cultural epidemiology, in ‟Annals of the New York Academy of Sciences", 1989, DLXX, pp. 268-282.
Gey, K. F., Puska, P., Jordan, P., Moser, U. K., Inverse correlation between plasma vitamin E and mortality from ischemic heart disease in cross-cultural epidemiology, in ‟American journal of clinical nutrition", 1991, LIII, suppl., pp. 326-334.
Gey, K. F., Stahelin, H. P., Puska, P., Evans, A., Relationship of plasma level of vitamin C to mortality from ischemic heart disease, in ‟Annals of the New York Academy of Sciences", 1987, CDXCVIII, pp. 110-123.
Goldstein, J. L., Ho, Y. K., Basu, S. K., Brown, M. S., Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition, in ‟Proceedings of the National Academy of Sciences", 1979, LXXVI, pp. 333-337.
Goss-Sampson, M. A., Muller, D. P. R., Studies on the neurobiology of vitamin E (alpha-tocopherol) and some other antioxidant systems in the rat, in ‟ Neuropathology and applied neurobiology", 1987, XIII, pp. 289-296.
Greenwald, R. A., Therapeutic usages of oxygen radical scavengers in human diseases: myths and realities, in ‟Free radical research communications", 1991, XII-XIII, pp. 531-538.
Grootveld, M., Henderson, E. B., Farrell, A. e altri, Oxidative damage to hyaluronate and glucose in synovial fluid during exercise of the inflamed rheumatoid joint, in ‟Biochemical journal", 1991, CCLXXIII, pp. 459-467.
Gutteridge, J. M. C., The membrane effects of vitamin E, cholesterol and their acetates on peroxidative susceptibility, in ‟Research communications in chemical pathology and pharmacology", 1978, XXII, pp. 563-571.
Gutteridge, J. M. C., The role of superoxide and hydroxyl radicals in phospholipid peroxidation catalysed by iron salts, in ‟FEBS letters", 1982, CL, pp. 454-458.
Gutteridge, J. M. C., Reactivity of hydroxyl and hydroxyl-like radicals discriminated by release of thiobarbituric acid-reactive material from deoxy sugars, nucleosides and benzoate, in ‟Biochemical journal", 1984, CCXXIV, pp. 761-767.
Gutteridge, J. M. C., Iron promoters of the Fenton reaction and lipid peroxidation can be released from haemoglobin by peroxides, in ‟FEBS letters", 1986, CCI, pp. 291-295.
Gutteridge, J. M. C., Bleomycin-detectable iron in knee-joint synovial fluid from arthritic patients and its relationship to the extracellular antioxidant activities of caeruloplasmin, transferrin and lactoferrin, in ‟Biochemical journal", 1987, CCXLV, pp. 415-421.
Gutteridge, J. M. C., Ferrous-salt promoted damage to deoxyribose and benzoate. The increased effectiveness of hydroxyl-radical scavengers in the presence of EDTA, in ‟Biochemical journal", 1987, CCXLIII, pp. 709-714.
Gutteridge, J. M. C., Free radicals in disease processes: a compilation of cause and consequence, in ‟Free radical research communications", 1993, XIX, pp. 141-158.
Gutteridge, J. M. C., Bannister, J. V., Copper-zinc and manganese superoxide dismutases inhibit deoxyribose degradation by the superoxide-driven Fenton reaction at two different stages: implications for the redox states of copper and manganese, in ‟Biochemical journal", 1986, CCXXXIV, pp. 225-228.
Gutteridge, J. M. C., Beard, A. P. C., Quinlan, G. J., Superoxide-dependent lipid peroxidation. Problems with the use of catalase as a specific probe for Fenton-derived hydroxyl radicals, in ‟Biochemical and biophysical research communications", 1983, CXVII, pp. 901-907.
Gutteridge, J. M. C., Cao, W., Chevion, M., Bleomycin-detectable iron in brain tissue, in ‟Free radical research communications", 1991, XI, pp. 317-320.
Gutteridge, J. M. C., Halliwell, B., The antioxidant proteins of extracellular fluids, in Cellular antioxidant defense mechanisms (a cura di C. K. Chow), Boca Raton, Flo., 1988, vol. II, pp. 1-23.
Gutteridge, J. M. C., Halliwell, B., Iron toxicity and oxygen radicals, in Clinical haematology. Iron chelating therapy (a cura di C. Hershko), London 1989, vol. II, pp. 195-256.
Gutteridge, J. M. C., Hou, Y., Iron complexes and their reactivity in the bleomycin assay for radical-promoting loosely-bound iron, in ‟Free radical research communications", 1986, II, pp. 143-151.
Gutteridge, J. M. C., Rowley, D. A., Halliwell, B., Superoxide-dependent formation of hydroxyl radicals in the presence of iron salts. Detection of ‛free' iron in biological systems by using bleomycin-dependent degradation of DNA, in ‟Biochemical journal", 1981, CIC, pp. 263-265.
Gutteridge, J. M. C., Smith, A., Antioxidant protection by hemopexin of haem-stimulated lipid peroxidation, in ‟Biochemical journal", 1988, CCLVI, pp. 861-865.
Gutteridge, J. M. C., Stocks, J., Caeruloplasmin: physiological and pathological perspectives, in ‟Critical reviews in clinical laboratory sciences", 1981, XIV, pp. 257-329.
Gutteridge, J. M. C., Winyard, P. G., Blake, D. R., Lunec, J., Brailsford, S., Halliwell, B., The behaviour of caeruloplasmin in stored human extracellular fluids in relation to ferroxidase II activity, lipid peroxidation and phenanthroline-detectable copper, in ‟Biochemical journal", 1985, CCXXX, pp. 517-523.
Halliwell, B., Superoxide-induced generation of hydroxyl radicals in the presence of iron salts. Its role in degradation of hyaluronic acid by a superoxide-generating system, in ‟FEBS letters", 1978, XCVI, pp. 238-242.
Halliwell, B., Free radicals, oxygen toxicity and aging, in Age pigments (a cura di R. S. Sohal), Amsterdam 1981, pp. 1-62.
Halliwell, B., Chloroplast metabolism. The structure and function of chloroplasts in green leaf cells, Oxford 19842.
Halliwell, B., Gutteridge, J. M. C., Lipid peroxidation, oxygen radicals, cell damage and antioxidant therapy, in ‟The lancet", 1984, n. 1, pp. 1396-1397.
Halliwell, B., Gutteridge, J. M. C., Oxygen toxicity, oxygen radicals, transition metals and disease, in ‟Biochemical journal", 1984, CCXIX, pp. 1-14.
Halliwell, B., Gutteridge, J. M. C., Oxygen radicals and the nervous system, in ‟Trends in neuroscience", 1985, VIII, pp. 22-26.
Halliwell, B., Gutteridge, J. M. C., The importance of free radicals and catalytic metal ions in human diseases, in ‟Molecular aspects of medicine", 1985, VIII, pp. 89-193.
Halliwell, B., Gutteridge, J. M. C., Free radicals in biology and medicine, Oxford 19892.
Halliwell, B., Wasil, M., Grootveld, M., Biologically-significant scavenging of the myeloperoxidase-derived oxidant hypochlorous acid by ascorbic acid. Implications for antioxidant protection in the inflamed rheumatoid joint, in ‟FEBS letters", 1987, CCXIII, pp. 15-17.
Harrison, J. E., Watson, B. D., Schultz, J., Myeloperoxidase and singlet oxygen: a reappraisal, in ‟FEBS letters", 1978, XCII, pp. 327-332.
Heiskala, H., Gutteridge, J. M. C., Westermarck, T. e altri, Bleomycin-detectable iron and phenanthroline-detectable copper in the cerebrospinal fluid of patients with neuronal ceroid-lipofuscinoses, in ‟American journal of medical genetics", 1988, V, suppl., pp. 193-202.
Hershko, C., Graham, G., Bates, G. W., Rachmilewitz, E. A., Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity, in ‟British journal of haematology", 1978, XL, pp. 255-263.
Hildebrandt, A. G., Roots, I., NADPH-dependent formation and breakdown of hydrogen peroxide during mixed function oxidation reaction in liver microsomes, in ‟Archives of biochemistry and biophysics", 1975, CLXXI, pp. 385-397.
Jacobs, A., Low molecular weight intracellular iron transport compounds, in ‟Blood", 1977, L, pp. 433-439.
Kanofsky, J. R., Wright, J., Miles-Richardson, G. E., Tauber, A. I., Biochemical requirements for singlet oxygen production by purified human myeloperoxidase, in ‟Journal of clinical investigation", 1984, LXXIV, pp. 1489-1495.
Kulig, M. J., Smith, L. L., Sterol metabolism XXV. Cholesterol oxidation by singlet molecular oxygen, in ‟Journal of organic chemistry", 1973, XXXVIII, pp. 3639-3642.
Lengfelder, E., Cadenas, E., Sies, H., Effect of DABCO (1,4-diazabicyclo[2,2,2]-octane) on singlet oxygen monomol (1270 nm) and dimol (634 and 703 nm) emission, in ‟FEBS letters", 1983, CLXIV, pp. 366-370.
McCord, J. M., Free radicals and inflammation. Protection of synovial fluid by superoxide dismutase, in ‟Science", 1974, CLXXXV, pp. 529-531.
Maddipati, K. R., Marnett, L. J., Characterization of the major hydroperoxide-reducing activity of human plasma. Purification and properties of a selenium-dependent glutathione peroxidase, in ‟Journal of biological chemistry", 1987, CCLXII, pp. 17398-17403.
O'Connell, M. J., Baum, H., Peters, T. J., Haemosiderin-like properties of free-radical-modified ferritin, in ‟Biochemical journal", 1986, CCXL, pp. 297-300.
O'Connell, M. J., Halliwell, B., Moorhouse, C. P., Aruoma, O. I., Baum, H., Peters, T. J., Formation of hydroxyl radicals in the presence of ferritin and haemosiderin. Is haemosiderin formation a biological protective mechanism?, in ‟Biochemical journal", 1986, CCXXXIV, pp. 724-731.
Osaki, S., Johnson, D. A., Frieden, E., The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum, in ‟Journal of biological chemistry", 1966, CCXLI, pp. 2746-2751.
Oshino, N., Jamieson, D., Chance, B., The properties of hydrogen peroxide production under hyperoxic and hypoxic conditions of perfused rat liver, in ‟Biochemical journal", 1975, CXLVI, pp. 53-65.
Packer, J. E., Mahood, J. S., Mora-Arellano, V. O., Slater, T. F., Willson, R. L., Wolfenden, B. S., Free radicals and singlet oxygen scavengers: reaction of a peroxy-radical with β-carotene, diphenyl furan and 1,4-diazobicyclo[2,2,2]-octane, in ‟Biochemical and biophysical research communications", 1981, XCVIII, pp. 901-906.
Pall, H. S., Williams, A. C., Blake, D. R. e altri, Raised cerebrospinal-fluid copper concentration in Parkinson's disease, in ‟The lancet", 1987, n. 2, pp. 238-241.
Pathak, M. A., Joshi, P. C., Production of active oxygen species (1O2 and O−2) by psoralens and ultraviolet radiation (320-400 nm), in ‟Biochimica et biophysica acta", 1984, DCCXCVIII, pp. 115-126.
Perl, D. P., Brody, A. R., Alzheimer's disease: X-ray spectrometric evidence of aluminum accumulation in neurofibrillary tangle-bearing neurons, in ‟Science", 1980, CCVIII, pp. 297-299.
Piersanti, P., Tesco, G., Latorraca, S. e altri, Alzheimer skin fibroblasts susceptibility to oxygen radical damage, in ‟Neurobiology of aging", 1992, XIII, pp. S1-S111.
Quinlan, G. J., Coudray, C., Hubbard, A., Gutteridge, J. M. C., Vanadium and copper in clinical solutions of albumin and their potential to damage protein structure, in ‟Journal of pharmaceutical science", 1992, LXXXI, pp. 611-614.
Reunanen, A., Knekt, R., Aaran, R. K., Serum ceruloplasmin level and the risk of myocardial infarction and stroke, in ‟American journal of epidemiology", 1992, CXXXVI, pp. 1082-1090.
Riederer, P., Sofic, E., Rausch, W. D. e altri, Transition metals, ferritin, glutathione and ascorbic acid in parkinsonian brains, in ‟Journal of neurochemistry", 1989, LII, pp. 515-520.
Roskams, A. J., Connor, J. R., Aluminum access to the brain: a role for transferrin and its receptor, in ‟Proceedings of the National Academy of Sciences", 1990, LXXXVII, pp. 9024-9027.
Rowley, D. A., Halliwell, B., Superoxide-dependent formation of hydroxyl radicals in the presence of thiol compounds, in ‟FEBS letters", 1982, CXXXVIII, pp. 33-36.
Rowley, D. A., Halliwell, B., Superoxide-dependent formation of hydroxyl radicals from NADH and NADPH in the presence of iron salts, in ‟FEBS letters", 1982, CXLII, pp. 39-41.
Rowley, D. A., Halliwell, B., Formation of hydroxyl radicals from hydrogen peroxide and iron salts by superoxide- and ascorbate-dependent mechanisms: relevance to the pathology of rheumatoid disease, in ‟Clinical science", 1983, LXIV, pp. 649-653.
Runge, P., Muller, D. P. R., McAllister, J. e altri, Oral vitamin E supplements can prevent the retinopathy of abetalipoproteinaemia, in ‟British journal of ophthalmology", 1986, LXX, pp. 166-173.
Samuni, A., Chevion, M., Czapski, G., Unusual copper-induced sensitization of the biological damage due to superoxide radicals, in ‟Journal of biological chemistry", 1981, CCLVI, pp. 12632-12635.
Saran, M., Bors, W., Oxygen radicals acting as chemical messengers: a hypothesis, in ‟Free radical research communications", 1989, VII, pp. 213-220.
Schraufstatter, I. U., Hinshaw, D. B., Hyslop, P. A., Spragg, R. G., Cochrane, C. G., Oxidant injury of cells. DNA strand-breaks activate polyadenosine diphosphate-ribose polymerase and lead to depletion of nicotinamide adenine dinucleotide,t; in ‟Journal of clinical investigation", 1986, LXXVII, pp. 1312-1320.
Singh, A., Singh, H., Kremers, W., Koroll, G. W., Involvement of singlet oxygen in biochemical systems, in ‟Bulletin européen de physiopathologie respiratoire", 1981, XVII, suppl., pp. 31-41.
Slawinski, J., Galezowski, W., Elbanowski, M., Chemiluminescence in the reaction of cytochrome c with hydrogen peroxide, in ‟Biochimica et biophysica acta", 1981, DCXXXVII, pp. 130-137.
Steinberg, D., Parthasarthy, S., Carew, T. E. e altri, Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity, in ‟New England journal of medicine", 1989, CCCXX, pp. 915-924.
Stocks, J., Gutteridge, J. M. C., Sharp, R. J., Dormandy, T. L., The inhibition of lipid autoxidation by human serum and its relation to serum proteins and alpha-tocopherol, in ‟Clinical science molecular medicine", 1974, XLVII, pp. 223-233.
Storz, G., Tartaglia, L. A., Ames, B. N., Transcriptional regulator of oxidative stress-inducible genes: direct activation by oxidation, in ‟Science", 1990, CCXLVIII, pp. 189-194.
Sullivan, J. L., The iron paradigm of ischemic heart disease, in ‟American heart journal", 1989, CXVII, pp. 1177-1188.
Sunderman, F. W. Jr., Lipid peroxidation as a mechanism of acute nickel toxicity, in ‟Toxicology and environmental chemistry", 1987, XV, pp. 59-69.
Symons, M. R., Gutteridge, J. M. C., Free radicals and iron. Chemistry, biology, and medicine, Oxford-New York 1998.
Tauber, A. I., Borregaard, N., Simons, E., Wright, J., Chronic granulomatous disease: a syndrome of phagocyte oxidase deficiencies, in ‟Medicine", 1983, LXII, pp. 286-309.
Walling, C., The nature of the primary oxidants in oxidations mediated by metal ions, in Proceedings of the 3rd International symposium on oxidases and related redox systems (a cura di T. E. King, H. S. Mason e M. Morrison), Oxford 1982, pp. 85-97.
Weiss, S. J., Oxygen, ischemia and inflammation, in ‟Acta physiologica scandinavica", 1986, DXLVIII, suppl., pp. 9-37.
Williams, M. D., Leigh, J. S., Chance, B., Hydrogen peroxide in human breath and its probable role in spontaneous breath luminescence, in ‟Annals of the New York Academy of Sciences", 1983, XLV, pp. 478-483.
Winterbourn, C. C., Evidence for the production of hydroxyl radicals from the adriamycin semiquinone and H, in ‟FEBS letters", 1981, CXXXVI, pp. 89-94.
Winterbourn, C. C., Hydroxyl radical production in body fluids. Roles of metal ions, ascorbate and superoxide, in ‟Biochemical journal", 1981, CXCVIII, pp. 125-131.
Youdim, M. B. H., Iron in the brain: implications for Parkinson's and Alzheimer's diseases, in ‟Mount Sinai journal of medicine", 1988, LV, pp. 97-101.
Zafiriou, O. C., Is sea water a radical solution?, in ‟Nature", 1987, CCCXXV, pp. 481-482.
Zaman, Z., Roche, S., Fielden, P. e altri, Plasma concentrations of vitamins A and E and carotenoids in Alzheimer's disease, in ‟Age and ageing", 1992, XXI, pp. 91-94.
Zigler, J. S. Jr., Goosey, J. D., Photosensitized oxidation in the ocular lens: evidence for photosensitizers endogenous to the human lens, in ‟Photochemistry and photobiology", 1981, XXXIII, pp. 869-874.
Chimica di Francesco Minisci
sommario: 1. Introduzione e cenni storici. 2. Sorgenti e caratterizzazione dei radicali liberi: a) decomposizione termica; b) fotolisi; c) radiolisi; d) processi redox; e) caratterizzazione spettroscopica. 3. Reattività dei radicali liberi: a) processi radicalici a catena; b) processi a catena redox; c) struttura, reattività e selettività. 4. Applicazioni di reazioni radicaliche: a) reazioni di radicali liberi con olefine e acetilenici; b) sostituzione radicalica aromatica; c) sostituzione radicalica alifatica. 5. Conclusioni. □ Bibliografia.
1. Introduzione e cenni storici
Si definisce radicale libero un atomo o un gruppo di atomi con un elettrone non accoppiato. Un biradicale è una specie con due elettroni non accoppiati. I poliradicali sono rari. Composti importanti della chimica inorganica, come il monossido e il diossido di azoto, sono radicali liberi. L'ossigeno, l'elemento più diffuso sulla Terra, allo stato molecolare è un biradicale. Il grande sviluppo della chimica dei radicali liberi ha riguardato però la chimica organica.
Ci sono alcune date storiche fondamentali nello sviluppo della chimica dei radicali liberi. Il primo evento importante riguarda la scoperta, effettuata da M. Gomberg (v. Triphenylmethyl, ein Fall..., e On the preparation..., 1900), del trifenilmetile: nel tentativo di preparare l'esafeniletano da cloruro di trifenilmetile e argento, egli ottenne un solido bianco che, sciolto in benzene, dava una colorazione gialla; egli spiegò la reazione come un equilibrio in soluzione tra esafeniletano e trifenilmetile:
(C6H5)3C−Cl + Ag → (C6H5)3C• + AgCl
2(C6H5)3C• ⇌ (C6H5)3C−C(C6H5)3.
Questa interpretazione era sostanzialmente corretta, anche se nel 1968 è stato dimostrato che la struttura del dimero è leggermente diversa, ma incontrò grande scetticismo nell'ambiente scientifico dell'epoca, in cui la razionalizzazione della chimica organica si basava sul ‛dogma' del carbonio tetravalente: una specie organica con il carbonio trilegato era considerata alla stregua di un'eresia. Nei decenni successivi si accumularono evidenze sulla reale formazione del trifenilmetile e vennero preparati altri radicali liberi persistenti, che però rappresentavano sempre poco più di una curiosità marginale nell'ambito della chimica organica. Un altro esperimento significativo fu effettuato da F. Paneth (v. Paneth e Hofeditz, 1929): facendo passare una corrente di azoto e idrogeno saturato con vapori di Pb (CH3)4 in un tubo, che veniva scaldato in alcuni punti, si osservava che su quei punti si depositava uno specchio di piombo; questo fenomeno venne correttamente interpretato come la rottura dei legami C−Pb con formazione di Pb metallico e radicali metile:
Pb(CH3)4 → Pb + 4 • CH3.
Il 1937 costituisce una pietra miliare nello sviluppo della chimica dei radicali liberi. D. H. Hey e W. A. Waters (v., 1937) pubblicarono una rassegna memorabile in cui spiegavano mediante meccanismi radicalici una grande varietà di reazioni in soluzione. Nello stesso anno, M. S. Kharasch (v. Kharasch e altri, 1937) razionalizzava l'effetto perossido nell'addizione anti-Markovnikov dell'acido bromidrico alle olefine e P. J. Flory (v., 1937), sviluppava la cinetica della polimerizzazione radicalica dei monomeri vinilici e inoltre introduceva il concetto di trasferimento di catena. Dal dopoguerra in poi è stato un continuo crescendo di scoperte, sfociato, nell'ultimo ventennio, in una vera e propria esplosione di sviluppo della chimica dei radicali liberi nella sintesi organica e in processi biologici di grande rilevanza.
2. Sorgenti e caratterizzazione dei radicali liberi
Tutti i composti organici sono suscettibili di subire rotture omolitiche dei legami covalenti ad alta temperatura (~ 800 °C) generando così radicali liberi. Il cracking puramente termico delle paraffine è un classico esempio di generazione termica di radicali liberi, che danno però origine a un sistema molto complesso di reazioni. Per ottenere processi più controllati e selettivi per via termica occorrono condizioni più blande.
a) Decomposizione termica
È possibile avere una scissione omolitica in condizioni blande per via termica utilizzando sorgenti che presentino energie di legame comprese tra 20 e 40 kcal/mol. Le più importanti, ottenute con perossidi e azoderivati, consentono di generare radicali liberi tra 40 e 150 °C:
b) Fotolisi
La fotolisi è un altro metodo molto generale per ottenere radicali liberi in condizioni blande. L'assorbimento di radiazioni ultraviolette può portare all'eccitazione di elettroni n o π in uno stato di singoletto π*, che può evolversi mediante vari processi, due dei quali portano alla formazione di radicali liberi. Può verificarsi una rottura omolitica di legami relativamente deboli, come negli esempi seguenti:
RO−OR → 2RO•
RO−NO → RO• + NO
RO−Cl → RO• + Cl•
R2N−Cl → R2N• + Cl•
Br−Br → 2Br•
Ar−I → Ar• + I•
R3Sn−SnR3 → 2R3Sn•.
La omolisi fotochimica è più specifica di quella termica, in quanto alla molecola viene fornita energia di valore ben definito. Lo stato π* può trasformarsi in uno stato eccitato di tripletto (intersystem crossing) e quindi agire direttamente come biradicale, come ad esempio il benzofenone:
I vapori di mercurio nello stato eccitato 63P1 possono decomporre gli alcani in fase gassosa per via radicalica:
Hg6 (3P1) + R−H → HgH + R•
HgH → Hg6 (1S0) + H•.
c) Radiolisi
La rottura omolitica dei legami covalenti può essere ottenuta anche mediante radiolisi, utilizzando sorgenti come 60Co, raggi X ed elettroni ad alta energia. Può verificarsi in questi casi l'espulsione di un elettrone con formazione di un radicale catione, che si evolve ulteriormente a radicale neutro; in questo modo, per esempio, l'acqua può essere un'utile sorgente di radicale ossidrile:
H2O → H2O•+ + e
H2O•+ → HO• + H+.
d) Processi redox
Sorgenti radicaliche molto versatili sono quelle che coinvolgono processi di ossidoriduzione con trasferimento monoelettronico, che spesso può consistere anche in un trasferimento di legante. In tal modo, i perossidi reagiscono con vari sali metallici generando radicali all'ossigeno:
RO−OR'+ Mn+ → RO• + R'O- + M(n+1)+ (Mn+ = Cu+, Fe2+, ecc.)
ROOH+M(n+1)+ → ROO• + H+ + Mn+ (Mn+ = Co2+, Ce3+, Mn3+, ecc.).
Radicali alcossilici e fenossilici possono originarsi per ossidazione di alcoli e fenoli,
RO−H+Ag2+ → RO• + H+ + Ag+,
mentre radicali benzilici si ottengono facilmente da alchilbenzeni, e radicali alchilici da acidi carbossilici,

RCOOH + Ag2+ → R• + CO2 + H+ + Ag+;
è stato possibile arrivare alla formazione di radicali amminici da N-aloammine e idrossilammine,
R2N−X + Mn+ → R2N• + MXn+ (X = Cl, Br, OH; Mn+ = Fe2+, Cu+, Ti3+, ecc.).
Composti carbonilici possono essere ridotti a radicali anioni mediante metalli alcalini o alcalino-terrosi:
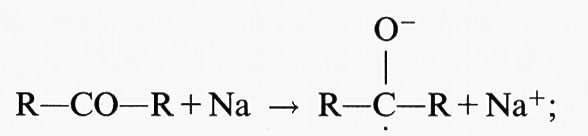
gli alogenoderivati, invece, possono essere ridotti per via radicalica da sali di metalli di transizione a basso stato di ossidazione:
R−Br+Cr2+ → R• + CrBr2+.
I sali di diazonio costituiscono agevoli sorgenti di radicali arilici,
ArN+2 + Cu+ → Ar• + N2 + Cu2+.
L'ossidazione anodica e la riduzione catodica di molte classi di composti organici procedono attraverso radicali liberi. La classica reazione di Kolbe rappresenta uno dei primi esempi di generazione di radicali per via elettrochimica:
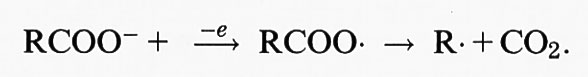
Questi, tuttavia, sono solo alcuni dei numerosi processi di ossidoriduzione che procedono attraverso radicali liberi.
e) Caratterizzazione spettroscopica
Il metodo di gran lunga più utile per determinare e caratterizzare un radicale libero è la risonanza di spin elettronico (ESR o EPR, risonanza paramagnetica elettronica), un metodo di sensibilità molto elevata (fino a 10-8 molare). La spettroscopia ESR si basa sul fatto che un elettrone non accoppiato ha uno spin e un momento magnetico e può pertanto esistere in due stati di spin in assenza di un campo magnetico esterno. In presenza di un campo magnetico applicato, l'elettrone può disporsi in modo parallelo o antiparallelo a questo campo. La differenza di energia è data dall'equazione seguente:
ΔE = gβH,
dove g è il fattore di Landé (v. atomo, vol. I), β è il magnetone di Bohr e H l'intensità del campo magnetico applicato. La frequenza associata con il salto da un livello di energia a un altro è data da:
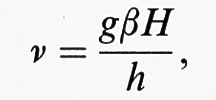
dove h è la costante di Planck.
La caratteristica più utile di uno spettro ESR ai fini della determinazione della struttura di un radicale, nota come hyperfine splitting, ha origine dall'interazione tra l'elettrone non accoppiato e i nuclei magnetici circostanti (1H, 13C, 14N, 18O, ecc.). Così lo spettro di un atomo di idrogeno è costituito da due linee di intensità 1 : 1, poiché il protone con spin 1/2 può disporsi in modo parallelo o antiparallelo al campo magnetico applicato. Lo spettro ESR del radicale metile consta di 4 linee di intensità, nei rapporti 1 : 3 : 3 : 1, dovute all'interazione dell'elettrone non accoppiato con 3 protoni magneticamente equivalenti. Gli spettri ESR sono abbastanza complessi, in quanto l'accoppiamento con i protoni in posizione β è spesso altrettanto importante di quello con i protoni in posizione α: lo spettro del radicale etile, per esempio, consta di 12 linee, in quanto lo spin dell'elettrone non accoppiato è scisso in un quartetto dai 3 protoni del metile, e ognuna di queste linee è scissa a sua volta in 3 linee dai protoni del gruppo metilenico.
I radicali liberi sono spesso molto reattivi ed è sperimentalmente difficile registrare gli spettri ESR; così talvolta si ricorre alla tecnica dello spin trapping, che consiste nel trasformare il radicale labile in uno stabile mediante un'apposita trappola, formata, ad esempio, da un nitrosoderivato o da un nitrone che reagiscono con una grande varietà di radicali originando radicali nitrossidi stabili:
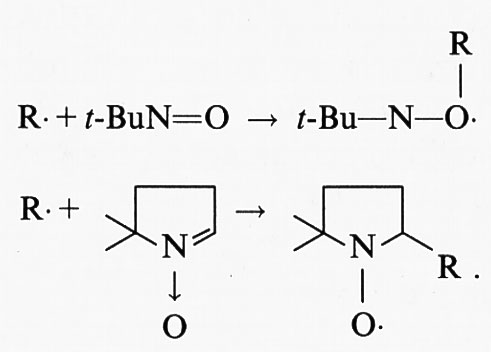
È agevole registrare gli spettri dei nitrossidi e da questi si può spesso risalire alla struttura del radicale iniziale.
Anche le spettroscopie IR, UV, NMR e di massa possono essere impiegate per la caratterizzazione di radicali liberi, ma sono di gran lunga meno efficaci della spettroscopia ESR.
3. Reattività dei radicali liberi
I radicali liberi possono dare origine a una grande varietà di reazioni inter- e intramolecolari, che vanno dall'addizione ai più svariati sistemi insaturi, al distacco di atomi (essenzialmente idrogeno o alogeni), a sostituzioni, ossidazioni, riduzioni, dimerizzazioni, trasposizioni, frammentazioni, ecc. Di particolare interesse sono i processi a catena, che possono raggrupparsi in due ampie categorie.
a) Processi radicalici a catena
I processi radicalici a catena, oltre ad avere grande rilevanza nella sintesi organica, nei processi industriali e nei processi biologici, hanno anche implicazioni ecologiche (relativamente, ad esempio, al buco dell'ozono); le caratteristiche cinetiche che essi hanno in comune verranno illustrate in relazione a uno dei processi di maggior rilievo della chimica: l'autossidazione. Questo processo è legato essenzialmente alla struttura dell'ossigeno molecolare allo stato fondamentale, 3 Σ−g; questo simbolo sta a indicare uno stato di tripletto, cioè due elettroni della molecola di ossigeno non sono accoppiati (si tratta quindi di un biradicale) ma occupano orbitali di antilegame con spins paralleli.
Tutti i processi a catena sono caratterizzati da tre fasi: iniziazione, propagazione, terminazione. Il processo di iniziazione può essere il più disparato, casuale o controllato; molte delle sorgenti radicaliche di cui si è parlato in precedenza possono essere responsabili, casualmente o con opportuna programmazione, della fase di iniziazione, che consiste nel generare una specie radicalica in grado di innescare un processo di propagazione.
Per autossidazione si intende l'ossidazione di composti organici con ossigeno molecolare che ha luogo in condizioni di temperatura relativamente blande (da temperatura ambiente a non oltre i 200 °C).
Nella fase preliminare della reazione la stechiometria riguarda l'inserimento di una molecola di ossigeno in un legame C−H:
R−H+O2 → RO−O−H.
L'idroperossido può essere stabile nelle condizioni operative oppure decomporsi in prodotti ossigenati vari (alcoli, aldeidi, chetoni, acidi carbossilici). Gli atti elementari che caratterizzano il processo globale sono mostrati nello schema 1.

L'impostazione dell'equazione cinetica è semplificata dal fatto che le due ultime interazioni di terminazione dello schema 1 sono trascurabili a causa dell'elevata velocità - praticamente controllata dalla diffusione - con cui i radicali al carbonio reagiscono con l'ossigeno molecolare (la concentrazione stazionaria del radicale R• è pertanto trascurabile rispetto a quella del radicale ROO•). La velocità di formazione dell'idroperossido che si ricava dallo schema 1 è data dall'espressione seguente:

in cui kp è la costante di propagazione.
La concentrazione del radicale ROO• si può ricavare dall'assunto dello stato stazionario,
vi + kp [RH] [ROO•] = k0 [O2] [R•]
k0 [O2] [R•] = kp [RH] [ROO•] + 2 kt [ROO•]2,
dove vi è la velocità di iniziazione, da cui si ricava
[ROO•] = (vi/2kt)1/2.
Di conseguenza l'espressione generale della velocità assume la forma seguente:
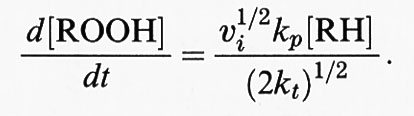
Questa equazione è analoga a quelle che si ricavano nello stesso modo per altri processi radicalici a catena di grande importanza, quali la polimerizzazione radicalica di monomeri vinilici che è alla base della produzione di importanti materiali plastici (polietilene, poliacrilonitrile, polivinilcloruro, polibutadiene, polistirene; v. polimeri, vol. V; v. materiali: Materiali polimerici, vol. VIII), l'alogenazione radicalica, l'addizione radicalica a catena di numerose classi di composti a sistemi insaturi, ecc. La caratteristica peculiare dell'autossidazione consiste nel fatto che il prodotto iniziale della reazione, l'idroperossido, può agire a sua volta da iniziatore per decomposizione termica, fotochimica o redox.

Il processo di autossidazione ha implicazioni importanti in aree di grande rilevanza, come la produzione di prodotti chimici di largo consumo e il deterioramento di sostanze organiche (manufatti, alimenti); esso, inoltre, è responsabile di svariati danni ai tessuti degli organismi viventi, che sono causa di molte patologie.
In alcuni casi, come in quello della produzione di fenolo da cumene o di idroperossido di butile terziario da isobutano, bisogna operare in condizioni tali da evitare, o limitare al massimo, la decomposizione dell'idroperossido inizialmente formatosi; in altri casi (produzione di acido acetico, acido benzoico, acidi ftalici, naftoli, ecc.) conviene invece impiegare la catalisi metallica redox per accelerare i processi di autossidazione, considerando che la velocità di iniziazione vi) compare nell'equazione cinetica generale. La prevenzione dei danni indotti per effetto dell'autossidazione nei composti organici e negli organismi viventi costituisce pertanto una problematica molto importante sia nell'ambito dell'economia industriale che in quello medico-biologico; in questi ultimi anni tale problema ha interessato in misura crescente gli studiosi di radicali liberi che si occupano dello sviluppo di antiossidanti industriali e biologici.
Gli antiossidanti si dividono in due grandi categorie: quelli primari o preventivi, che mirano a distruggere l'idroperossido man mano che si forma senza generare nuovi radicali, oppure a chelare tracce di sali metallici e renderli inerti verso iniziazioni redox, e quelli secondari, che agiscono interrompendo il processo a catena mediante intervento nella fase di propagazione. Tra questi ultimi occupano un posto di rilievo i fenoli, il cui meccanismo di azione si basa essenzialmente su due caratteristiche cinetiche: la prima riguarda la velocità con cui queste sostanze reagiscono verso i radicali perossidici, che attaccano molto più velocemente i fenoli che i legami C−H,
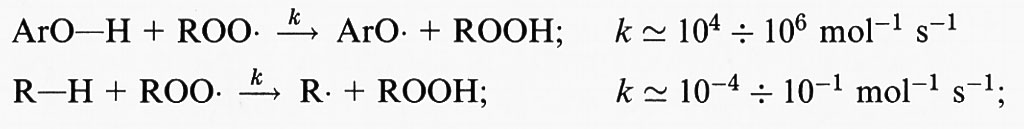
la seconda è legata all'inerzia del radicale fenossilico verso i legami C−H:

La rottura della catena di propagazione è la conseguenza di questo comportamento. Il meccanismo con cui opera il BHT (2,6-di-t-butil-4-metilfenolo), uno dei più importanti antiossidanti industriali, illustra l'attività antiossidante dei fenoli:

Una molecola di antiossidante riesce in questo modo a rompere due catene di propagazione.
Negli organismi viventi gravi danni sono provocati proprio dall'aggressione dei radicali perossidici ai legami C−H dei tessuti. È da tempo noto che i fenoli, come la vitamina E, o strutture analoghe, come la vitamina C e l'acido urico, esercitano un'azione antiossidante in vivo simile a quella menzionata per il BHT. La ricerca medico-biologica tiene sempre maggior conto di questa attività antiossidante non solo attiva contro molti processi patologici quali cirrosi epatica, cardiopatie e tumori, ma che esplica un ruolo rilevante anche nell'invecchiamento (v. radicali liberi: Biologia e patologia, vol. XI).
b) Processi a catena redox
Anche questi processi hanno notevole importanza nella chimica organica e spesso coinvolgono una catalisi metallica ossidoriduttiva, che può essere, in linea generale, quella indicata nello schema 3.

Numerose sintesi radicaliche rientrano in questo schema generale. Per esempio, due delle più classiche reazioni dei sali di diazonio, quella di Sandmeyer (v. schema 4) e di Meerwein (v. schema 5), sono caratterizzate da una catena redox.
La sostituzione radicalica aromatica è spesso caratterizzata da catalisi ossidoriduttiva. Per esempio, l'amminazione dell'anisolo con idrossilammina catalizzata da sali di titanio e l'ammidazione di piridine con formammide e perossido di idrogeno catalizzata da sali di ferro si basano sulle catene redox degli schemi 6 e 7.

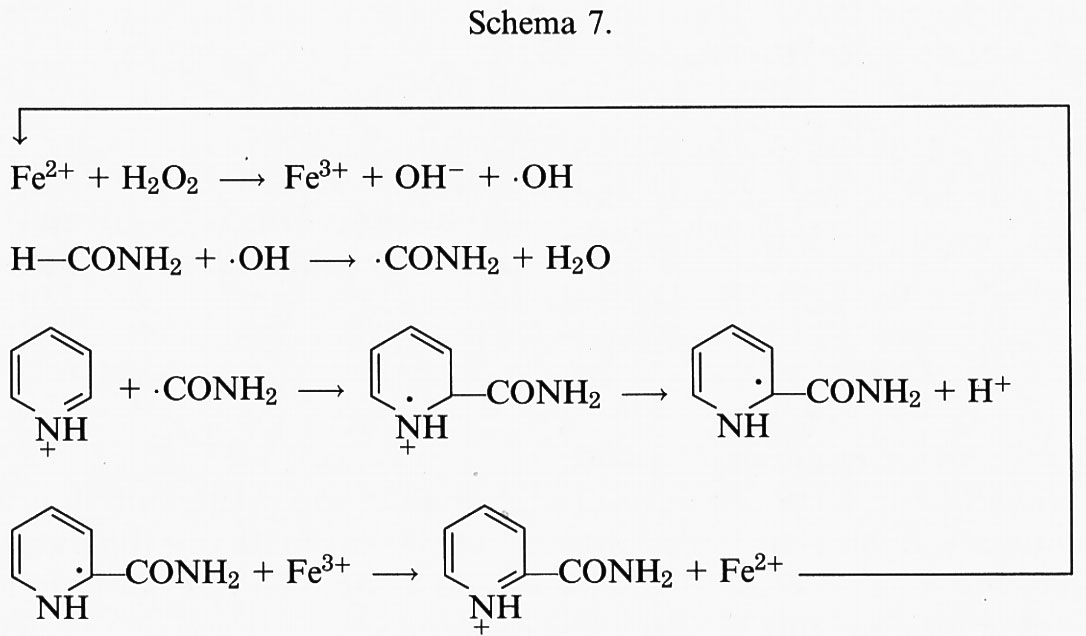
La catalisi redox può talvolta modificare la regioselettività dei processi radicalici a catena: è questo il caso, per esempio, dell'addizione di cloroformio alle olefine, che nel processo radicalico a catena avviene secondo la seguente propagazione:
se la reazione è catalizzata da sali di rame, si hanno invece una differente propagazione redox e differenti prodotti di reazione:
c) Struttura, reattività e selettività.
In condizioni relativamente blande di temperatura, la reattività e la selettività delle reazioni radicaliche in soluzione sono generalmente controllate dalla cinetica, tranne pochi casi di processi reversibili in cui il controllo è termodinamico. A temperature più elevate in fase gassosa, il controllo termodinamico diventa sempre più importante. Comunque, l'entalpia dei processi elementari condiziona spesso in modo determinante l'energia di attivazione e quindi la selettività globale. Un esempio classico è fornito dalla clorurazione e bromurazione di alcani,
R−H + X2 → R−X + HX (X = Cl, Br),
in cui la selettività è determinata dal distacco di un atomo di idrogeno a opera di un atomo di alogeno:
R−H + X• → R• + H−X.
I dati della tab. I mostrano come il fattore entalpico abbia un ruolo molto importante nella bromurazione, in cui il distacco di idrogeno è sempre endotermico e la selettività tra C−H primario, secondario e terziario è molto elevata; nella clorurazione, invece, il distacco di idrogeno è sempre esotermico e la selettività è molto bassa. Nel caso della bromurazione la velocità di reazione cresce di 2.000 volte passando da un C−H primario a un C−H terziario, ma solo di 4,5 volte nel caso della clorurazione. Analogamente, l'effetto entalpico governa la selettività di distacco di idrogeno da parte di due radicali apparentemente simili, RO• e ROO• (v. tab. II):


R−H + RO• → R• + RO−H
R−H + ROO• → R• + ROO−H.
Anche in questo caso, con i radicali alcossilici il processo di distacco di idrogeno è sempre esotermico, la reattività è elevata e la selettività è bassa, mentre con i radicali perossidici il processo è sempre endotermico, la velocità è molto più bassa e la selettività più elevata.
Il fattore entalpico non è però l'unico fattore che governa la selettività nel distacco di idrogeno, come mostra la regioselettività nell'alogenazione dell'1-cloroeptano e dell'esanoato di metile con N-aloammine in ambiente acido,
dove i numeri posti sotto gli atomi di C indicano le velocità relative di alogenazione. In questi casi la selettività tra il gruppo metilenico più lontano dal sostituente polare e il metile è sempre determinata dal fattore entalpico (più elevata energia dei legami C−H primari), mentre la grande differenza di reattività tra i vari gruppi metilenici, che hanno la stessa energia dei legami C−H, è dovuta all'effetto polare induttivo e di campo, per cui reagisce più velocemente il gruppo metilenico più lontano dal sostituente polare. La polarizzazione dei legami C−Cl e C−O si trasmette attraverso i legami σ (effetto induttivo) o attraverso lo spazio (effetto di campo), per cui la disponibilità elettronica dei legami C−O aumenta con la distanza dal gruppo polare. La selettività è determinata dal distacco di un atomo di idrogeno dai legami C−H a opera di radicali amminici protonati secondo il seguente processo a catena:
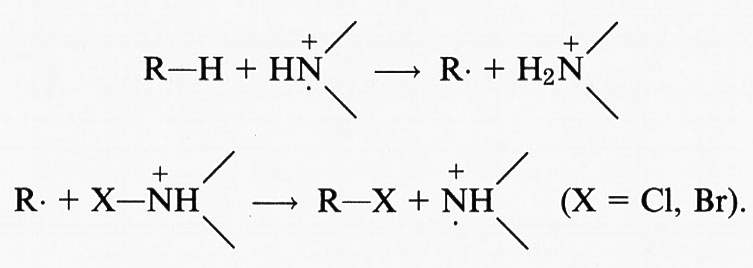
La selettività è identica con cloro e bromo, perché è identica la specie (radicale amminico) che la determina. L'effetto polare si manifesta come un contributo di trasferimento di carica nello stato di transizione,
il che determina un abbassamento dell'energia di attivazione tanto più elevato quanto più alta è la disponibilità elettronica dei legami C−H. Questo tipo di alogenazione, oltre a fornire una elevata regioselettività, è caratterizzato anche da grande chemioselettività, nel senso che l'introduzione di un atomo di alogeno nella catena paraffinica genera una forte disattivazione, per cui è possibile convertire tutto il substrato arrestando il processo alla monosostituzione. Questo comportamento è in netto contrasto con l'alogenazione radicalica tradizionale con cloro e bromo; quest'ultima, infatti, produce un effetto polare molto modesto, e quindi l'introduzione di un atomo di alogeno non modifica sostanzialmente la reattività dei legami C−H della catena paraffinica, per cui col procedere della conversione si formano miscele estremamente complesse di prodotti di polisostituzione in tutte le posizioni disponibili, che fanno perdere ogni interesse sintetico al processo stesso. L'effetto polare ha un ruolo fondamentale nella sostituzione radicalica aromatica ed è il risultato della polarità e polarizzabilità sia del radicale che del substrato con cui reagisce; pertanto, radicali poco polari, come quelli arilici, hanno scarso interesse applicativo nella sostituzione aromatica, in quanto caratterizzati da regio- e chemioselettività molto basse, mentre i radicali elettrofili reagiscono con elevata selettività con anelli aromatici elettron-ricchi e il comportamento dei radicali nucleofili è del tutto opposto. Per esempio, i radicali amminici in ambiente acido reagiscono con elevatissima selettività con benzene o anisolo, mentre non reagiscono del tutto con piridine protonate (v. schema 8).
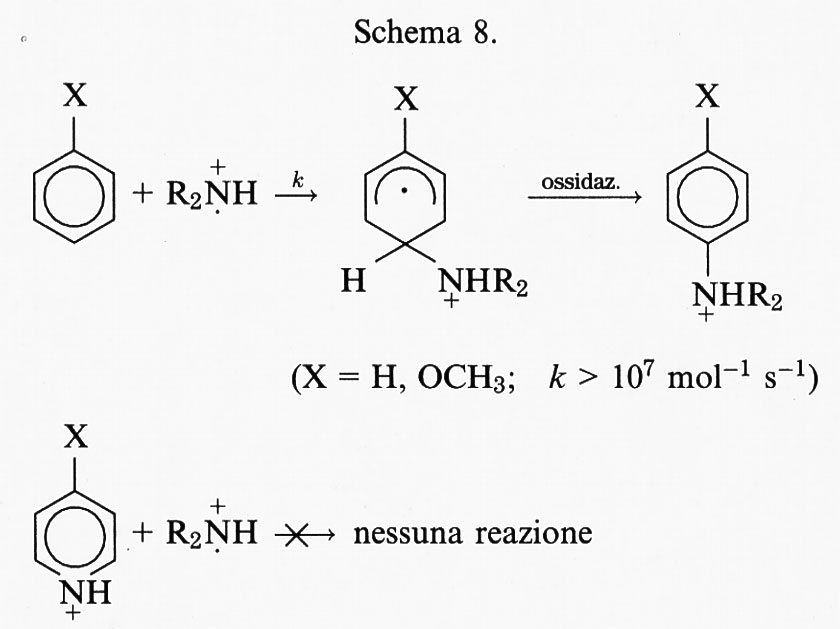
Al contrario, radicali nucleofili (R = t-Bu o MeCO) reagiscono con elevata velocità e selettività con piridine protonate, ma non reagiscono con derivati benzenici (v. schema 9).
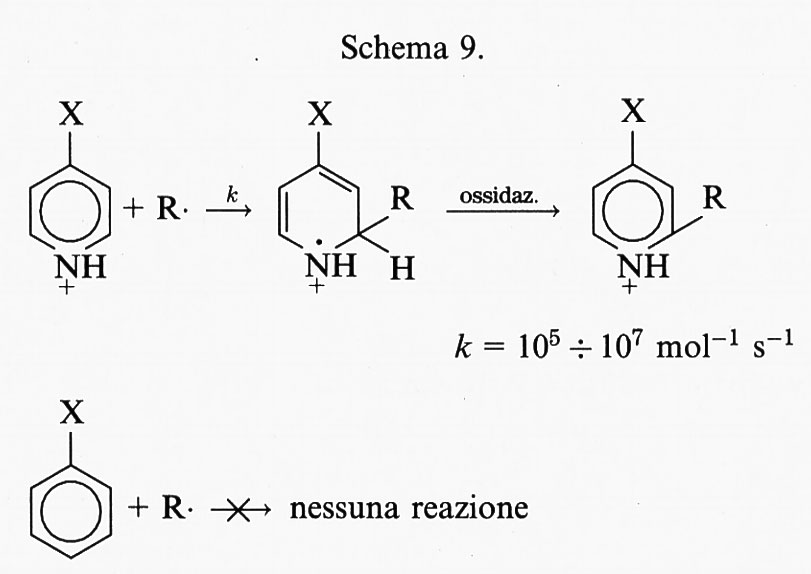
Anche in questi casi l'effetto polare si manifesta come un contributo di trasferimento di carica nello stato di transizione assimilabile alle strutture seguenti:

Più alte sono la disponibilità o la deficienza di elettroni dell'anello aromatico e il carattere elettrofilo o nucleofilo del radicale, più elevate saranno reattività e selettività nella sostituzione.
Un altro fattore importante che influenza la cinetica, e quindi la selettività, delle reazioni radicaliche è quello sterico, al quale si deve in gran parte l'orientamento anti-Markovnikov nell'addizione radicalica alle olefine, cioè l'attacco preferenziale sul carbonio etilenico meno sostituito:
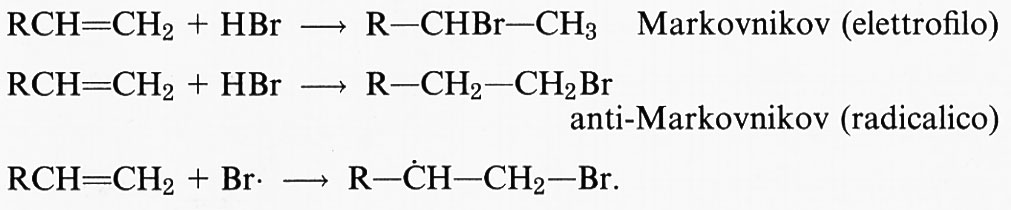
L'effetto sterico influenza non solo l'orientamento, ma anche la reattività (molto minore nel caso delle olefine interne che in quello delle α-olefine) con molti radicali, sempre per la maggior difficoltà sterica di accedere al carbonio olefinico.
Nella sostituzione aromatica, reattività e selettività sono fortemente influenzate dall'ingombro sterico sia dell'anello aromatico che del radicale. Così, per esempio, il radicale dimetilamminico protonato si addiziona con completa selettività nella posizione para dell'acetanilide, senza che venga attaccata la posizione orto che è egualmente attivata per effetto polare (v. schema 10).
Il radicale amminico semplice protonato, •+NH3, non ingombrato stericamente, attacca invece in quantità confrontabili sia la posizione orto che quella para. L'effetto sterico può addirittura cambiare il decorso stesso della reazione, come ad esempio nel caso dei radicali amminici con toluene (v. schema 11).
Quando R è poco ingombrante (ad es., R = metile) l'attacco avviene esclusivamente al nucleo aromatico; se R ha invece un ingombro sterico rilevante (ad es., R = isopropile) si verifica esclusivamente l'attacco al metile del toluene, che ha una richiesta sterica inferiore rispetto all'attacco nucleare. Con gruppi alchilici di ingombro sterico intermedio tra metile e isopropile i due processi possono essere competitivi.
Gli effetti sterici sono particolarmente importanti per quanto riguarda la persistenza dei radicali liberi e la loro capacità di dimerizzare o disproporzionare. Lo scontro tra due radicali alchilici dà origine generalmente a due processi competitivi, dimerizzazione e disproporzionamento, il cui rapporto è determinato essenzialmente dall'ingombro sterico dell'alchile: più alto è quest'ultimo, più prevale il disproporzionamento, che ha minore richiesta sterica:
L'effetto sterico è quello che determina la struttura del dimero del trifenilmetile di Gomberg, che non è l'esafeniletano, come da lui stesso suggerito originariamente, ma un dimero asimmetrico, la cui struttura è stata stabilita molti anni dopo la sua scoperta (v. schema 12).

Anche la tecnica dello spin trapping, di cui si è parlato in precedenza, è legata a effetti sterici. Il nitrosoderivato di t-butile funziona perché genera un radicale nitrossido persistente per effetto sterico, mentre i nitroso-derivati di metile ed etile, non essendo in grado di rendere persistenti i corrispondenti radicali nitrossido, non risultano utili. Anche l'attività antiossidante del BHT, cui si è accennato in precedenza, è in larga misura da mettere in relazione alla stabilità e persistenza del corrispondente radicale fenossilico dovute all'effetto sterico dei gruppi alchilici nelle posizioni orto e para, che ne impedisce la dimerizzazione e permette di raggiungere concentrazioni stazionarie sufficientemente elevate da agire da scavenger (‛spazzino') di radicali perossidici. I radicali fenossilici non sostituiti dimerizzano molto facilmente (v. schema 13) e hanno di conseguenza un'attività antiossidante molto più attenuata.

4. Applicazioni di reazioni radicaliche
I radicali liberi, oltre a reagire per via mono- o bimolecolare tra loro, interagiscono praticamente con tutte le classi di composti organici e possono, pertanto, dare origine a innumerevoli trasformazioni che non è possibile riassumere in breve spazio. Negli ultimi decenni non solo si è assistito a uno sviluppo esplosivo dell'impiego di queste reazioni nella sintesi organica, ma è stata anche riconosciuta la grande importanza delle reazioni radicaliche nei processi biologici. Accenneremo ad alcune applicazioni di reazioni radicaliche a derivati olefinici, acetilenici, aromatici e paraffinici, che rappresentano solo qualche esempio della grande potenzialità di queste reazioni.
a) Reazioni di radicali liberi con olefine e acetilenici
La reattività radicalica delle olefine, con la razionalizzazione dell'addizione anti-Markovnikov, ha rappresentato uno dei punti di partenza della chimica dei radicali liberi, e ha costituito l'area di maggiore impegno e progresso nell'ambito delle reazioni radicaliche; essa è caratterizzata, oltre che dalla polimerizzazione radicalica di monomeri vinilici, da vari processi di tipo generale.
L'addizione radicalica a catena consente di addizionare una grande varietà di gruppi al legame olefinico, come pure a quello acetilenico:

La catena radicalica può essere semplice, come ad esempio nel caso di addizione di acido bromidrico o molti altri derivati:
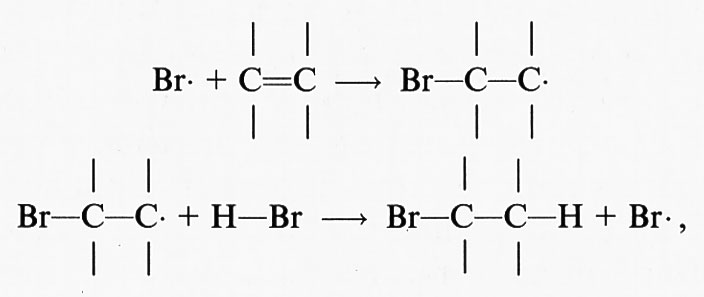
oppure può divenire più complessa attraverso una sequenza selettiva di reazioni radicaliche:

L'addizione può essere costituita da una catena redox, che consente di ottenere prodotti non preparabili mediante semplice addizione radicalica a catena. Una delle reazioni fondamentali delle olefine è infatti l'addizione di molecole di alogeni o pseudoalogeni (Cl−Cl, SCN−NCS): per esempio, la molecola dello pseudoalogeno azidico (N3−N3) non esiste e non si può quindi addizionare al doppio legame, ma si è ovviato a questo inconveniente mediante una catena redox, che costituisce il miglior metodo per ottenere le diazidi e quindi le diammine vicinali:
la stechiometria globale della reazione è data dall'equazione:
Negli ultimi anni ha ricevuto grande attenzione lo studio dell'addizione radicalica intramolecolare finalizzata alla sintesi selettiva di molecole complesse. Tale studio ha permesso di evidenziare le grandi potenzialità di questo metodo, un esempio delle quali è illustrato nello schema 14.

I radicali liberi, oltreché addizionarsi ai doppi legami, possono funzionalizzare le posizioni alliliche delle olefine. La bromurazione allilica con N-bromosuccinimmide è un esempio classico di questo tipo di reazione, nella quale i processi di addizione e sostituzione radicalica sono competitivi, ma la selettività è determinata dalla combinazione di controlli cinetici e termodinamici. Il meccanismo della reazione è illustrato nello schema 15.
L'atomo di bromo può addizionarsi al doppio legame (a) oppure determinare il distacco di un idrogeno allilico (b); il processo di addizione è molto più veloce, ma anche molto più reversibile rispetto al processo di distacco di idrogeno, per cui se la concentrazione di Br2 è elevata si minimizza la reversibilità dell'addizione e si ottiene il dibromoderivato. La funzione della N-bromosuccinimmide è proprio quella di tenere molto bassa la concentrazione stazionaria di Br2, favorendo lo spostamento dell'equilibrio verso la bromurazione allilica. La competizione tra addizione e attacco allilico dipende dalla struttura del radicale; in altri casi, come per esempio con radicali alcossilici, la velocità di strappo di idrogeno allilico è più elevata che l'addizione al doppio legame e quindi si forma preferenzialmente il monoalogenoderivato:
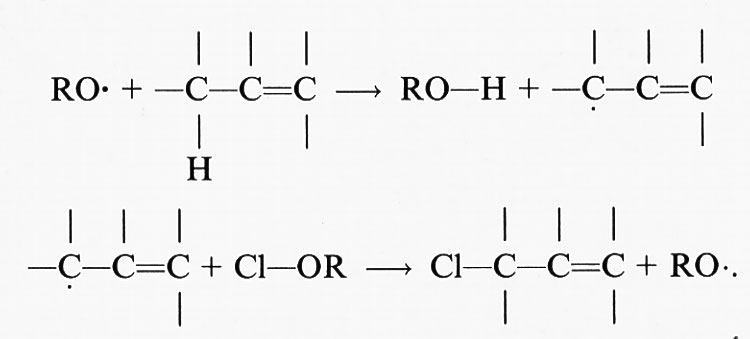
Funzionalizzazioni radicaliche di olefine possono realizzarsi anche mediante processi di trasferimento monoelettronico chimico o elettrochimico. Le olefine elettron-ricche danno facilmente radicali cationi e quelle elettron-povere radicali anioni. Così, la riduzione elettrochimica del nitrile acrilico, la cui prima fase è costituita proprio dalla formazione di un radicale anione,
è alla base del processo industriale di dimerizzazione riduttiva a nitrile adipico (NC−(CH2)4−CN), intermedio base per la produzione del nailon 6,6.
b) Sostituzione radicalica aromatica
Dal punto di vista sintetico, per lungo tempo la sostituzione radicalica aromatica è stata considerata un aspetto marginale della reattività aromatica; tale considerazione era basata principalmente sul fatto che lo studio si era concentrato sulla arilazione radicalica aromatica, una reazione caratterizzata da regio- e chemioselettività molto basse. Questo stato di cose è chiaramente illustrato dai fattori parziali di velocità nelle posizioni orto, meta e para di derivati benzenici sostituiti (v. tab. III).
La trasposizione ingiustificata di questo comportamento a tutte le sostituzioni radicaliche ha rappresentato il principale ostacolo allo sviluppo di queste reazioni. La consapevolezza che gli effetti polari nelle sostituzioni radicaliche possono essere di entità non inferiore a quelli delle reazioni ioniche ha portato negli ultimi trent'anni allo sviluppo di sostituzioni radicaliche aromatiche di elevata selettività e di grande interesse sintetico. L'amminazione radicalica aromatica con N-cloroammine è una reazione caratterizzata da elevatissime regio- e chemioselettività, confrontabili con le più selettive sostituzioni elettrofile aromatiche. La stechiometria della reazione è indicata dall'equazione seguente:

il meccanismo di reazione è costituito da un processo a catena di addizione-eliminazione (v. schema 16).
Radicali del carbonio contenenti gruppi elettron-attrattori direttamente legati al centro radicalico hanno carattere elettrofilo e possono essere utilizzati per la sostituzione di aromatici omociclici ed eterociclici elettron-ricchi. I radicali del carbonio, in assenza di sostituenti elettron-attrattori legati al carbonio radicalico, hanno invece carattere nucleofilo; essi reagiscono con elevata selettività - una caratteristica che li rende di grande interesse sintetico - con aromatici fortemente elettron-deficienti, in particolare basi eteroaromatiche protonate, e riproducono i numerosi aspetti della sostituzione aromatica di Friedel-Crafts, ma con opposta reattività e selettività. Il meccanismo generale della reazione è simile a quello illustrato nello schema 7. Il parallelismo con le sostituzioni di Friedel-Crafts va posto in relazione al fatto che più stabile è un carbocatione, più nucleofilo sarà il corrispondente radicale, e di conseguenza tutte le specie elettrofile utili nelle sostituzioni di Friedel-Crafts possono essere utilizzate, allo stato di radicali liberi, per la sostituzione selettiva di basi eteroaromatiche. È stata utilizzata una grande varietà di sorgenti radicaliche, che interessano le più importanti classi di composti organici.
Tutte le basi eteroaromatiche - inclusi composti di grande interesse biologico, quali possono essere ad esempio nucleosidi, pteridine, basi puriniche, ecc. - sono suscettibili di sostituzioni selettive. La grande influenza degli effetti polari, che poi sono la causa prima dell'elevato interesse sintetico, è mostrata dai dati della tab. IV che, al confronto con quelli riportati nella tab. III, rivelano l'enorme differenza di sensibilità agli effetti polari tra radicali arilici e alchilici.

c) Sostituzione radicalica alifatica
Le paraffine sono così chiamate a causa della loro bassa reattività (parum affinis), che in realtà riguarda le reazioni ioniche ed è dovuta alla polarizzazione molto piccola dei legami C−H e C−C. Le paraffine possono invece essere molto reattive verso i radicali liberi. Così una miscela di paraffina e cloro reagisce in pochi secondi a temperatura ambiente, in presenza di iniziazione radicalica chimica o fisica, mediante un processo a catena (la lunghezza cinetica della catena è di circa 106), mentre una miscela di paraffina e fluoro reagisce in modo esplosivo. La metodologia di gran lunga più versatile per la funzionalizzazione di paraffine è pertanto quella radicalica. Una grande varietà di radicali liberi (del carbonio, dell'ossigeno, dell'azoto, atomi di alogeno, ecc.) è in grado di strappare un atomo di idrogeno da legami C−H generando radicali alchilici:
R−H + •X → R• + H−X.
Abbiamo già menzionato (v. cap. 3, §§ a, c) i processi di autossidazione e di alogenazione e quali siano i fattori che governano reattività e selettività. La funzionalizzazione selettiva delle paraffine rimane tuttavia una delle sfide di maggior rilevanza in campo chimico, a causa delle differenze di energia - spesso molto piccole - tra i legami C−H: è una sfida che la natura in qualche caso è riuscita a superare mediante la catalisi enzimatica, come per esempio in quello delle reazioni catalizzate da monossigenasi, basate sul citocromo P-450, che è largamente diffuso in tutti gli organismi viventi, dai Batteri ai Mammiferi, uomo incluso. La tendenza della ricerca in questo campo è quella di riuscire a mimare la natura: ci si propone, cioè, di arrivare a sintetizzare catalizzatori che presentino caratteristiche analoghe a quelle degli enzimi.
5. Conclusioni
Le reazioni radicaliche sono state per lungo tempo dominio pressoché incontrastato della chimica fisica e dell'industria chimica di base (polimerizzazione di monomeri vinilici, ossidazioni con ossigeno molecolare, clorurazione di idrocarburi, ecc.), dove l'uso di molecole semplici e la possibilità di conversioni parziali, senza i notevoli inconvenienti rappresentati dalla separazione dei prodotti, rende meno drammatici i problemi di regio- e chemioselettività. Le reazioni radicaliche, come sinonimo di processi poco selettivi, erano considerate invece di scarso interesse nell'industria della chimica fine e nella sintesi di composti sofisticati o in processi biologici, dove una elevata selettività è condizione pregiudiziale per il successo. Negli ultimi 25 anni si è assistito a uno sviluppo davvero esplosivo nelle applicazioni di reazioni radicaliche a sintesi selettive e nello stesso tempo è stata riconosciuta la grande importanza delle reazioni radicaliche nei processi biologici e nel metabolismo dei farmaci.
La ricerca è in pieno fermento e coinvolge un numero sempre più ampio di ricercatori, per cui si possono prevedere ulteriori, interessanti sviluppi tanto nella comprensione dei fenomeni quanto nelle applicazioni.
BIBLIOGRAFIA
Barton, D. H. R., Half a century of free radical chemistry, Cambridge 1993.
Chanon, M., Juilliard, M., Poite, J.-C. (a cura di), Paramagnetic organometallic species in activation/selectivity, catalysis, Dordrecht 1989.
Flory, P. J., The mechanism of vinyl polymerization, in ‟Journal of the American Chemical Society", 1937, LIX, pp. 241-253.
Giese, B., Radicals in organic synthesis: formation of carbon-carbon bonds, Oxford 1986.
Gomberg, M., On the preparation of triphenylchloromethane, in ‟Journal of the American Chemical Society", 1900, XXII, pp. 757-771.
Gomberg, M., Triphenylmethyl, ein Fall von dreiwerthigem Kohlenstoff, in ‟Berichte der Deutschen Chemischen Gesellschaft", 1900, XXXIII, pp. 3150-3163.
Hey, D. H., Waters, W. A., Some organic reactions involving the occurrence of free radicals in solution, in ‟Chemical reviews", 1937, XXI, pp. 169-208.
Huyser, E. S. (a cura di), Methods in free-radical chemistry, voll. I-IV, New York-Basel 1969-1973.
Huyser, E. S., Free-radical chain reactions, New York 1970.
Kharasch, M. S., Engelmann, H., Mayo, F. R., The peroxid effect in the addition of reagents to unsaturated compounds. XV. The addition of hydrogen bromide to 1- and 2- bromo- and chloropropenes, in ‟Journal of organic chemistry", 1937, n. 2, pp. 288-302.
Kochi, J. K. (a cura di), Free radicals, 2 voll., New York-Toronto 1973.
Minisci, F., Recent aspects of homolytic aromatic substitutions, in ‟Topics in current chemistry", 1976, LXII, pp. 1-48.
Minisci, F. (a cura di), Free radicals in synthesis and biology, Dordrecht 1989.
Nonhebel, D. C., Walton, J. C., Free-radical chemistry: structure and mechanism, Cambridge 1974.
Paneth, F., Hofeditz, W., Über die Darstellung von freiem Methyl, in ‟Berichte der Deutschen Chemischen Gesellschaft" (Abteilung B), 1929, LXII, pp. 1335-1347.
Pryor, W. A., Free radicals, New York 1966.
Viehe, H. G., Janousek, Z., Merényi, R. (a cura di), Substituent effects in radical chemistry, Dordrecht 1986.
Walling, C., Free radicals in solution, New York 1957.
Williams, G. H. (a cura di), Advances in free-radical chemistry, 6 voll., New York 1965-1980.