
Reazioni chimiche
Reazioni chimiche
di Gabriello Illuminati
Reazioni chimiche
sommario: 1. Introduzione e cenni storici. 2. Perché avviene una reazione chimica. 3. Come avviene una reazione chimica. 4. Metodi di studio: a) metodi non cinetici; b) metodi cinetici; c) intermedi attivi delle reazioni chimiche. 5. Reazioni dei composti del carbonio (reazioni organiche): a) sostituzioni nucleofile al carbonio saturo; b) reazioni di β-eliminazione; c) trasposizioni dei carbocationi; d) idrolisi basica degli esteri; e) addizioni elettrofile; f) reazioni radicaliche. 6. Reazioni dei composti degli elementi diversi dal carbonio: a) sostituzioni tetraedriche; b) sostituzioni nei complessi tetracoordinati piani; c) sostituzioni presso centri a struttura ottaedrica. 7. Considerazioni e prospettive sullo stato delle conoscenze delle reazioni chimiche. □ Bibliografia.
1. Introduzione e cenni storici
I problemi della chimica gravitano intorno a due argomenti di fondo: la struttura delle molecole e le loro reazioni. La struttura delle molecole si riferisce alle caratteristiche di composizione e di forma che si conservano entro un intervallo di tempo sufficientemente grande da permetterne l'osservazione sotto certe condizioni sperimentali. Ma le molecole, a seconda della loro natura e delle condizioni in cui si trovano, possono subire modifiche che sono generalmente associate a scambi di energia con le altre molecole o, più in generale, con l'ambiente circostante. Queste modifiche possono essere di entità molto variabile. In linea di principio ogni modifica, sia pur lieve (per es. un passaggio conformazionale attraverso una rotazione intramolecolare), potrebbe essere considerata una reazione chimica, ma comunemente questa espressione si riferisce alle trasformazioni che comportano la rottura e/o la formazione di legami chimici principali, la cui energia è dell'ordine di 50 kcal/mole o più. Le reazioni comportano quindi trasformazioni più o meno profonde delle molecole; di tali trasformazioni interessano comunemente quelle che conducono da certe specie molecolari ad altre di diversa composizione e struttura. A ogni reazione chimica corrisponde un'equazione stechiometrica che, rispecchiando la legge della conservazione della massa (di Lavoisier), contiene la prima essenziale informazione sperimentale di cui si deve tener conto per ogni ulteriore approfondimento. Per es., per la formazione di ammoniaca
3H2 + N2 = 2NH3
l'equazione stechiometrica registra il fatto sperimentale che due moli di ammoniaca si formano da tre moli di idrogeno e una di azoto; per la combustione completa del glucosio
C6H12O6 + 6O2 = 6CO2 + 6H2O
ci dice quanto ossigeno viene globalmente consumato per la conversione completa di una mole di carboidrato a diossido di carbonio e acqua.
Ma l'equazione stechiometrica è ben lungi dall'esaurire la descrizione delle caratteristiche di una reazione chimica. I quesiti fondamentali che riguardano una trasformazione chimica sono ‛perché avviene' e ‛come avviene'. Questi quesiti hanno sempre appassionato lo scienziato fin dagli albori della chimica, ma solo negli ultimi decenni si sono avute risposte attendibili alla seconda domanda, cioè sulla natura delle reazioni chimiche, cui sarà dedicato questo articolo. È tuttavia opportuno un breve cenno sull'evoluzione del pensiero chimico riguardo alle reazioni chimiche in generale (v. Solov'ev, 1976).
La risposta al quesito sul perché avvengano le reazioni chimiche è venuta a conclusione di un processo evolutivo del concetto di affinità chimica durato oltre duecento anni. A Newton (1704) si deve la suggestiva ipotesi che la combinazione chimica delle sostanze è l'effetto di una forza che agisce nell'ambito dei sistemi materiali in modo analogo a quella di gravitazione universale. Sebbene a noi questa possa sembrare un'ipotesi meccanica e ovviamente generica, essa contiene la geniale intuizione che le reazioni che decorrono spontaneamente in determinate condizioni lo fanno con la stessa fatalità della caduta dei gravi e adombra, in tal senso, la moderna condizione dell'energia libera (ΔG 〈 0) per i processi spontanei.
All'affinità chimica è stata sempre rivolta grande attenzione da illustri scienziati, tra cui A.-L. Lavoisier e P.-S. de Laplace nel Settecento. Un'importante tappa fu segnata da G. I. Hess (1840), che individuò nel calore di reazione una misura dell'affinità. L'inadeguatezza di questa correlazione apparve manifesta in seguito, con H. L. F. von Helmholtz (1882) e con J. H. van't Hoff (1884), quando si riconobbe che non il calore di reazione ma l'energia libera (J. W. Gibbs, 1875) fornisce una misura dell'affinità delle sostanze reagenti. Il chiarimento definitivo sull'importanza e sul ruolo dell'energia libera venne dal consolidamento della termodinamica, la cui base si completò con il 3° principio per merito di W. H. Nernst (1906) e di M. Planck (1912) (v. Lewis e Randall, 1923; v. Glasstone, 1947). Giungiamo in tal modo quasi ai giorni nostri e non c'è da sorprendersi se lo sviluppo della chimica è avvenuto così rapidamente solo in tempi recenti. Accanto all'avvento della termodinamica, altri processi di maturazione del pensiero chimico furono decisivi nel determinare l'odierno sviluppo delle conoscenze dei processi dinamici in chimica, primi fra tutti i progressi sulla struttura delle molecole.
Naturalmente, per rispondere al quesito ‛come avviene' una reazione chimica, è essenziale riconoscere l'importanza del parametro tempo; in altre parole, è essenziale lo sviluppo della cinetica chimica e della catalisi. Può sorprendere come questa branca della chimica abbia anch'essa radici piuttosto lontane nel tempo. Di cinetica chimica si occuparono acutamente A. W. Williamson (1851), L. F. Wilhelmy (1850), P.-E.-M. Berthelot e L. Péan de Saint-Gilles (1862-1863). Van't Hoff diede un contributo decisivo all'argomento (1884) non solo formulando le leggi fondamentali della cinetica chimica, ma anche introducendo il concetto di molecolarità, di cui riconobbe il significato in termini di meccanismo di reazione. Qualche tempo dopo S. A. Arrhenius (1889) scopriva la relazione empirica tra velocità di reazione e temperatura. Di catalisi si occuparono, tra gli altri, J. J. Berzelius e J. von Liebig nella prima metà del secolo scorso, ma solo C. W. W. Ostwald (1909) fornì una prima corretta interpretazione del ruolo di un catalizzatore come specie chimica capace di influire sulla velocità di reazione.
Dalla fine del secolo scorso fino agli anni venti non sono mancati studi cinetici al fine di scoprire i meccanismi di reazione, ma l'impulso maggiore allo studio dei fondamenti delle reazioni chimiche si è avuto in seguito allo sviluppo delle teorie delle reazioni chimiche, quella delle collisioni e, soprattutto, quella dello stato di transizione (v. Glasstone e altri, 1941). Una nuova generazione di chimici appariva all'orizzonte, stimolata dal lavoro di alcuni pionieri, tra i quali C. N. Hinshelwood, C. K. Ingold, E. A. Moelwyn-Hughes e L. P. Hammett. Questi chimici hanno dischiuso un immenso campo interpretativo dei fenomeni dinamici, favorendo molti degli attuali successi della chimica e dei suoi sviluppi verso i grandi problemi interdisciplinari che sono oggi al centro dell'attenzione delle varie scienze.
2. Perché avviene una reazione chimica
Le reazioni chimiche avvengono spontaneamente solo a condizione che la trasformazione comporti una diminuzione dell'energia libera del sistema materiale che vi prende parte. L'energia libera (G) si compone di due parti, l'entalpia (H) e l'entropia (S), e soddisfa l'equazione
ΔG = ΔH − T ΔS.
Sebbene il contributo del termine entropico, T ΔS, spesso sia modesto e la reazione sia dominata da una diminuzione dell'entalpia (reazioni esotermiche), esistono casi in cui il termine entropico è determinante perché la reazione avvenga spontaneamente. Ciò che conta è che sia ΔG 〈 0, qualunque sia la combinazione algebrica dei due termini. Le reazioni avvengono perché perdono energia potenziale, come fanno gli oggetti quando cadono e le acque che scorrono nel letto dei fiumi. È certo possibile sollevare un oggetto o fare tornare indietro una reazione chimica, ma in tali casi occorre fornire energia potenziale all'oggetto o al sistema chimico.
La componente entalpica tiene conto dei trasferimenti di energia legati alla rottura e/o alla formazione dei legami chimici, ai fenomeni associativi o dissociativi tra molecole, agli effetti conformazionali e agli effetti di repulsione sterica. La componente entropica riflette invece lo stato di disordine molecolare e tende ad aumentare quando il disordine aumenta: risente delle variazioni di stato di aggregazione, delle variazioni di numero di molecole tra reagenti e prodotti, della riorganizzazione o meno delle molecole del solvente. Attraverso il termine entropico, T ΔS, la trasformazione risente infine della temperatura, al crescere della quale essa può anche subire un'inversione di senso.
3. Come avviene una reazione chimica
Anche se una reazione può avvenire in modo spontaneo, altre condizioni debbono essere soddisfatte perché essa proceda entro un tempo sufficientemente breve da essere osservabile. Una miscela di metano e ossigeno si conserva per un tempo praticamente indeterminato a temperatura ambiente anche se la combustione dell'idrocarburo va nel senso della reazione spontanea
CH4 + 2O2 → CO2 + 2H2O.
Tali condizioni riguardano il modo in cui la reazione avviene, cioè il ‛meccanismo di reazione.
Il tempo di reazione è una variabile essenziale delle trasformazioni chimiche ed è la spia del meccanismo di reazione. Nel corso di una reazione chimica avviene una ridistribuzione degli atomi di una o più specie molecolari mediante la rottura e/o la formazione di legami chimici. Una idea immediata di che cosa s'intenda per meccanismo si può ottenere immaginando di assistere, come in una sequenza cinematografica, al movimento delle molecole, ai loro incontri, alla rottura dei legami, al trasferimento di frammenti molecolari, al comporsi di nuove specie molecolari. È nella particolare sequenza di questi eventi che il sistema trova il percorso energeticamente più favorevole perché la trasformazione avvenga. In ogni caso occorre aggiungere energia perché la reazione abbia luogo. Secondo la teoria delle velocità assolute di reazione (v. Glasstone e altri, 1941), il sistema dei reagenti si trasforma nei prodotti passando attraverso una speciale condizione critica detta ‛stato di transizione', nella quale l'energia libera del sistema raggiunge un massimo (v. fig. 1). Le molecole che partecipano allo stato di transizione costituiscono il ‛complesso attivato', che non è un vero composto chimico poiché ha una vita media comparabile ai periodi di vibrazione dei legami chimici. La differenza di energia libera tra complesso attivato e sistema dei reagenti, detta ‛energia libera di attivazione', ΔG≠ fornisce una misura della reattività chimica. Quanto più alto è i ΔG≠ tanto più lenta è la reazione. La teoria prevede che anche l'energia libera di attivazione abbia una componente entalpica e una entropica secondo l'equazione
ΔG≠ = ΔH≠ − T ΔS≠,
nella quale i parametri di attivazione indicano differenze tra lo stato di transizione e i reagenti.
Il caso della fig. 1 è quello di una trasformazione a un solo stadio. Quando la reazione avviene in più stadi vi è uno stato di transizione per ciascuno di essi. Il passaggio da uno stadio all'altro è caratterizzato da un prodotto intermedio cui corrisponde un minimo nel diagramma dell'energia libera.
Le condizioni sperimentali, quali la natura dei reagenti, del solvente e di altre sostanze aggiunte, possono avere effetti notevoli sull'energia libera di attivazione, così da provocare abbassamenti di ΔG≠ fino a varie kcal/mole, che corrispondono a incrementi di velocità di reazione di varie potenze di 10. Lo studio della velocità di reazione (cinetica chimica) è alla base dello studio dei meccanismi di reazione.
Una conoscenza soddisfacente del meccanismo di una reazione chimica comporta l'individuazione degli eventuali prodotti intermedi e un'ipotesi sufficientemente accurata sulla configurazione del complesso attivato di ciascuno stadio. Le caratteristiche generali di meccanismo sono oggi note per molte reazioni chimiche, ma la comprensione dettagliata di esso è impresa assai ardua. Le nostre conoscenze nel campo vengono continuamente aggiornate man mano che nuovi dati sperimentali sono raccolti dai ricercatori. Un meccanismo attendibile deve poter spiegare tutti i fatti sperimentali noti, che spesso sono compatibili con più varianti diverse.
L'obiettivo di questi studi non è meramente teorico, ma si propone di individuare regole generali del comportamento chimico, che permettano di guidare il chimico alla ricerca di nuove reazioni, di migliorare i metodi di sintesi, di produrre prodotti industriali a minor costo o di migliori prestazioni, di approfondire le conoscenze nel campo dei processi biologici. E oggetto di questi studi anche la catalisi (v. catalisi eterogenea; v. catalisi omogenea), un fenomeno che oggi possiede un significato ben preciso in termini di meccanismo di reazione.
4. Metodi di studio
In questo capitolo consideriamo gli aspetti generali dello studio delle reazioni chimiche dal punto di vista delle moderne metodologie in uso per chiarire i meccanismi di reazione. Poiché le reazioni organiche, per la loro adattabilità a ricerche sistematiche, hanno contribuito in modo rilevante allo sviluppo della parte dinamica della chimica, per comodità ricorreremo di frequente a casi tipici di reazioni di questo gruppo senza tuttavia voler implicare alcuna delimitazione di confini precisi tra sistemi chimici, ma, al contrario, intendendo che gli stessi concetti generali si possono applicare nello studio di tutte le reazioni chimiche. Ciò sarà evidente nei successivi capitoli, quando si illustreranno reazioni tipiche dei diversi elementi. Naturalmente, dalle proprietà delle sostanze chimiche in gioco dipenderà il ricorso a determinate tecniche di lavoro anziché ad altre. Faremo riferimento a reazioni omogenee soprattutto in soluzione, cioè a condizioni di uso molto generale nelle sintesi chimiche.
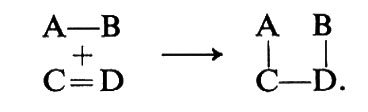
Le molecole reagiscono essenzialmente in tre modi dal punto di vista della rottura e/o della formazione dei legami chimici. Se un legame si rompe in modo che entrambi gli elettroni passino a uno dei frammenti risultanti,
A:B → A: + B, (1)
e se un legame si forma in modo che i due elettroni provengano da uno stesso atomo,
A: + C → A:C. (2)
la reazione si chiama ‛eterolitica' o ‛ionica', perché spesso, ma non sempre, comporta intermedi ionici.
Se la trasformazione è il risultato dell'interazione tra due diverse sostanze, prende il nome di ‛substrato' la sostanza il cui comportamento si intende studiare, e di ‛reagente' la sostanza con cui il substrato reagisce. Se il reagente interviene nella formazione dei legami chimici (per es. A nell'eq. 2) mettendo a disposizione i due elettroni, prende il nome di ‛nucleofilo'; viceversa, se accetta detti elettroni (per es. C nell'eq. 2), si chiama ‛elettrofilo'.
Un secondo modo di reagire delle molecole consiste nella rottura omolitica dei legami, per la quale ciascun elettrone si associa a un frammento risultante da tale rottura:
A:B → A• +B•. (3)
In tal modo si generano radicali liberi, cioè specie fornite di elettroni non condivisi. Le reazioni ‛omolitiche' o ‛radicaliche' sono appunto quelle nelle quali i legami si rompono in modo omolitico e si formano per combinazione di radicali liberi. I reagenti sono in questo caso radicali o precursori di radicali.
Infine esistono reazioni, dette ‛pericicliche', nelle quali non vi sono intermedi, siano essi ioni o radicali liberi, e gli elettroni si ridistribuiscono muovendosi lungo un anello di atomi:
a) Metodi non cinetici
Per studiare il meccanismo di una reazione chimica non si può prescindere dalla conoscenza completa dei prodotti di reazione, date le sostanze chimiche di partenza e le condizioni di esperienza. Ciò sottintende una parte importante del lavoro sperimentale, che comprende l'individuazione delle equazioni chimiche atte a descrivere globalmente la trasformazione in esame e l'identificazione di tutti i prodotti, alcuni dei quali, anche se presenti in piccole quantità, danno utili indizi sul percorso della trasformazione. Per es., qualsiasi ipotesi di meccanismo per la reazione fotocatalizzata
CH4 + Cl2 → CH3Cl + HCl
che fosse incapace di spiegare la presenza di piccole quantità di etano tra i prodotti sarebbe errata.
Molte reazioni avvengono in due o più stadi successivi e passano attraverso la formazione di prodotti intermedi che, essendo spesso molto labili, subiscono ulteriori trasformazioni verso i prodotti finali della reazione.
Una parte importante dello studio consiste nello stabilire se e quali prodotti intermedi si formano durante la reazione. Può accadere che l'intermedio si accumuli in una certa misura nel corso della reazione e prima che questa abbia termine. Se s'interrompe la reazione e/o si cerca di farla avvenire in condizioni più moderate, si può favorire tale accumulo al punto da poterlo isolare, ovvero da poterlo ‛intercettare' nella miscela nella quale si forma attraverso qualche sua proprietà spettrale (spettri UV, vis., IR, RMN, ecc.). Per es., nella conversione di un'ammide ad ammina per opera di ipobromito in ambiente basico
RCONH2 + BrO- → RNH2 + CO2 + Br-
è possibile isolare una N-bromoammide, RCONHBr. Se si tratta questo composto nelle stesse condizioni sperimentali, si ottengono gli stessi prodotti finali di reazione in un tempo non superiore a quello richiesto dalla reazione in esame. Tale risultato rende plausibile che la N-bromoammide sia un intermedio della reazione.
Se un composto intermedio non può essere isolato come tale, si può ‛catturare' come derivato per mezzo di qualche reattivo specifico capace di consumarlo rapidamente e trasformarlo in un derivato isolabile. Per es., in certe reazioni nucleofile di composti aromatici, si forma un intermedio labile detto benzino, la cui presenza è stata dimostrata per cattura con un diene in presenza del quale il benzino forma prontamente un addotto caratteristico e stabile.
Informazioni univoche sui particolari atomi che prendono parte a una reazione si possono ottenere marcando le molecole con isotopi. Per es., nella trasposizione di Claisen, dall'analisi dei prodotti non è possibile dedurre se nella migrazione del gruppo allilico si lega all'anello aromatico il carbonio a o il carbonio c (eq. 4).
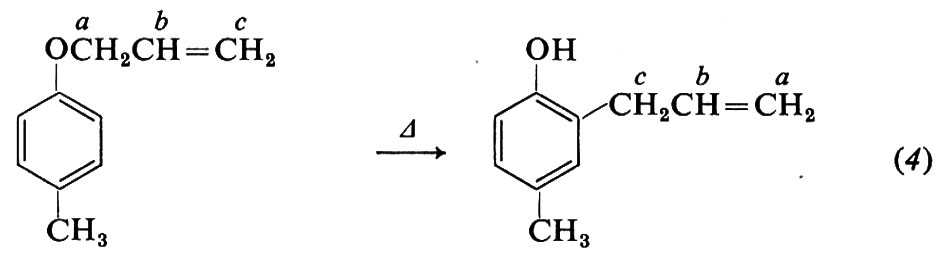
Ma se si parte da un etere allilico marcato nel carbonio c con l'isotopo radioattivo 14C e si ricerca la posizione di tale atomo nel prodotto con misure di radioattività su opportuni prodotti di decomposizione, si trova che nella migrazione della catena allilica è il carbonio c che forma il legame con il carbonio aromatico in posizione orto all'ossigeno. In altri problemi si può fare anche uso di isotopi non radioattivi, come l'ossigeno 18O, la cui presenza nella molecola può essere individuata mediante misure di spettrometria di massa.
Un metodo molto valido per lo studio delle reazioni chimiche è quello stereochimico, che consiste nel seguire le eventuali variazioni di configurazione degli atomi nel corso della reazione. Per es., se un dato atomo di carbonio del substrato è asimmetrico ed è sede di una trasformazione chimica, nel prodotto esso si può ritrovare con la configurazione invertita, racemizzata o conservata. Questi risultati hanno implicazioni significative sul percorso stereochimico della reazione e, in particolare, sulla direzione di attacco del reagente e sulla configurazione del complesso attivato. Essi si sono rivelati di grande utilità nello studio di molte reazioni organiche ed elemento-organiche. Tecniche stereochimiche si possono applicare anche allo studio di reazioni fra composti inorganici dove, per esempio, la sede della reazione può essere un metallo di transizione tetracoordinato (a configurazione piana o tetraedrica) o esacoordinato (a configurazione ottaedrica) e può subire cambiamenti di configurazione.
b) Metodi cinetici
Accanto ai metodi di studio non cinetici finora citati, ne esistono altri basati sul comportamento cinetico che, come già si è detto, dipende direttamente dal meccanismo e, pertanto, fornisce uno strumento essenziale per lo studio delle reazioni chimiche (v. Frost e Pearson, 1961). Lo studio cinetico consiste nel determinare la velocità di reazione a una o più temperature e nell'individuare le leggi che la governano per la reazione in esame in funzione delle variabili sperimentali (concentrazione, solvente, struttura, catalizzatori, ecc.).
La velocità di una reazione chimica ha una definizione corrispondente a quella della meccanica: è la quantità di materia che si trasforma nell'unità di tempo e, in generale, va trattata come derivata, poiché varia istante per istante. Vi è un'importante legge che regola le trasformazioni più semplici, la legge ideale dell'azione di massa, secondo la quale la velocità di reazione è proporzionale al prodotto delle concentrazioni delle sostanze che effettivamente prendono parte alla reazione e solo a quello. Per es., se la sostanza S subisce una trasformazione unimolecolare e si trasforma in P, ne risulta una velocità v che segue l'equazione del 1° ordine
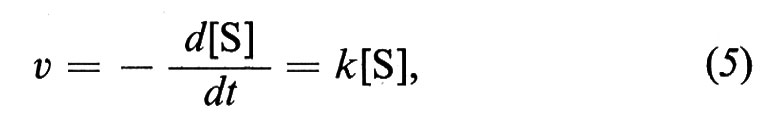
dove k è la costante specifica di velocità. Se avviene una trasformazione bimolecolare della sostanza S o una reazione tra due sostanze S′ e S″, si ottiene un'equazione cinetica del 2° ordine
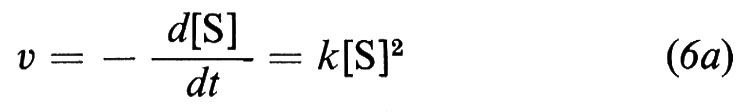
ovvero
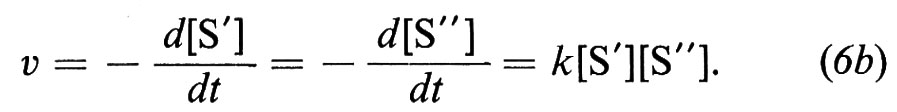
La molecolarità è riferita a una reazione semplice, che avviene cioè in un solo stadio; essa è il numero delle molecole che partecipano alla formazione del complesso attivato ed è pertanto un aspetto molto importante del meccanismo di reazione.
Ma molte reazioni chimiche non sono trasformazioni semplici e decorrono attraverso una successione di più stadi. In tali casi l'ordine cinetico, che si determina sperimentalmente e si riferisce all'intero processo, non è un criterio per stabilire la molecolarità della reazione, che va stabilita stadio per stadio. La complessità del meccanismo di reazione si può dedurre da eventi diversi: per es., la velocità può non dipendere dalla concentrazione di una delle sostanze reagenti; ovvero può dipendere dalla concentrazione di una sostanza che non appare come reagente nell'equazione chimica della reazione in esame. Queste deviazioni apparenti dalla legge dell'azione di massa significano che la reazione procede in più stadi, ciascuno dei quali (ma non la trasformazione nel suo insieme) obbedisce alla legge dell'azione di massa. Il meccanismo della reazione è definito dalla sequenza dei vari stadi e dalla natura dei prodotti intermedi che si formano tra uno stadio e l'altro. Poiché lo stadio più lento condiziona la velocità dell'intero processo, qualunque sia la sua posizione nella sequenza, esso è, insieme alla sua molecolarità, una delle caratteristiche della reazione.
Per determinare la velocità di reazione si deve disporre di un metodo accurato di analisi per misurare la concentrazione di uno o più componenti della miscela di reazione. I metodi di analisi variano da caso a caso, ma spesso si ricorre alla variazione dello spettro di assorbimento UV o visibile che accompagna la scomparsa delle sostanze di partenza e/o la formazione dei prodotti. Possono essere impiegati anche altri metodi spettrali (IR, NMR, ESR), come pure altre tecniche analitiche, quali l'analisi volumetrica, l'analisi potenziometrica, ecc. Se l'analisi richiede il prelevamento di campioni, la reazione in ciascun campione va fermata al tempo prescelto, aggiungendo un opportuno reagente o con un altro metodo. Talvolta si ricorre a metodi di analisi non chimici, quali quelli che misurano la variazione di volume (dilatometria) della miscela, la variazione di pressione (manometria), se si tratta di reazioni in fase gassosa, lo svolgimento di calore (calorimetria). Reazioni molto rapide possono essere seguite con tecniche speciali: per es., la tecnica stopped-flow (v. catalisi enzimatica) consente di studiare reazioni che hanno tempi di dimezzamento dell'ordine dei millisecondi. La valutazione dei risultati delle misure cinetiche per l'identificazione dell'equazione cinetica è un compito di difficoltà variabile a seconda dei casi. Non sono infrequenti cinetiche complesse, di ordine variabile al variare della concentrazione di uno dei componenti e anche di ordine frazionario. Sebbene lo studio cinetico sia importante ai fini del meccanismo di reazione, l'interpretazione dei dati, anche di cinetiche semplici, può essere non univoca e va confermata con altri metodi d'indagine.
Oltre all'identificazione della legge cinetica, studi cinetici possono fornire vari tipi di informazioni preziose. Lo studio dell'influenza della temperatura sulle costanti di velocità conduce alla determinazione delle entalpie e delle entropie di attivazione, i cui contributi all'energia libera di attivazione gettano luce, per esempio, sull'importanza relativa degli effetti legati alla rottura (e alla formazione) dei legami chimici e degli effetti di repulsione sterica (ΔH≠) e di solvatazione (ΔS≠).
Lo studio delle proprietà del solvente e dei suoi effetti cinetici, che è stato portato avanti con grande interesse negli ultimi anni, ha permesso di individuare varie categorie di solventi e di chiarire la natura delle interazioni soluto-solvente sia nello stato fondamentale sia nello stato di transizione dei sistemi in reazione. In tal modo l'effetto del solvente può fornire un importante criterio diagnostico di meccanismo (v. Coetzee e Ritchie, 1969).
Un notevole grado di approfondimento su particolari aspetti di una reazione si può raggiungere attraverso lo studio dell'effetto che provoca sulla velocità di reazione la sostituzione di un determinato atomo con un suo isotopo. La teoria degli effetti isotopici cinetici ha permesso interpretazioni di notevole valore dei risultati sperimentali, specialmente nel caso degli isotopi dell'idrogeno, molto frequentemente usati per stabilire se e in che misura la rottura, per es., del legame C−H sia avvenuta nello stadio lento di un processo (v. Melander, 1960).
Gli studi cinetici si sono rivelati utili per apprezzare con precisione l'effetto delle modifiche di struttura del substrato. Ciò ha permesso di scoprire correlazioni empiriche, aventi grande generalità e potere di previsione, tra energia libera di attivazione e struttura. La prima di queste correlazioni, l'equazione di Hammett
log(k/k0) = σρ, (7)
fu proposta per correlare gli effetti dei sostituenti R, situati nelle posizioni meta e para di un anello benzenico, con la reattività di un gruppo funzionale Y in catena laterale, in serie del tipo m- o p-R−C6H4−Y. Nell'equazione (7) la reattività di un composto sostituito di una serie (log k) è confrontata con quella del capostipite (non sostituito) della serie (log k0) e la variazione dovuta alla modifica di struttura (effetto del sostituente) è uguagliata al prodotto di due parametri, l'uno caratteristico del sostituente (σ) e l'altro caratteristico della reazione in esame (ρ). Il valore e il segno di ρ riflettono la natura del meccanismo della reazione chimica. Il segno corrisponde al carattere nucleofilo (+) o elettrofilo (−) del reagente, mentre il valore assoluto sta a indicare l'intensità con cui la perturbazione elettronica del sostituente si trasmette - in funzione della natura del substrato iniziale e del complesso attivato - al centro di reazione Y. Decine di migliaia di reazioni seguono l'equazione di Hammett o correlazioni analoghe proposte in seguito e dotate di diversi gradi di affinamento (v. Chapman e Shorter, 1972). Va aggiunto che l'equazione di Hammett è solo un tipo di una intera famiglia di correlazioni di energia libera che coprono un campo fenomenologico ancora più vasto delle relazioni tra reattività e struttura, comprendente certi aspetti della catalisi omogenea, gli effetti del solvente sulle reazioni solvolitiche, ecc.
c) Intermedi attivi delle reazioni chimiche
Negli esempi di reazioni chimiche che saranno illustrati nelle parti successive si vedrà che uno dei problemi chiave è quello di dimostrare se il sistema in reazione passa attraverso intermedi che differiscono dal centro di reazione iniziale (atomo di carbonio o altro atomo) per un numero di coordinazione inferiore o superiore. Il primo caso si verifica in seguito alla dissociazione di un legante, MXn → MXn-1 + X; il secondo quando un reagente Y si addiziona al substrato legandosi al centro di reazione, MXn + Y → MXnY. Questi intermedi ricorrono frequentemente nelle reazioni chimiche, ma per la loro elevata reattività se ne può dimostrare la presenza con metodi generalmente indiretti: cinetici, stereochimici e spettroscopici. Solo in condizioni particolari è stato possibile in alcuni casi isolarli. Gli intermedi maggiormente studiati sono quelli che derivano dal carbonio tetraedrico per rottura di un legame chimico e contengono pertanto un carbonio tricoordinato (v. McManus, 1973). Quelli che si formano per eterolisi sono specie ioniche positive (carbocationi), per es. CH3+, o negative (carbanioni), per es. C6H5CH2-. In seguito a omolisi si formano invece radicali liberi, per es. CH3•. I carbocationi sono resi più stabili dai gruppi alchilici o arilici che sono in grado, con meccanismi diversi di trasmissione elettronica, di disperdere in qualche misura la carica del centro positivo rilasciando elettroni verso di esso. Così il catione (CH3)3C+ (terz-butilico) è molto più stabile del catione CH3+. Al contrario, i carbanioni sono stabilizzati da gruppi che disperdono la carica del centro negativo attraendola verso di sé. L'anione −CH2NO2 è molto più stabile di −CH2C6H5 per il forte effetto di attrazione elettronica del nitro-gruppo.
Altri importanti intermedi attivi delle reazioni organiche sono i radicali ioni, i carbeni e i nitreni (v. McManus, 1973).
5. Reazioni dei composti del carbonio (reazioni organiche)
Sebbene la chimica dei composti del carbonio sia molto vasta, le reazioni organiche si possono ricondurre a un numero limitato di tipi: le sostituzioni, le addizioni, le eliminazioni e le trasposizioni. A queste si aggiungono le polimerizzazioni, le ossidazioni e le riduzioni, che si possono anche far rientrare nei tipi precedenti o in combinazioni di essi. I primi tre tipi sono schematicamente definiti qui di seguito. Le trasposizioni hanno la caratteristica di dar luogo a cambiamenti nello scheletro molecolare del substrato, le polimerizzazioni quella di permettere lo sviluppo di strutture macromolecolari mediante la ripetizione indefinita di un processo e le ossidoriduzioni quella di trasferire elettroni fra reagente e substrato.
Sostituzione
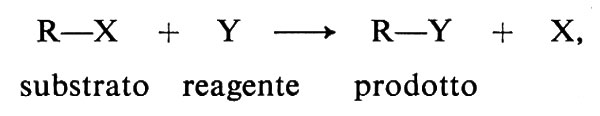
addizione
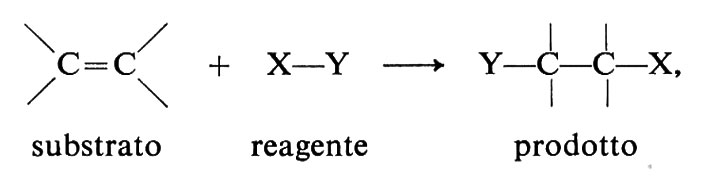
eliminazione
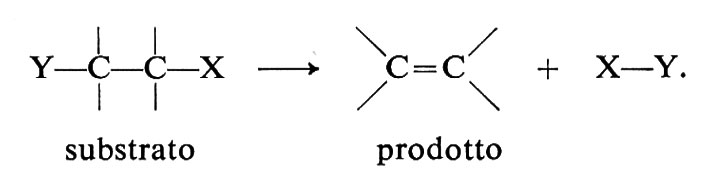
In questo capitolo illustriamo alcune reazioni di grande generalità e importanza nelle sintesi (v. Illuminati, 1971; v. March, 1977).
a) Sostituzioni nucleofile al carbonio saturo
Numerose classi di derivati alchilici subiscono la sostituzione eterolitica del gruppo X da parte di un reagente nucleofilo Y-, secondo la seguente equazione:
R−X + Y- → R−Y + X-. (8)
Nel derivato alchilico, RX, si intende che la reazione abbia luogo presso un atomo di carbonio tetraedrico (o saturo) al quale è inizialmente legato il gruppo uscente X, quindi sostituito da Y. Queste reazioni comprendono un gran numero di substrati diversi, tra cui gli alogenuri, gli esteri di acidi solfonici, gli esteri di acidi carbossilici, gli alcoli, gli eteri, gli ioni solfonio, le ammine, gli ioni ammonio. Per reazione con una grande varietà di reagenti nucleofili (Cl-, OH-, RO-, RS-, CN-, ROH, RNH2, ecc.) si formano alogenuri, alcoli, eteri, solfuri, nitrili, ammine, ecc. Poiché molte di queste reazioni hanno importanza come metodi di sintesi, sono tra le reazioni organiche meglio studiate dal punto di vista del meccanismo.
Qualsiasi trasformazione corrispondente all'equazione (8) comprende la rottura di un legame covalente (C−X) e la formazione di un nuovo legame (C−Y). In che ordine avvengono questi processi elementari e in che modo le molecole in reazione si dispongono perché la reazione abbia luogo? A questi interrogativi è stata data risposta con un notevole grado di attendibilità. La risposta non è la stessa in tutti i casi, poiché il meccanismo di reazione è sensibile alla struttura del substrato e alle condizioni sperimentali alle quali esso è sottoposto. Al chiarimento di queste reazioni un contributo determinante fu dato dal lavoro di C. K. Ingold e dei suoi collaboratori, che furono i primi a proporre l'esistenza di due meccanismi generali capaci di spiegare un grande insieme di fatti sperimentali. Tali meccanismi furono designati con i simboli SN2 e SN1, che si riferiscono, rispettivamente, alle sostituzioni nucleofile bimolecolari e monomolecolari; i loro modelli tipici sono illustrati nelle figg. 3 e 4.
Il meccanismo SN2 è un processo bimolecolare e concertato, nel quale le molecole in reazione, del derivato alchilico e del nucleofilo, si trasformano nei prodotti finali in uno stadio unico, secondo lo schema seguente:
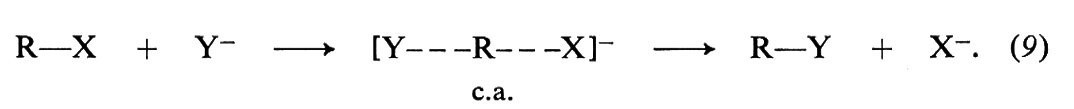
Il processo è bimolecolare, perché entrambe le molecole dei reagenti fanno parte del complesso attivato (c.a.) nello stato di transizione, ed è concertato, perché la formazione del legame C−Y e la rottura del legame C−X avvengono simultaneamente. Un'altra caratteristica di questo fenomeno è la stereospecificità, cioè la capacità di dare prodotti a configurazione spaziale specifica, come è illustrato nella fig. 3 per l'idrolisi basica del 2-bromobutano, dove il reagente nucleofilo, OH-, si suppone provenire dal lato opposto a quello di uscita di Br-, in modo che nel c.a. gli atomi che partecipano alla sostituzione siano allineati nell'ordine Y---C---X e gli altri tre legami del carbonio si appiattiscano nel piano passante per C, perpendicolare a detto allineamento. Alla fine del processo la configurazione dell'atomo di carbonio risulta invertita.
Il meccanismo SN1 consiste invece in un processo in due stadi, come indicato nel seguente schema:

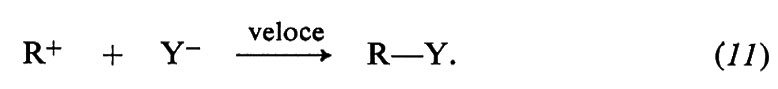
Nel primo stadio avviene la ionizzazione del legame C−X e conseguente formazione di un carbocatione come intermedio altamente reattivo. Nel secondo stadio il carbocatione reagisce con il nucleofilo trasformandosi nel prodotto finale. Lo stadio chiave di questo meccanismo è la lenta ionizzazione, cui corrisponde un complesso attivato comprendente soltanto una delle molecole reagenti; da questo punto di vista questo meccanismo si può considerare unimolecolare. L'intero processo SN1 non è, almeno in linea di principio, stereospecifico, poiché il carbocatione ha forma trigonale piana e può essere successivamente attaccato dal nucleofilo con uguale probabilità da entrambi i lati del piano. La fig. 4 illustra l'idrolisi del bromuro di terzbutile in solvente acquoso in assenza di basi forti.
I meccanismi SN2 e SN1 sono modelli ideali che derivano dal comportamento sperimentale, coerente con essi, osservato in condizioni appropriate. Tali modelli si sono rivelati utili per inquadrare una notevole quantità di informazioni.
Ora descriveremo alcune tipiche prove sperimentali a favore dei due meccanismi descritti, che permettono di illustrare l'applicazione di alcuni metodi di studio (v. sopra, cap. 4) alle reazioni di questo gruppo. Innanzitutto vi sono prove di cinetica chimica. Se una reazione avviene con un meccanismo SN2 (bimolecolare), deve essere possibile riscontrare cinetiche del secondo ordine in condizioni appropriate. Ciò è avvenuto in un gran numero di casi. Un esempio è la reazione degli alogenuri alchilici con etossido di sodio in alcool etilico,
CH3CH2Br + C2H2O- → CH3CH2OC2H5 + Br-,
per la quale v k[CH3CH2Br][C2H5O-]. La velocità di reazione è proporzionale alla concentrazione di ciascuna delle due sostanze reagenti. In particolare, un effetto caratteristico per una reazione di tipo SN2 è la dipendenza della velocità di reazione dalla natura del nucleofilo.
Se il meccanismo è SN1, la reazione è del primo ordine a condizione che il secondo stadio sia molto veloce rispetto al primo (eqq. 10 e 11); la velocità di reazione è proporzionale alla concentrazione del derivato alchilico, ma non dipende da quella del nucleofilo. La solvolisi (idrolisi) del 2-bromobutano in acqua-alcool,
CH3CH2CHBrCH3 + 2H2O → CH3CH2CH(OH)CH3 + H3O+ + Br-, (13)
segue infatti la legge cinetica v = k[CH3CH2CHBrCH3]. Tuttavia questo solo risultato, pur essendo coerente con l'ipotesi del meccanismo SN1, non è diagnostico nelle reazioni in cui il solvente è uno dei reagenti. Occorrono altre prove sperimentali.
Se la reazione avviene con un meccanismo SN2, la sua velocità dipende non solo dalla concentrazione, ma anche dalla natura del nucleofilo impiegato, perché nel c.a. vi è un incipiente legame tra nucleofilo e atomo di carbonio e l'energia di attivazione del processo bimolecolare è tanto più bassa quanto maggiore è la capacità del reagente di agire come donatore di elettroni. Tale capacità è detta nucleofilicità ed è una caratteristica del reagente impiegato, espressa dalla costante specifica del 2° ordine k. Esempi di reagenti ad alta nucleofilicità sono i solfuri, RS-, e gli ioni ioduro, I-, e cianuro, CN-. Debolmente nucleofili sono invece lo ione acetato, CH3CO2-, e l'acqua. Al contrario, nelle reazioni SN1 la velocità di reazione è indipendente sia dalla concentrazione sia dalla natura del nucleofilo.
Il solvente ha un ruolo importante nel determinare la velocità di una reazione eterolitica e può avere effetti diversi a seconda del meccanismo di reazione. Nelle reazioni nucleofile bimolecolari tra substrati neutri e anioni, condizioni particolarmente favorevoli sono quelle che impiegano solventi polari non protici (per es. acetonitrile, dimetilsolfossido), mentre i solventi protici (per es. acqua, alcool metilico) sono meno efficaci, perché si associano fortemente con il nucleofilo anionico (solvatazione specifica da legame a idrogeno) riducendone la nucleofilicità. Al contrario, i solventi protici promuovono la ionizzazione dei substrati favorendo il distacco dell'anione X- per solvatazione specifica (eq. 10) e fanno sì che il meccanismo SN1 possa prevalere su quello bimolecolare, specie in assenza di nucleofili forti. Nelle solvolisi, oltre al potere ionizzante, ha influenza anche la nucleofilicità del solvente (v. Illuminati, 1976).
Importanti differenze di comportamento si osservano in funzione della natura di R nel derivato alchilico R−X a seconda del meccanismo di reazione. Se la reazione avviene in condizioni favorevoli al meccanismo SN2 (solventi non ionizzanti, presenza di nucleofili forti), essa è molto sensibile agli effetti sterici. Questi si osservano confrontando la reattività (misurata dalla costante specifica del 2° ordine) dei composti di una serie nella quale si aumentano progressivamente i requisiti di ingombro spaziale del gruppo alchilico nelle vicinanze del centro di reazione. Per esempio, se vengono sostituiti atomi di idrogeno con gruppi −CH3 di un alogenuro di metile, la reattività diminuisce nell'ordine
CH3Br > CH3CH2Br > (CH3)2CHBr > (CH3)3CBr.
L'effetto è così forte nel bromuro terziario che non si riesce a misurarne la velocità di reazione. In che consiste l'effetto sterico nel caso del modello SN2? Innanzitutto, per una reazione bimolecolare, al crescere delle dimensioni delle molecole reagenti nelle vicinanze del sito di reazione, via via che tali molecole si avvicinano tra loro, parti non legate di esse finiscono per interferire e respingersi mutuamente. Il risultato cinetico è il rallentamento della reazione, se l'energia di repulsione sterica è maggiore nello stato di transizione che nello stato iniziale: ciò è in accordo con il modello SN2, in base al quale da una configurazione tetraedrica (tetracoordinata) del carbonio allo stato iniziale si passa a quella pentacoordinata, più ‛affollata', del c.a. Se la reazione avviene in condizioni favorevoli al meccanismo SN1 (solventi ionizzanti, assenza di nucleofili forti), essa risulta insensibile a effetti sterici di questo tipo e, al contrario, è fortemente accelerata dai gruppi alchilici in virtù di effetti di natura elettronica. L'ordine di reattività è pertanto esattamente l'opposto di quello che si osserva nelle reazioni SN2: i derivati alchilici terziari reagiscono più facilmente dei secondari e dei primari, cioè nell'ordine
(CH8)3CBr > (CH3)2CHBr > CH3CH2Br > CH3Br,
come avviene nell'idrolisi dei bromuri alchilici in acido formico. In questa reazione il bromuro di butile terziario reagisce 108 volte più rapidamente del bromuro di metile. Si noti che questo ordine di reattività segue la relativa stabilità dei carbocationi (v. sopra, cap. 4, § c), in accordo con il meccanismo in due stadi che presuppone un carbocatione come intermedio attivo.
Un'altra importante prova a favore della formazione di carbocationi intermedi tipica del meccanismo SN1 è data dai fenomeni di trasposizione (v. sotto, § c).
Un procedimento essenziale per definire il meccanismo della sostituzione nucleofila nei suoi particolari stereochimici consiste nel partire da un derivato alchilico in cui l'atomo di carbonio presso il quale avviene la sostituzione è asimmetrico. Certe peculiarità del destino stereochimico delle sostituzioni hanno importanza storica. P. Walden (1896) scoprì cicli di reazioni che inequivocabilmente dimostravano il fenomeno dell'inversione di configurazione (inversione di Walden) nelle sostituzioni. Un classico ciclo di Walden è quello dell'acido malico, che attraverso quattro passaggi alternati di clorurazione con SOCl2 e di idrolisi con KOH ritorna ad acido malico con la configurazione invertita. Non è facile tuttavia dimostrare il cambiamento o meno di configurazione in ogni singola reazione del ciclo. Occorre infatti determinare la configurazione del prodotto di reazione attraverso cicli di reazione privi di ambiguità (Kenyon-Phillips) ovvero con metodi basati su reazioni che non coinvolgano i legami del carbonio asimmetrico. Con l'aiuto di questi metodi, sia pure laboriosi, è stato accertato che le sostituzioni che avvengono con meccanismo SN2 sono invariabilmente accompagnate da inversione del carbonio asimmetrico (v. fig. 3). Un tipico esempio è la reazione del 2-bromopropionato di metile con CH3O- in alcool metilico,
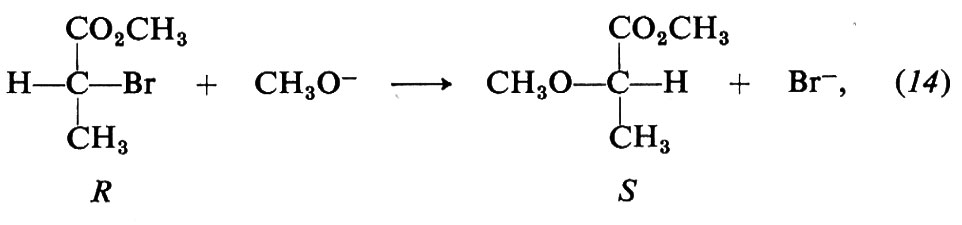
nella quale dalla configurazione R del substrato si passa alla S del prodotto, con evidente inversione.
Passando alla reazione del tipo SN1 trova invece che il prodotto si ottiene in forma racemica più o meno completa. La racemizzazione avviene completamente se il carbocatione è ‛libero' e capace di reagire con il nucleofilo da entrambi i lati della sua struttura piana. Per esempio, nella solvolisi del cloruro di 1-feniletile otticamente attivo in acetone acquoso si ha un prodotto racemizzato al 98%.
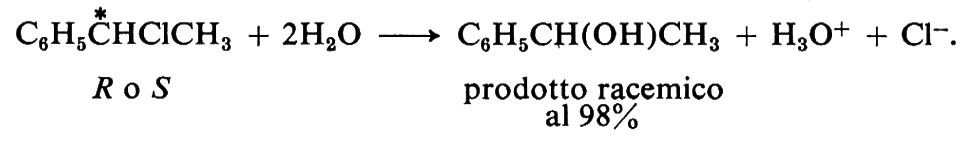
Spesso il prodotto della reazione è in parte racemizzato e per il resto invertito. Anche questo risultato è considerato caratteristico di reazioni di tipo SN1. Si interpreta ammettendo che il reagente nucleofilo (Y-) attacchi il carbocatione intermedio prima che il gruppo uscente (X) si sia allontanato sufficientemente dall'atomo di carbonio. Quando ciò avviene, il lato più disponibile per l'attacco è quello opposto al sito ancora occupato dal gruppo uscente, mentre il carbocatione non ha ancora acquistato la struttura piana. Situazioni di questo tipo possono essere causate sia da una reattività particolarmente elevata (bassa stabilità) del carbocatione, sia da un solvente poco polare (per es. alcool), sia, naturalmente, da entrambe queste circostanze. Se un solvente è poco polare (cioè non è un buon dielettrico), gli ioni che derivano dalla ionizzazione del legame R−X tendono a rimanere associati sotto forma di coppie ioniche, R+H-, o di più alti aggregati ionici, anziché separarsi liberamente nel mezzo. Come si vede, il meccanismo SN1 può diventare complesso non solo dal punto di vista cinetico, ma anche da quello stereochimico.
Quanto sopra mostra che il meccanismo di una sostituzione nucleofila dipende sia dalla struttura sia dalle condizioni sperimentali. La natura dei prodotti può dipendere dalle condizioni sperimentali anche se si parte da uno stesso substrato; inoltre uno stesso prodotto si può ottenere in condizioni sperimentali diverse tra loro. La scelta delle condizioni più moderate per la sintesi di una sostanza chimica è un obiettivo di grande interesse che può essere perseguito con l'aiuto di una conoscenza approfondita delle reazioni chimiche.
I due modelli di meccanismo sopra illustrati non esauriscono i modi in cui i derivati alchilici subiscono la Sostituzione nucleofila. Particolarmente i meccanismi di tipo dissociativo cui appartiene il modello SN1 ora descritto presentano notevoli complessità che si riflettono nella cinetica chimica e nella stereochimica. Vi sono inoltre condizioni sperimentali in cui il meccanismo sembra avere caratteri intermedi tra quelli corrispondenti ai due modelli SN2 e SN1 e nelle quali, in alcuni casi ma non sempre, i due diversi meccanismi coesistono. Numerose sono infine le sostituzioni che seguono altri corsi caratteristici che dipendono dalla particolare struttura del substrato e/o del reagente. Citiamo le sostituzioni assistite da partecipazione di un gruppo vicinale e quelle, designate con il simbolo SNi, che avvengono in modo stereospecifico con la completa conservazione, anziché inversione, di configurazione (v. March, 1977).
b) Reazioni di β-eliminazione
Queste reazioni sono rappresentate dallo schema generale (15) e consistono nell'eliminazione del gruppo uscente X insieme a un protone dalla posizione β in presenza di una base :B:
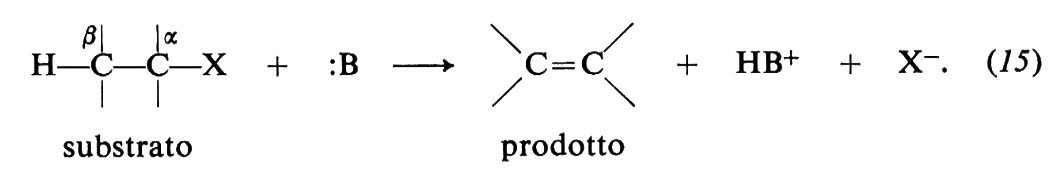
Esse sono importanti nelle sintesi, perché servono alla preparazione delle olefine. Esempi sono le deidroalogenazioni (da alogenuri alchilici) e le disidratazioni (da alcoli).
Esse competono con le sostituzioni nucleofile accompagnandosi a queste ultime in proporzioni variabili che dipendono dalle condizioni sperimentali. Una conoscenza approfondita dei meccanismi di reazione è di grande interesse per orientare i processi verso una delle due reazioni a seconda delle esigenze di sintesi.
Per le reazioni di β-eliminazione di tipo eterolitico sono stati riconosciuti diversi meccanismi. I principali, designati con i simboli E2, E1 ed Elcb, sono modelli di riferimento per un ampio spettro di varianti che dipendono dall'ordine in cui i legami C−H e C−X si rompono e in cui la base :B si associa con il substrato e il doppio legame si forma. I più noti sono i primi due.
Il meccanismo E2 corrisponde al modello dello schema seguente:
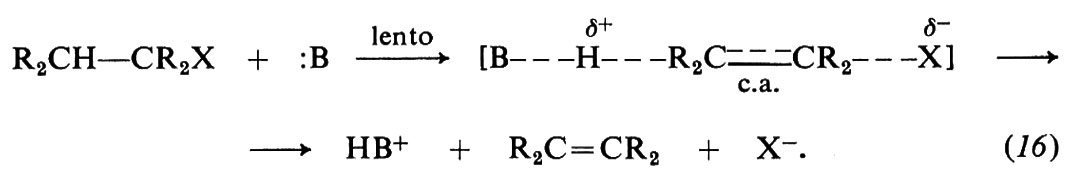
La reazione è bimolecolare e consiste in un solo stadio nel quale tutti i cambiamenti di legame avvengono in modo concertato attraverso 5 atomi facenti parte del substrato e della base.
Il meccanismo El si svolge in due stadi secondo le equazioni seguenti:
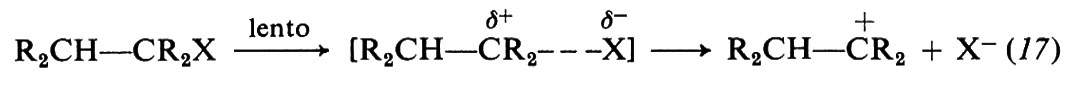

Nel 1° stadio avviene la ionizzazione (lenta) del legame Cα−X; nel 2° il trasferimento rapido del protone del legame Cβ−H alla base. Il simbolo E1 indica che la ionizzazione è un processo unimolecolare.
Vediamo alcuni dei principali tipi di prove sperimentali che sono alla base dei due meccanismi. Dei numerosi metodi di studio applicati a queste reazioni, ci soffermiamo solo su due: l'effetto isotopico cinetico e il corso stereochimico. Il primo è basato sulla teoria degli effetti isotopici. Poiché nello stato di transizione del meccanismo E2 avviene la rottura del legame Cβ−H, la teoria prevede una diminuzione della costante di velocità se si usa un substrato in cui al posto del β-H vi è il deuterio (effetto isotopico primario). Effetti di questo genere sono stati effettivamente osservati nelle eliminazioni bimolecolari (E2). L'effetto isotopico si esprime come rapporto tra le costanti di velocità per il substrato ‛leggero' e per il substrato deuterato preso come riferimento. Il rapporto kH/kD in queste reazioni è spesso compreso tra 3 e 8, il che va considerato come un risultato molto netto che non lascia dubbi interpretativi.
Le reazioni E1 non sono accompagnate da effetti isotopici se corrispondono al modello tipico, perché in tal caso la rottura del legame C−H non avviene nello stadio lento del processo (formazione del carbocatione ‛libero', eq. 17), ma solo nel successivo stadio rapido.
Quanto alla stereochimica della reazione, l'esperienza suggerisce due percorsi distinti, le eliminazioni anti (o trans) e le eliminazioni sin (o cis). Le prime sono più comuni perché, se la struttura lo consente, sono energeticamente più favorite.
Il modello stereochimico dell'eliminazione anti può essere rappresentato con l'aiuto di formule prospettiche come nello schema (19). Nel c.a. i quattro atomi del derivato alchilico che partecipano alla reazione, cioè H, Cβ, Cα e X, giacciono su uno stesso piano. Inoltre la conformazione del derivato alchilico è tale da tenere H e X il più possibile lontani l'uno dall'altro (conformazione alternata) ed è chiamata anti-peripiana. Con questa disposizione l'energia del sistema è resa minima e l'eliminazione anti costituisce un percorso stereochimico preferenziale nei riguardi di altri possibili.
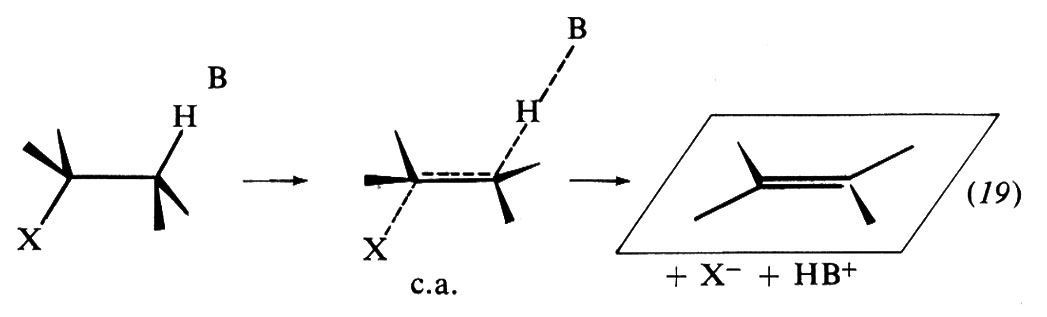
Le prove sperimentali dell'eliminazione anti si basano sull'esame del comportamento stereochimico di substrati di struttura appropriata. Per esempio, nei composti a catena aperta, se Cα e Cβ sono entrambi asimmetrici, la disposizione anti-peripiana conduce univocamente a uno solo dei due possibili isomeri geometrici dell'olefina prodotta. Cosi, dalla deidrobromurazione dello stereoisomero meso-2,3-dibromobutano si ottiene esclusivamente il cis-2-bromo-2-butene; dall'analoga reazione del d,l-2,3-dibromobutano si ottiene invece l'isomero trans.
I derivati cicloesilici forniscono altri casi importanti per accertare il corso stereochimico delle eliminazioni. Si può, per esempio, dimostrare che il percorso stereochimico anche qui è essenzialmente anti-peripiano, come illustrato nello schema (20). L'eliminazione bimolecolare richiede che l'idrogeno β e il gruppo uscente X siano entrambi in posizione assiale. Un esempio spettacolare di eliminazione
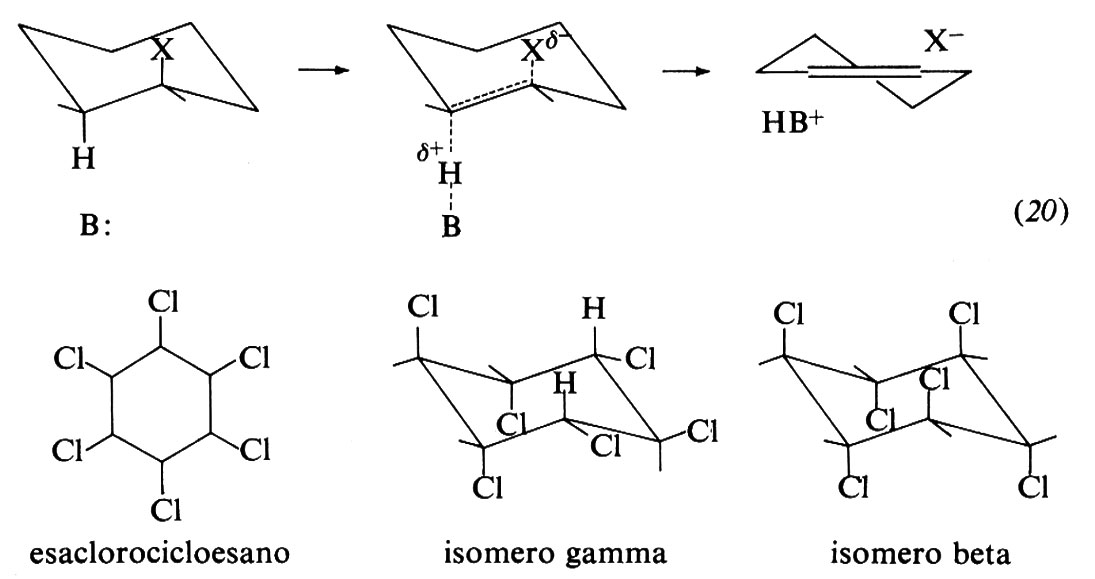
preferenziale anti si ha nella deidroclorurazione degli esaclorocicloesani. Oli esaclorocicloesani sono in tutto otto isomeri geometrici, di cui l'isomero ‛gamma' (gammaesano) è un noto insetticida: tutti questi isomeri, a eccezione di uno, il cosiddetto ‛beta', subiscono eliminazione con analoga facilità. L'isomero beta, che non contiene alcun atomo di idrogeno β in posizione trans rispetto ad atomi di cloro α e non può, pertanto, acquistare una posizione anti-peripiana nel modo illustrato, reagisce 104 volte più lentamente degli altri isomeri (nelle formule degli isomeri gamma e beta sono indicate le conformazioni a sedia utili ai fini della discussione, anche se meno stabili; sono inoltre indicati gli atomi di idrogeno assiali che sono in posizione peripiana con gli atomi di cloro assiali adiacenti, condizione che non si verifica nell'isomero beta).
Le eliminazioni bimolecolari di tipo sin si possono incontrare nei derivati ciclopentilici, dove una vera disposizione anti-peripiana non è ottenibile anche se l'idrogeno β e il gruppo uscente X sono disposti in posizione relativa trans. Di conseguenza l'eliminazione da posizioni trans è solo mediocremente favorita. Un caso è quello dei tosilati di 2-fenilciclopentile (cis e trans). Da questi composti si elimina acido p-toluensolfonico per azione della base terz-BuO-; si forma l'1-fenilciclopentene. Ciò può avvenire solo attraverso un'eliminazione di tipo sin se si parte dall'isomero trans e una di tipo anti se si parte dall'isomero cis; in questo secondo caso, la reazione è solo 9 volte più veloce che nel primo.
c) Trasposizioni dei carbocationi
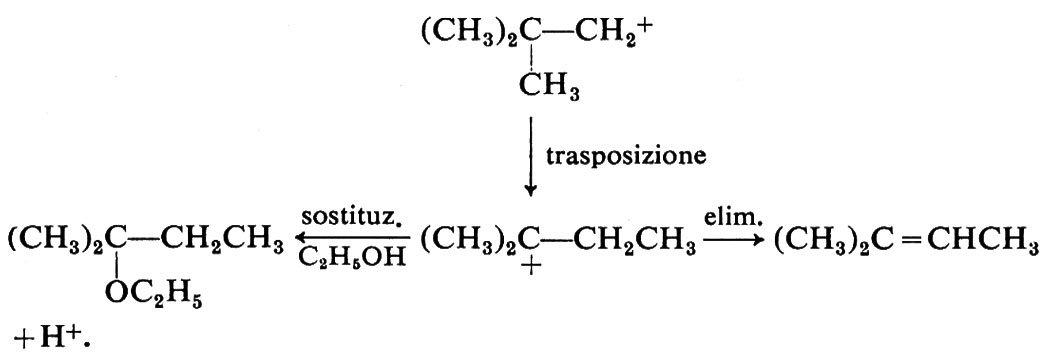
Le modificazioni dello scheletro molecolare, o trasposizioni, formano un vasto capitolo della chimica: possono essere reazioni eterolitiche od omolitiche e comprendono strutture e reazioni di ogni tipo. Come esempi ci limitiamo a considerare alcune reazioni che comportano carbocationi come intermedi, quali le sostituzioni e le eliminazioni che avvengono con meccanismi SN1 e, rispettivamente, E1, e fanno parte di un gruppo noto con il nome di trasposizioni di Wagner-Meerwein. Un caso tipico è la solvolisi del bromuro di neopentile in alcool etilico:

I carbocationi tendono a trasformarsi in carbocationi più stabili in accordo con l'ordine di stabilità che si deduce da diversi risultati sperimentali indipendenti. In particolare, i carbocationi terziari sono più stabili dei secondari e questi dei primari. Di conseguenza la reazione (21) si può interpretare con la seguente variante di meccanismo SN1:
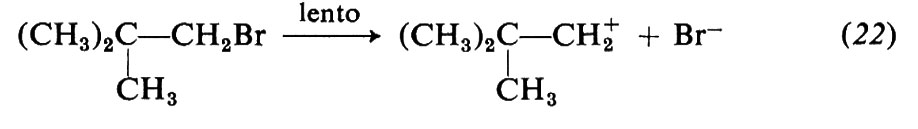
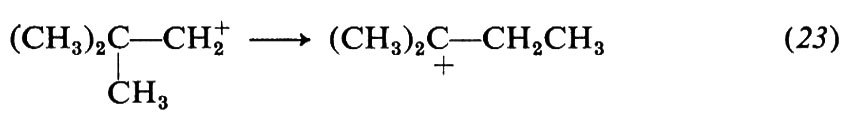
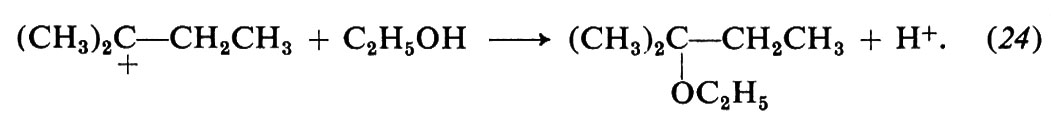
La trasposizione ha luogo nel secondo stadio e consiste in una migrazione di un gruppo −CH3 da un atomo di carbonio a uno adiacente, sede della carica positiva (migrazione 1,2); il gruppo si sposta con gli elettroni del vecchio legame e il sito positivo va al suo posto. In definitiva il carbocatione primario si è trasformato in uno terziario più stabile.
Il prodotto di sostituzione può essere accompagnato da olefina in seguito a eliminazione, che risulta anch'essa dal carbocatione trasposto. Nel caso della solvolisi del bromuro di neopentile l'olefina è il 2-metil-2-butene:
d) Idrolisi basica degli esteri
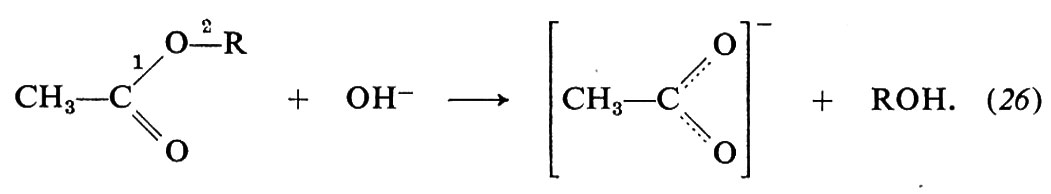
Vi è un importante gruppo di reazioni degli acidi carbossilici e dei loro derivati, di formula generale RCOX, che consistono nella sostituzione del gruppo X con un reagente nucleofilo, Y-. Tra questi processi sono l'idrolisi basica degli esteri,
R−COX + Y- → R−COY + X-, (25)
quale la saponificazione dei grassi, l'idrolisi delle ammidi - che si ricollega alle reazioni dei legami peptidici delle sostanze proteiche -, la condensazione di Claisen e quella di Dieckmann - di notevole interesse nelle sintesi -, per citarne solo alcune. Consideriamo in particolare l'idrolisi basica degli esteri, che offre interessanti esempi di metodi di studio delle reazioni chimiche:
Vi sono vari modi di idrolisi degli esteri. Due alternative derivano dal punto di rottura dell'ossigeno del gruppo OR, che può avvenire in 1 o in 2, cioè nel legame acile-ossigeno o, rispettivamente, nel legame alchile-ossigeno. I numerosi studi su questa reazione portano a concludere che, negli esteri più semplici, il meccanismo di reazione comporta i seguenti stadi:

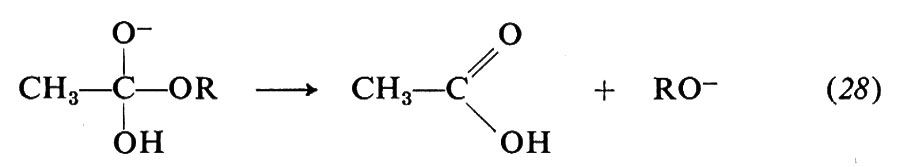
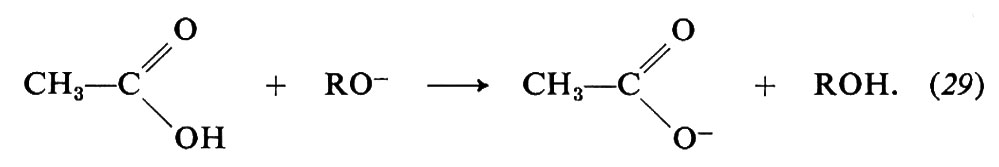
La sostituzione avviene quindi attraverso un'addizione che dà luogo a un intermedio in cui il carbonio acilico diventa tetraedrico, seguita da un'eliminazione dell'anione RO-, quindi, dalla dissociazione dell'acido carbossilico formatosi.
Il problema del punto di rottura dell'estere (legame acile-ossigeno) che si ha nello stadio (28) è stato elegantemente risolto con le tecniche isotopiche. Secondo la classica esperienza di J. C. Polanyi e Z. G. Szabo (1934), facendo reagire l'estere in acqua arricchita in 18O si trova che dei due prodotti della reazione soltanto l'acido carbossilico, e non l'alcool, risulta arricchito in 18O. Partendo dall'acetato di amile, CH3COOC5H11, si ottiene
CH3COOC5H11 + 18OH- → CH3CO18O- + C5H11OH.
Ciò prova che nel corso della reazione (eq. 28) si rompe il legame acetile-ossigeno, ma non quello pentile-ossigeno.
e) Addizioni elettrofile
Tra le reazioni dei reagenti elettrofili va ricordata l'addizione al doppio legame C=C:
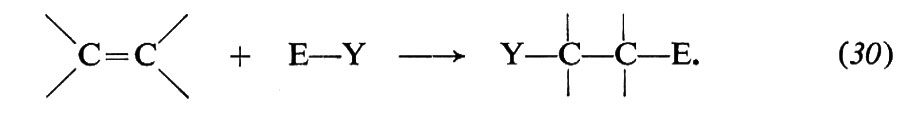
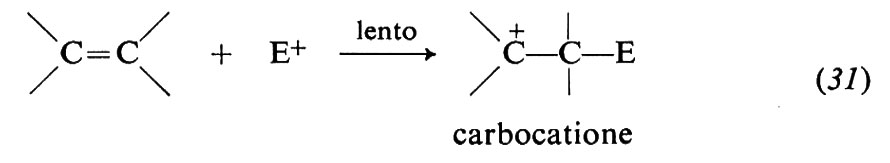
Il doppio legame degli alcheni ha particolare tendenza a reagire con questo tipo di reagenti, perché il legame π, relativamente debole (paragonato al legame σ, si comporta come donatore di elettroni ed è, pertanto, vulnerabile all'azione degli accettori. Molte sono le reazioni di questo gruppo: addizioni di acidi, di acqua, di alcoli, di alogeni, di acidi ipoalogenosi, di perossidi, ecc. Di particolare interesse sono le polimerizzazioni ioniche, perché impiegate nei processi industriali per la sintesi di alti polimeri. Alcuni aspetti caratteristici di queste reazioni possono essere illustrati nel caso dell'addizione di alogeni. Le reazioni avvengono in più stadi, di cui almeno due ben accertati in generale: a) attacco di una specie elettrofila E+ e conseguente formazione di un carbocatione (eq. 31); b) combinazione del carbocatione con un nucleofilo Y-(eq. 32):
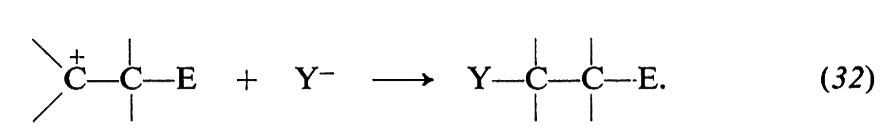
Se il reagente è un alogeno (per es. Cl2, Br2) i prodotti della reazione sono dialogenoderivati. La natura elettrofila di questo reagente dipende dallo stato di polarizzazione della molecola. La polarizzazione,
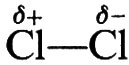
, è indotta dalle interazioni substrato-reagente, in base alle quali la parte del reagente rivolta verso il substrato (donatore di elettroni) acquista la polarità positiva. Nel 1° stadio della reazione (31), che è bimolecolare, si ha un complesso attivato caratterizzato da un'incipiente formazione del legame olefina-alogeno e da rottura dei legami alogeno-alogeno e carbonio-carbonio in modo concertato.
Che il meccanismo di addizione dell'alogeno avvenga in più stadi e sia del tipo generale ora illustrato è dimostrato dal fatto che in presenza di nucleofili diversi si ottengono prodotti diversi. Ciò è possibile solo se le due parti del reagente X−X (o X−Y) si legano ai due atomi di carbonio del doppio legame in stadi separati, dei quali il primo è lo stadio lento dell'attacco dell'elettrofilo e il secondo quello rapido di combinazione con il nucleofilo.
Per esempio, nell'addizione di cloro a un'olefina in solvente acquoso si ottiene una miscela di dicloroderivato e di cloridrina:
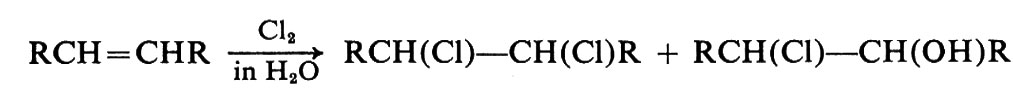
Questo risultato si spiega ammettendo la formazione di un carbocatione intermedio che può reagire con entrambi i nucleofili disponibili, Cl- H2O:
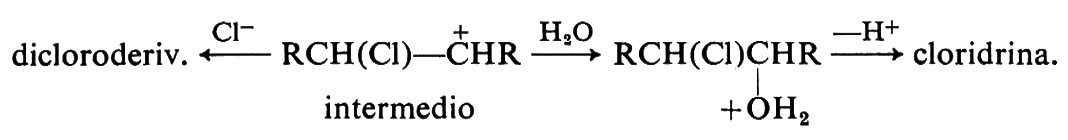
Un'altra prova del carattere elettrofilo dell'addizione dell'alogeno si ottiene esaminando gli effetti dei sostituenti. Infatti, poiché il doppio legame agisce come donatore di elettroni nello stadio lento della reazione, i sostituenti a rilascio elettronico (per es., −CH3) facilitano la reazione, quelli ad attrazione elettronica (per es., −CO2Et) la rendono più difficile. Nella bromurazione dei 3 R-acrilati di etile, il metilderivato reagisce 25 volte più velocemente del composto non sostituito, l'etossicarbonilderivato 1.000 volte più lentamente.
La stereochimica dell'addizione suggerisce un modello più accurato del suo meccanismo. Queste reazioni con l'alogeno sono generalmente stereospecifiche. L'addizione globale al doppio legame può avvenire in due modi, dal punto di vista stereochimico: o i due frammenti dell'addendo si uniscono agli atomi di carbonio insaturo dallo stesso lato del piano del doppio legame (addizione sin) ovvero si uniscono l'uno dal lato opposto a quello dell'altro (addizione anti). Questi modi stereochimici si possono individuare esaminando i prodotti che si ottengono se la struttura dell'olefina di partenza è tale da generare isomeri ottici (sistemi aciclici) o isomeri geometrici (sistemi ciclici). Le addizioni anti sono molto più frequenti delle addizioni sin. Un esempio è la bromurazione del cis-2-butene, che dà luogo al (±) 2,3-dibromobutano e non all'isomero meso.
L'addizione anti non sarebbe prevista se il carbocatione intermedio avesse la struttura CH2CH(Br)−+CHCH3 che rende possibile la rotazione intorno al legame C2−C3. Essa si spiega ammettendo che nel carbocatione avvenga un'interazione attrattiva intramolecolare tra l'alogeno e il centro positivo adiacente. In condizioni favorevoli si forma un vero e proprio legame che porta alla formazione di uno ione alonio nel quale la carica positiva si trasferisce sull'atomo di alogeno bicovalente:
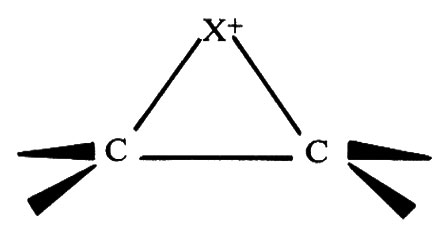
Una struttura del genere impedisce la rotazione intorno al legame C2−C3 prima che il nucleofilo X- abbia modo di attaccare il carbonio dalla parte opposta alla posizione dell'alogeno positivo.
Altre reazioni elettrofile di grande interesse sono le diffusissime sostituzioni aromatiche, che pure avvengono in più stadi, consistenti di solito in un'addizione lenta seguita da eliminazione:
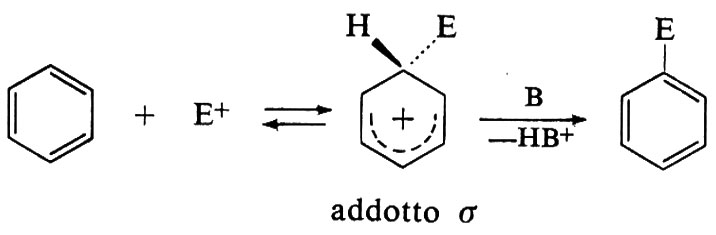
La stabilità dell'addotto σ intermedio è un punto chiave che ora permette di spiegare agevolmente i vistosi fenomeni di orientamento orto, meta e para dei derivati del benzene, che appassionarono i chimici fin dal secolo scorso. Gli intermedi, benché labili, sono stati intercettati o addirittura isolati in un buon numero di casi per opera del gruppo di G. A. Olah.
f) Reazioni radicaliche
Le reazioni radicaliche mostrano i tipi fondamentali di trasformazioni di tutte le reazioni organiche: sostituzione, addizione, eliminazione e trasposizione. La loro natura si può diagnosticare dalle condizioni sperimentali. Queste reazioni sono generalmente provocate dalla luce o dal riscaldamento e catalizzate dai perossidi. Sono insensibili all'azione degli acidi e delle basi. Possono avvenire ugualmente bene in fase gassosa e in soluzione. Poiché le reazioni ioniche sono spesso fortemente accelerate da solventi polari e protici, la velocità delle reazioni radicaliche è spesso sopravanzata da quella delle reazioni ioniche in detti solventi. Infine, le reazioni radicaliche sono rallentate o soppresse da sostanze dette inibitori, che sono capaci di catturare rapidamente i radicali liberi e, pertanto, di bloccare un processo radicalico; tra queste sono l'ossigeno molecolare e il benzochinone.
Nelle reazioni radicaliche si possono distinguere tre diversi tipi di eventi: la formazione dei radicali liberi, la distruzione dei radicali liberi e il trasferimento del sito radicalico. La formazione dei radicali consiste nella omolisi unimolecolare del legame covalente. Esempi sono la formazione di due atomi di cloro da una molecola di cloro e quella di due radicali RO• da una di perossido:
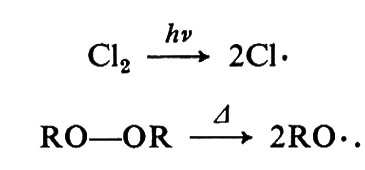
Nell'omolisi i radicali liberi si formano necessariamente a coppie. La distruzione dei radicali avviene per combinazione di due radicali liberi come, per es., nella formazione dell'etano da due radicali metilici e del cloruro di metile da un radicale metilico e un atomo di cloro:
2CH3• → CH3−CH3
CH3• + Cl• → CH3−Cl.
Essendo un evento inverso rispetto alla formazione dei radicali, anche la distruzione dei radicali avviene a coppie. Nel trasferimento del sito radicalico ha luogo una reazione bimolecolare di un radicale libero con una molecola di substrato e la conseguente formazione di altre specie radicaliche. Importanti reazioni di questo tipo sono le estrazioni degli atomi di idrogeno dai legami C−H, come è illustrato nei due esempi seguenti:
R3C−H + R′O• → R3C• + R′OH
R3C−H + CI• → R3C + HCl.
Un processo radicalico si può concludere con i soli eventi di formazione e distruzione dei radicali liberi. Tuttavia i più caratteristici processi radicalici sono reazioni a catena. Il meccanismo di queste reazioni comprende tre stadi: l'inizio del processo, nel quale avviene una reazione di formazione di radicali; la propagazione dei radicali, che consiste in reazioni di trasferimento del sito radicalico nel corso delle quali si formano le molecole del prodotto della reazione globale e si rigenera il radicale libero un numero indefinito di volte, dando così luogo a una ‛catena' di reazioni; infine, la terminazione del processo, comprendente tutte le possibili reazioni di distruzione dei radicali che provocano l'arresto delle catene di reazione. Questo meccanismo è illustrato nel seguente schema per la clorurazione foto-catalizzata degli alcani:
inizio

propagazione
R−H + Cl• → R• + HCl (34)
R• + Cl• → R−Cl + Cl•, (35)
terminazione
2Cl• → Cl2 (36)
2R• → R−R (37)
R• + Cl → R−Cl. (38).
Si noti che le due reazioni di propagazione, sommate membro a membro, danno l'equazione stechiometrica della reazione globale:
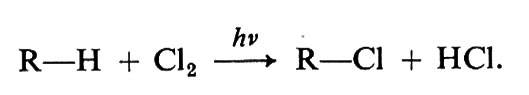
Se queste due reazioni si ripetono un gran numero di volte, cioè se la catena radicalica è ‛lunga', il numero di radicali liberi provenienti dal 1° stadio è piccolo e sufficiente a mantenere l'intero processo; inoltre, è piccola anche la quantità di prodotto che si forma per terminazione. Di conseguenza, praticamente tutto il prodotto si forma nello stadio della propagazione.
L'energia necessaria alla formazione dei radicali liberi per rottura omolitica di un legame covalente
R−R → R• + R•
per definizione è l'energia di dissociazione del legame. Le fonti principali di energia per ottenere i radicali liberi sono il calore (omolisi termica) e la luce (fotolisi).
La semplicità del fenomeno dell'omolisi consente di interpretare in opportune serie di composti i valori dell'energia di dissociazione del legame in funzione della struttura del radicale libero che si forma. Una serie importante è quella dei radicali alchilici, R., che si ottengono per dissociazione del legame C−H. Le energie di dissociazione di legame di alcuni idrocarburi sono raccolte nella tab. I. Questi valori corrispondono alle entalpie (ΔH) di reazioni del tipo
R−H → R• + H•.
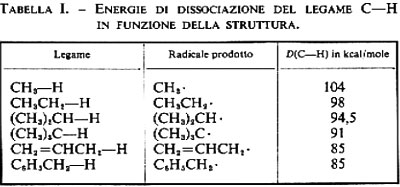
Poiché dei due radicali che si formano uno è sempre l'idrogeno, le differenze che si osservano sono imputabili a differenze di stabilità dei radicali alchilici formati. L'ordine di stabilità nella serie alchilica è il seguente:

dal che si deduce che i gruppi alchilici hanno effetto stabilizzante sul radicale libero. Se il carbonio radicalico è in posizione α rispetto a un doppio legame o a un anello aromatico, l'energia decresce ulteriormente. Questo effetto è evidente nella formazione dei radicali allilico e benzilico ed è dovuto a un effetto elettronico coniugativo.
Come esempio di reazione radicalica illustriamo la sostituzione con alogeno molecolare, che può avvenire per omolisi termica o per fotolisi. Il meccanismo è a catena ed è rappresentato dalle eqq. (33)-(38). La forma cinetica di queste reazioni è complessa e non può essere usata per descrivere in modo semplice le caratteristiche principali del meccanismo di reazione. Sono invece molto usate nelle reazioni radicaliche le determinazioni di velocità relativa, che servono a stabilire quanto un certo composto è più o meno veloce di un altro preso come riferimento. Queste determinazioni possono fornire informazioni sul meccanismo di reazione.
Vi sono analogie e differenze di comportamento tra clorurazione e bromurazione. In entrambe le reazioni il processo a catena è assicurato da due reazioni di propagazione, quella di estrazione dell'idrogeno da un legame C−H per opera dell'atomo di alogeno X• e quella di rigenerazione di X•:
R−H + X• → R• + HX (39)
R• + X2 → R−X + X•. (40)
Sia nella clorurazione sia nella bromurazione le catene sono abbastanza ‛lunghe' da rendere conto da sole della formazione del prodotto di reazione. Ciò avviene nella chimica radicalica tutte le volte che le reazioni di propagazione sono più veloci delle reazioni di terminazione. La clorurazione è caratterizzata da catene molto lunghe, pari a un milione di processi di propagazione o anche più. Per certe strutture, quali gli alcani contenenti atomi di idrogeno terziari, anche le catene della bromurazione sono lunghe (per es., nella reazione dell'isobutano a 127 °C, esse hanno lunghezze dell'ordine del miliardo), ma per altri alcani sono più corte che nella clorurazione (per es., nella reazione del metano a 297 °C, hanno lunghezza di circa 100).
L'estrazione dell'idrogeno è la più importante delle due reazioni di propagazione. Ciò risulta dalle velocità relative dei vari idrocarburi nelle reazioni con Cl2 e Br2. In entrambe le reazioni si trova che l'idrogeno terziario (3°) è sostituito più rapidamente del secondario (2°) e questo più rapidamente del primario (1°); è valido pertanto il seguente ordine di reattività:
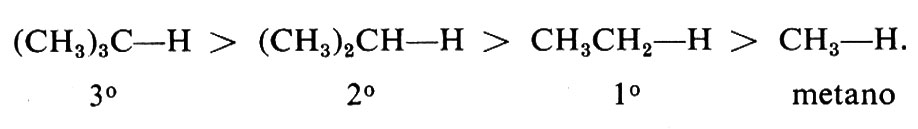
Alcuni dati di velocità relativa (etano = 1) sono riportati nella tab. Il.

Questi risultati si interpretano ammettendo che nello stadio lento di tali reazioni avvenga la rottura del legame C−H e che, pertanto, le entalpie dell'omolisi diano un contributo importante all'energetica del processo. Ora, le energie di dissociazione di questo legame, D(C−H), diminuiscono regolarmente dal metano al C−H terziario (v. tab. I) a causa degli effetti della struttura sulla stabilità dei radicali liberi in formazione. Per lo stesso motivo, oltre agli atomi di idrogeno terziario, sono molto reattivi gli atomi di idrogeno di tipo allilico e benzilico.
L'estrazione dell'idrogeno con X• (eq. 39) è un processo bimolecolare; essa passa per un complesso attivato avente la struttura indicata nell'equazione seguente:
In accordo con questo meccanismo, nel quale il legame C−H è parzialmente scisso, sono anche i risultati sugli effetti isotopici cinetici. Nelle reazioni di questo tipo il deuterio è trasferito dall'idrocarburo all'atomo di alogeno più lentamente dell'idrogeno (effetto isotopico cinetico primario). Si trova infatti che nelle alogenazioni è kH > kD, cioè kH/kD > 1. Una caratteristica delle sostituzioni radicaliche degli alcani è che l'attacco avviene di preferenza sugli atomi univalenti (in questo caso l'idrogeno) anziché sul carbonio tetraedrico come avviene invece nelle sostituzioni nucleofile bimolecolari (SN2).
La principale differenza tra clorurazione e bromurazione è la selettività del reagente radicalico (Cl• o Br•), cioè la capacità che ha detto reagente di discriminare tra i diversi tipi di idrogeno (primario, secondario, terziario, allilico, benzilico). La clorurazione con Cl2 è una reazione poco selettiva; la bromurazione è invece molto selettiva. Ciò risulta evidente dai dati della tab. Il. Nel passare da un atomo di idrogeno primario a uno terziario vi è un rapporto uguale a 6,7 nella clorurazione a 25 °C; il rapporto è invece pari a 4.713 nella bromurazione a 100 °C. Una conseguenza della diversa selettività è la natura dei prodotti di reazione. Nella clorurazione si formano miscele di tutti i possibili monocloroderivati (due cloropropani, due cloroisobutani, ecc.); al contrario, nella bromurazione si può facilmente ottenere un solo prodotto per sostituzione dell'idrogeno più reattivo. La α-bromurazione del toluene è 64.000 volte più rapida di quella dell'etano. Nella reazione con l'etilbenzene si forma solo 1-bromo-1-feniletano, C6H5CHBrCH3, senza alcuna traccia di prodotto β-sostituito.
La diversa selettività delle due reazioni si spiega in base ai diversi andamenti dell'energia potenziale in funzione della coordinata di reazione nel corso del 1° stadio di propagazione della catena. Questi andamenti sono influenzati dalle entalpie di reazione, riportate in tab. III. Nel caso della clorurazione, la cui entalpia scende dai reagenti ai prodotti per le reazioni (1) e (2) della tabella (reazioni esotermiche), le energie di attivazione sono piccole e differiscono di poco per i due substrati etano e isobutano. Ciò equivale a bassa selettività. Nel caso invece della bromurazione l'entalpia cresce dai reagenti ai prodotti per le reazioni (3) e (4) della tabella (reazioni endotermiche); le energie di attivazione e le loro differenze sono relativamente grandi per gli stessi substrati. Di conseguenza la bromurazione è più selettiva della clorurazione.

Tra le addizioni radicaliche va ricordata quella dell'HBr agli alcheni che, in certe condizioni, compete con l'addizione eterolitica mutandone la natura del prodotto finale. Solo dopo aver chiarito la distinzione dei due meccanismi, i chimici potevano razionalizzare i due diversi orientamenti (Markovnikov e anti-Markovnikov) che l'HBr produceva nell'addizione al propilene.
Un altro importante gruppo di reazioni radicaliche è quello delle autossidazioni, in base alle quali il legame R−H si trasforma in un legame idroperossidico R−O−OH per azione dell'ossigeno molecolare che si comporta come un diradicale.
Un grande sviluppo negli ultimi 20 anni ha avuto la fotochimica, ricchissima di processi radicalici, anche in relazione ai problemi dell'utilizzazione dell'energia solare (v. fotochimica).
6. Reazioni dei composti degli elementi diversi dal carbonio
Il grande sviluppo delle nostre conoscenze sulle reazioni dei composti del carbonio (reazioni organiche) ha contribuito in modo notevole a fornire i criteri e le metodologie allo studio della dinamica chimica in generale ed è servito di stimolo alle ricerche sui meccanismi di reazione dei composti degli elementi diversi dal carbonio (reazioni inorganiche e metallorganiche). Il campo di indagine si è arricchito di nuovi orizzonti offerti dalla grande varietà delle strutture elettroniche degli elementi del sistema periodico e dalla più elevata complessità della maggior parte di essi rispetto al carbonio e agli altri elementi leggeri del 2° periodo. Negli ultimi decenni le nostre conoscenze si sono estese praticamente a tutto il sistema periodico, sia agli elementi di transizione sia a quelli non di transizione (v. Basolo e Pearson, 1967; v. Tobe, 1972 e 1973). Si deve subito notare che, nonostante la grande diversità dei sistemi, le reazioni chimiche si possono inquadrare in un numero ristretto di tipi fondamentali, anche se ciascuno di essi sottintende una grandissima varietà di comportamenti che dipendono dalla natura delle molecole specifiche e dalle condizioni sperimentali.
Dal punto di vista dei metodi di studio, valgono sostanzialmente quelli già incontrati per le reazioni del carbonio, cioè i metodi cinetici, stereochimici e isotopici. Tuttavia il quadro conoscitivo di molti processi è ancora basato sulle dimostrazioni fornite da solo alcuni dei metodi possibili. Inoltre nelle reazioni non organiche sono molto frequenti i casi di processi estremamente rapidi che richiedono tecniche di flusso, rilassamento e risonanza magnetica nucleare. Una spinta determinante a questi studi è stata data dallo svilupparsi di queste tecniche. La risonanza magnetica nucleare si è rivelata di grande importanza per lo studio degli aspetti stereochimici, tra cui il destino delle configurazioni enantiomeriche e la pseudorotazione.
Ancbe nelle reazioni non organiche si possono distinguere due grandi gruppi di processi, eterolitici e omolitici, dipendenti dal modo in cui si rompono e/o si formano i legami di coordinazione intorno a un dato atomo. Un processo omolitico presso un atomo metallico conduce generalmente a un'ossidoriduzione. Per esempio, la reazione tra Cr2+(II) e un radicale organico,
Cr2+(II) + •CH2R → Cr3+(III) −CH2R,
va considerata come un'ossidazione monoelettronica del Cr2+(II) (sono sottintese le molecole di acqua coordinata sul cromo). Come già detto nel cap. 4, i reagenti possono essere nucleofili, elettrofili e radicalici.
Nella formulazione più generale della trasformazione che subisce un composto inorganico, è utile considerare la molecola come derivante da un atomo M coordinato da un certo numero di leganti L (MLx). In tal caso si possono distinguere: a) reazioni che riguardano la sfera di coordinazione di M; b) reazioni che interessano lo stato di ossidazione di M; e c) reazioni che riguardano i leganti. Queste ultime sono di grande interesse perché si ritrovano nei processi di molti sistemi catalitici importanti per sintesi di interesse industriale, nei quali i leganti sono organici. Tra le reazioni del gruppo a) possiamo riconoscere tipi di trasformazioni corrispondenti a quelli incontrati tra le reazioni organiche: sostituzione di legante, aumento di numero di coordinazione (addizione), diminuzione di numero di coordinazione (eliminazione), trasposizione di leganti. Inoltre, le molecole MLx possono subire modifiche di configurazione.
Il numero di coordinazione e la configurazione condizionano notevolmente il modo di reagire delle molecole. Pertanto forniscono un criterio di suddivisione delle reazioni dei composti inorganici. I principali gruppi di molecole sono costituiti da quelle a geometria tetraedrica (numero di coordinazione 4), cui appartiene anche il carbonio sp3, da quelle a geometria piana quadrata (n.c. 4) e da quelle a geometria ottaedrica (n.c. 6).
Le sostituzioni sono state oggetto di molti studi nelle diverse strutture. Secondo C. T. Langford e P. Gray si possono in generale distinguere tre tipi di meccanismi del substrato MLx: il meccanismo dissociativo (D), che consiste in un processo in due stadi il primo dei quali conduce, in seguito a eliminazione di un legante, all'intermedio attivo MLx-1; il meccanismo associativo (A), che prevede un intermedio reattivo MLxY in seguito ad addizione del reagente Y; e, infine, il meccanismo di interscambio (I), che non passa per un intermedio vero e proprio. Senza entrare nei dettagli della definizione di questi meccanismi, si fa notare che le sostituzioni presso il carbonio tetraedrico rientrano in questo quadro, dove le reazioni SN2 sono di tipo I e le reazioni SN1 che producono carbocationi ‛liberi' sono di tipo D. Nei paragrafi che seguono daremo uno sguardo d'insieme ai meccanismi di sostituzione tetraedrica, piana e ottaedrica per alcuni elementi, in relazione alla loro posizione nel sistema periodico.
a) Sostituzioni tetraedriche
Le sostituzioni che avvengono presso un atomo a coordinazione tetraedrica sono molto diffuse e interessano sia elementi dei gruppi principali del sistema periodico (non di transizione) sia i metalli di transizione. Rimanendo nello stesso periodo del carbonio, nei primi due gruppi il litio e il berillio sono caratterizzati da una geometria tetraedrica, mentre il legame assume un carattere elettrovalente; negli elementi dei gruppi successivi (boro, carbonio, azoto, ecc.) il legame ha invece carattere, almeno prevalentemente, covalente. La coordinazione tetraedrica si ritrova anche in molti importanti casi di composti di elementi dei periodi successivi, per es. alluminio, gallio, silicio, germanio, stagno, fosforo, arsenico e alogeni.
Tra i metalli di transizione si trovano sostituzioni su strutture tetraedriche caratterizzate sia da legami essenzialmente elettrovalenti (TiCl4, MnCl42-, FeC4-, CoCl42-, NiCl42-), sia da legami covalenti (Ni(CO)4, Pt(PPh3)4, Cu(PPh3)3I) aventi metalli a configurazione d10. A questi esempi si aggiungono importanti e diffusi substrati, quali gli ossianioni MnO4-, CrO42-, Cr2O72-.
Del carbonio tetraedrico si è trattato nel precedente capitolo. I meccanismi di sostituzione, secondo la definizione di Langford e Cray, avvengono presso il carbonio con meccanismi di tipo D o I. Nonostante la formale analogia tra carbonio e silicio, presso il silicio si trovano invece meccanismi di tipo I o A; presso il fosforo meccanismi di tutti e tre i tipi. Nei metalli di transizione, a seconda dello stato di ossidazione, si possono avere meccanismi D o I, ovvero I o A. Vediamo di illustrare alcuni modelli di meccanismo di sostituzione tetraedrica.
Vi sono varie differenze di proprietà tra carbonio e silicio, tali da giustificare un quadro di reattività ben distinto di questi due elementi al di là di analogie formali. Si hanno differenze significative nel più grande raggio covalente del silicio, nelle energie di dissociazione generalmente minori, nell'accessibilità energetica di orbitali d vacanti e, infine, nei numeri di coordinazione. Numeri di coordinazione inferiori a quattro sono estremamente improbabili nel silicio, a differenza del carbonio (v., per es., i carbocationi), mentre una covalenza superiore a quattro è frequente.
Una diretta conseguenza di queste caratteristiche della coordinazione è l'assenza di meccanismi dissociativi nella sostituzione al silicio tetraedrico. Laddove (C6H5)2CCl subisce la solvolisi con un meccanismo dissociativo, promosso dalla stabilità del carbocatione intermedio, (C6H5)3C+, l'analogo composto del silicio, (C6H5)3SiCl, reagisce con un meccanismo bimolecolare. Nel primo caso, l'aggiunta di basi non ha effetto sulla velocità di reazione, mentre nel secondo la fa aumentare. In sistemi particolarmente adatti sono stati tuttavia individuati meccanismi che presuppongono uno ione siliconio come intermedio, ma si tratta presumibilmente di specie cationiche a ponte di silicio piuttosto che di cationi R3Si+ liberi veri e propri. Un esempio è l'eliminazione solvolitica dell' 1,2-dibromo-1-trimetilsililpropano, per la quale è stato proposto il meccanismo illustrato nello schema (42), dove si forma un catione intermedio a ponte di silicio.
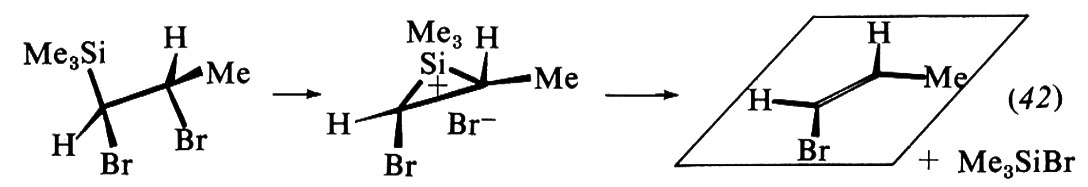
D'altra parte neanche il meccanismo bimolecolare che si ritrova di norma in tutte le sostituzioni nucleofile dei composti del silicio è generalmente analogo a quello indicato dal simbolo SN2 per i composti del carbonio. Vi possono essere diversi modi di reagire, che il substrato sceglie in base a differenze di condizioni, anche modeste.
Nei composti R1R2R3SiX (con il silicio asimmetrico), se X è un buon gruppo uscente (per es. Cl) la reazione di sostituzione bimolecolare conduce a inversione di configurazione (schema 43), a condizione che il nucleofilo Y- sia più basico di X-.

Se X non è un buon gruppo uscente (H, OCH3) la reazione avviene con la conservazione della configurazione. In questo caso si propone un meccanismo a 4 centri nel quale l'attacco di Y- avviene dallo stesso lato di uscita di X. Un esempio è la reazione di R3SiOMe con AlH4- (schema 44). Vi è infine un terzo meccanismo che conduce alla
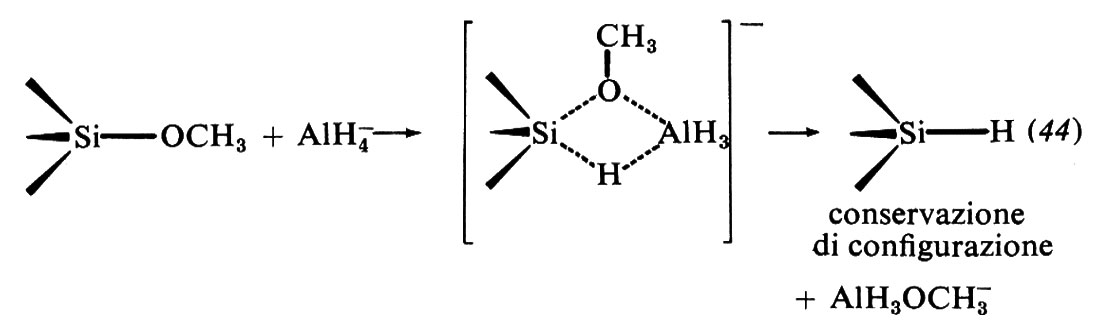
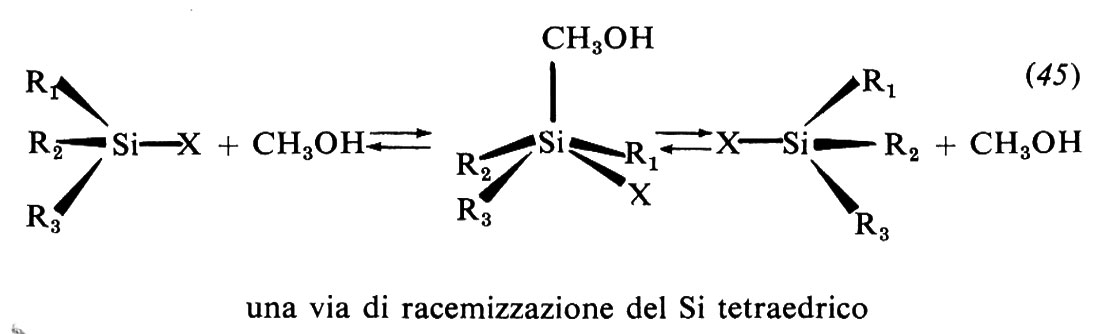
racemizzazione del prodotto di reazione (schema 45). Un esempio è l'effetto dell'alcool metilico sull'1 -naftilfenilmetilfluorosilano che, pur non concludendosi con la sostituzione del fluoro, illustra una terza via di sostituzione bimolecolare del silicio. Si noti che il meccanismo bimolecolare che conduce a tutti e tre i possibili esiti stereochimici (inversione, ecc.) non è necessariamente a un solo stadio. È anzi una caratteristica del silicio, che ha tendenza ad aumentare la sua coordinazione, il poter passare attraverso un vero composto intermedio pentacoordinato, come è illustrato negli schemi (43) e (45).
Un altro centro tetraedrico molto studiato è il fosforo, specie in molecole del tipo R3PO. Anche qui prevalgono meccanismi associativi e percorsi stereochimici che permettono inversione o conservazione a seconda dei casi. Tuttavia, anche se meno frequenti e meno ben dimostrati, esistono esempi di processi dissociativi, come negli ossianioni, quale Cl2PO2-.
Tra i derivati dei metalli di transizione, molti studi sono stati rivolti alle sostituzioni del nicheltetracarbonile, Ni(CO)4, e di composti analoghi, con opportuni leganti (fosfine) o a scambi con CO. I risultati indicano un meccanismo dissociativo (D) attraverso un intermedio a più basso numero di coordinazione; in accordo con questo tipo di sostituzione la velocità di trasformazione è indipendente dalla concentrazione e dalla natura del gruppo entrante. Ad analoghe conclusioni si giunge per le reazioni di composti tetracoordinati del palladio e del platino del tipo M(PPh3)4. Ciò si spiega con una non facile disponibilità di orbitali del metallo capaci di formare legami di coordinazione con i reagenti donatori di elettroni.
b) Sostituzioni nei complessi tetracoordinati piani
Un altro gruppo di interessanti reazioni è costituito dalle sostituzioni nucleofile presso centri metallici a tetracoordinazione piana. Le più importanti sono quelle in cui il metallo ha la configurazione d8, particolarmente Rh(I), Ir(I), Ni(II), Pd(II), Pt(II) e Au(III). Di gran lunga il più adatto a ricerche estese e approfondite si è rivelato il platino (II).
Le strutture tetracoordinate piane di questi metalli hanno carattere ‛insaturo', perché i centri a configurazione d8 sono in grado di utilizzare 5 orbitali per formare 5 legami di coordinazione con leganti donatori di coppie di elettroni. In tal modo questi centri raggiungono la configurazione a 18 elettroni del più vicino gas nobile che segue. Strutture pentacoordinate sono infatti molto comuni in questi metalli e suggeriscono l'ipotesi che le sostituzioni dei composti a struttura tetracoordinata piana con i reagenti nucleofili avvengano attraverso un incremento preliminare del numero di coordinazione da 4 a 5. Oli studi di queste reazioni avvalorano questa ipotesi.
La cinetica chimica si presenta generalmente complessa. L'equazione della velocità di reazione ha espressione binomiale, con un termine del primo ordine e uno del secondo. Si dimostra tuttavia che si tratta di due percorsi paralleli, ciascuno consistente in un processo di tipo associativo (bimolecolare), in accordo con le previsioni ricordate poc'anzi. Essi sono rappresentati dalle eqq. (46) e (47).

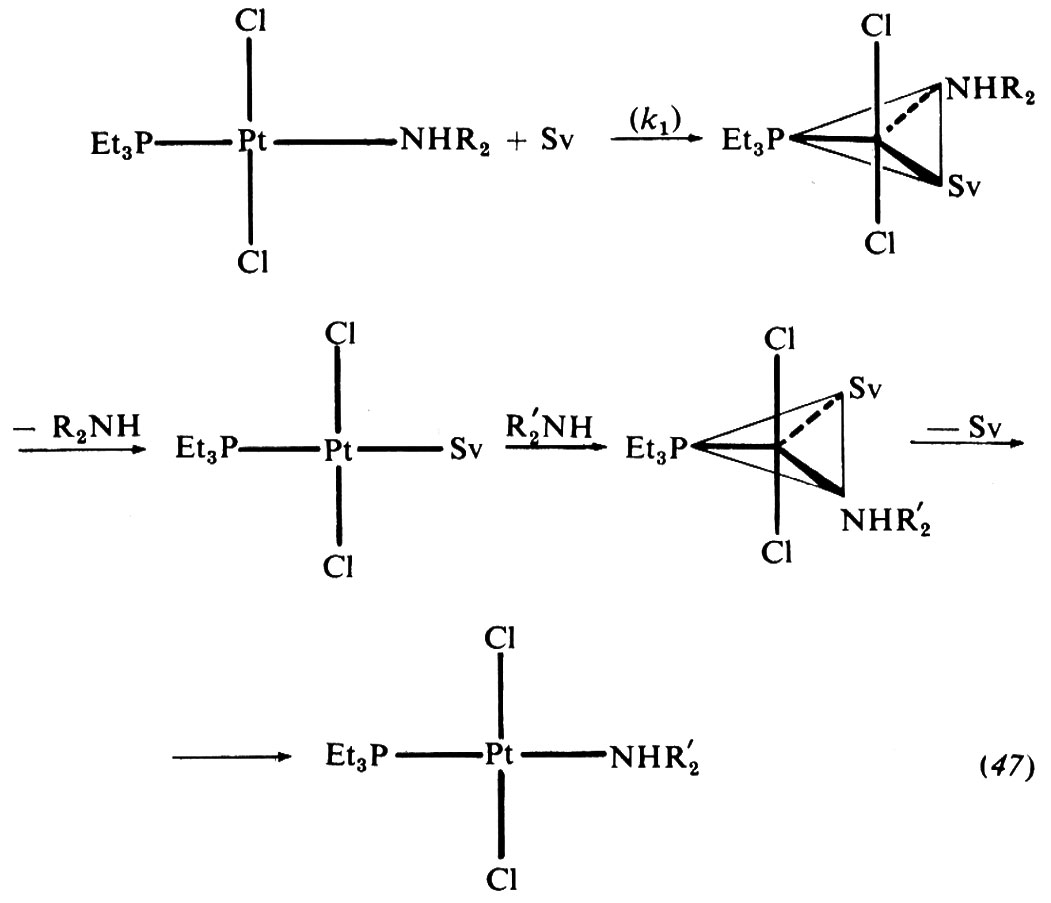
Nell'eq. (47) si nota che il percorso corrispondente alla reazione del primo ordine (k1) si interpreta ammettendo la partecipazione del solvente (Sv), che agisce da blando nucleofilo. Lo studio degli effetti del solvente è in accordo con questo suo ruolo. Nell'esempio illustrato, trans-[PtCl2(PEt3)(NHR2)] + R2′NH, la reazione può essere infatti esaminata con un'ampia scelta di solventi, poiché il sistema è costituito da molecole neutre e solubili anche in solventi non polari. Il carattere associativo di entrambi i percorsi è illustrato anche dalla loro sensibilità agli effetti sterici. L'aumento della coordinazione nello stato di transizione dello stadio lento della reazione conduce a un sistema più ‛affollato' e la reazione rallenta al crescere delle dimensioni dei gruppi in gioco per fattori fino a varie potenze di 10. Naturalmente nel percorso del secondo ordine la velocità di reazione dipende dalla concentrazione e dalla natura del nucleofilo. Determinando gli effetti cinetici di numerosi reagenti in diversi composti piani del platino(II), si è trovato che esiste una comune scala dei poteri nucleofili dei reagenti, che permette di prevedere la reattività chimica nell'ambito di questo tipo di composti del platino.
I due modelli di meccanismo proposti per la sostituzione del Pt(II) tengono conto anche della stereochimica di queste reazioni, che avvengono normalmente con conservazione di configurazione. Ciò è infatti possibile se, come e illustrato nelle eqq. (46) e (47), l'intermedio di reazione, sia che esso si formi per attacco diretto del nucleofilo sia che intervenga il solvente, assume la configurazione di una bipiramide trigonale nella quale le due posizioni assiali sono occupate dai gruppi (Cl, Cl), inizialmente cis rispetto al gruppo uscente (NHR2), e le posizioni sul piano trigonale sono occupate dai gruppi uscente ed entrante e dal gruppo inizialmente trans rispetto al gruppo uscente.
Le sostituzioni nucleofile dei composti piani del platino (II) sono anche interessanti per il cosiddetto effetto trans, che consiste nell'influenza che i leganti occupanti la posizione trans rispetto al gruppo uscente esercitano sulla velocità di reazione e si manifesta attraverso effetti di orientamento del reagente di notevole rilevanza preparativa. Per la sua origine cinetica, l'effetto trans fornisce un altro criterio di studio del meccanismo di queste reazioni.
c) Sostituzioni presso centri a struttura ottaedrica
La struttura ottaedrica è tra le più diffuse e frequenti di tutta la chimica. Essa caratterizza la distribuzione di atomi e gruppi atomici intorno agli elementi dei più diversi settori del sistema periodico, legati con legami chimici sia ionici (come nei cationi Mg(H2O)62+, Fe(H2O)63+), sia covalenti (come nelle molecole SF6 e Cr(CO)6 e nell'anione Fe(CN)64-), ma è particolarmente favorita nella configurazione d6 degli elementi di transizione, quali V(−I), Cr(0), Mo(0), Mn(I), Fe(II), Ru(II), Co(III), Rh(III), Ni(IV), Pd(IV), Pt(IV). In tutte le sostituzioni note dei composti ottaedrici, si nota una notevole uniformità di meccanismi di reazione, dove domina, sia pure con leggere variazioni, il carattere dissociativo, anche se molte configurazioni elettroniche sono ‛insature' e potrebbero permettere processi associativi. Le reazioni più studiate sono quelle dei complessi interni cosiddetti inerti (d2sp3, configurazioni da d3 a d6), tra i quali figura in primo luogo il Co(III), seguito da Cr(III), Rh(III), Ru(III), Ir(III). Le tecniche di studio dei meccanismi di reazione sono quelle classiche di cui abbiamo dato esempi in tutto l'articolo. Le reazioni generalmente dipendono poco dalla concentrazione e dalla natura del reagente nucleofilo: questa è una prima caratteristica sperimentale a favore del meccanismo dissociativo. Tuttavia solo in alcuni casi è stato possibile identificare un intermedio pentacoordinato labile; tra questi sono le reazioni della serie Co(NH3)5X2+, dove l'intermedio è stato dimostrato mediante criteri di cattura nel corso di esperienze di idrolisi basica.
Senza entrare nei dettagli della cinetica chimica, le reazioni possono andare da un meccanismo di interscambio (Id) a un meccanismo dissociativo (D). Al primo tipo appartiene la reazione del complesso Co(NH3)5H2O3+ con nucleofili anionici Y-; si ammette che in essa abbia luogo un equilibrio preliminare (rapido), nel quale il reagente prende posto nella sfera di solvatazione del complesso, cui segue lo scambio di posto tra il legante uscente e il legante entrante che si trova nelle immediate vicinanze:
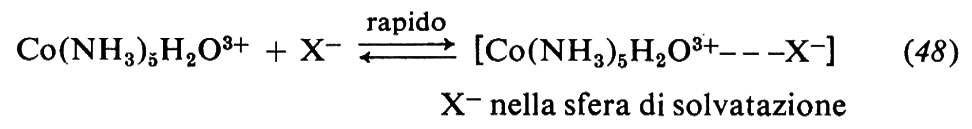
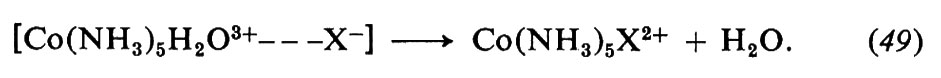
Del tipo D è la reazione del complesso Co(CN)5H2O2- con analoghi reagenti, dove nel preequilibrio avviene una dissociazione completa seguita dall'attacco del reagente sull'intermedio pentacoordinato:
Co(CN)5H2O2- ⇄ Co(CN)52- + H2O (50)
Co(CN)52- + X- → Co(CN)5X3-. (51)
La differenza tra i due modi di reagire è più di misura che di qualità, poiché nel secondo caso l'intermedio pentacoordinato ha un tempo di vita media finito e sufficientemente lungo da potersi equilibrare con la sfera di solvatazione circostante. Ciò conduce ad alcune conseguenze sperimentali aventi carattere diagnostico (per es. la forte capacità discriminante dell'intermedio nei confronti del nucleofilo e l'indipendenza del suo comportamento dalla natura del gruppo uscente).
Particolari caratteristiche cinetiche si osservano nell'idrolisi basica degli amminocomplessi del Co(III) e di alcuni altri elementi. Queste reazioni mostrano cinetiche del secondo ordine,
v = k[complesso][OH-],
che sono la risultante di un processo complesso costituito da un preequilibrio acido-base, in cui avviene il trasferimento di un protone da un gruppo amminico al reagente, seguito dalla dissociazione lenta della base coniugata (b.c.) formatasi e quindi dal rapido ripristino della struttura esacoordinata con una molecola di solvente:

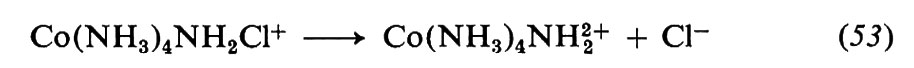
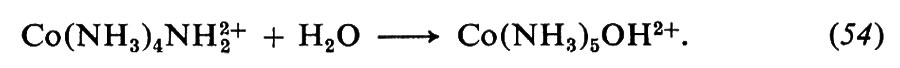
Una delle importanti prove a favore di questo meccanismo è la scarsa reattività dei complessi amminici mancanti di legami N−H, poiché in tal caso non si può formare la b.c. intermedia caratteristica di questa ipotesi. La formazione della b.c. è giustificata da un effetto coniugativo stabilizzante ammido-metallo.
In molti casi le sostituzioni ottaedriche avvengono con la completa conservazione della configurazione (cis o trans). Ciò si spiega ammettendo che l'intermedio pentacoordinato conservi la distribuzione dei gruppi secondo una piramide tetragonale, che risulta dall'ottaedro per allontanamento del gruppo uscente (eq. 55).
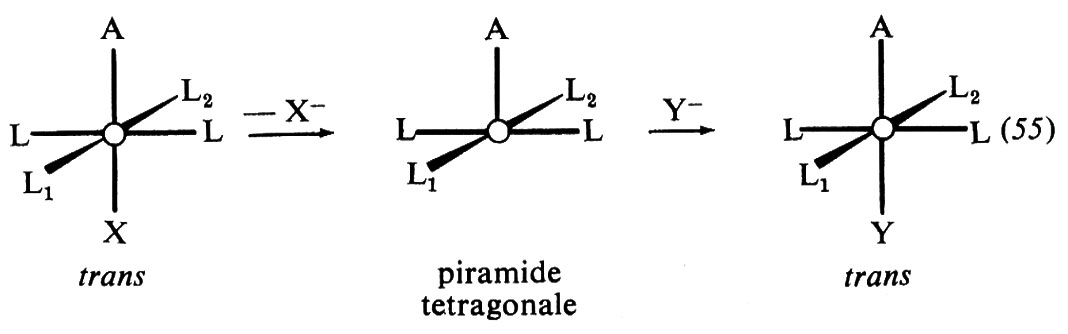
In alcune reazioni, quale l'idrolisi basica del cis- e trans-Co(NH3)4Cl+2, avviene un parziale cambiamento di configurazione e il prodotto di reazione è una miscela di due isomeri. Questo comportamento stereochimico si interpreta ammettendo che l'intermedio pentacoordinato assuma una configurazione di bipiramide trigonale in seguito a un assestamento di due leganti trans tra di loro (L1, L2) e in cis rispetto al gruppo uscente (eq. 56), cui segue l'attacco del reagente per generare i due isomeri.

7. Considerazioni e prospettive sullo stato delle conoscenze delle reazioni chimiche
Nei precedenti capitoli abbiamo notato che la ricerca di un approfondimento delle nostre conoscenze sulla natura delle reazioni chimiche ha creato una piattaforma culturale comune a gruppi di reazioni tradizionalmente rimasti separati nelle divisioni classiche della chimica (inorganica, organica, ecc.). Sebbene certe caratteristiche di struttura e di reattività siano prevalenti per determinati elementi e per i loro composti, non vi è un solo elemento, con la possibile eccezione dei gas nobili la cui tendenza a reagire è molto limitata, che abbia caratteristiche non condivise, per qualità se non per misura, da molti altri elementi. Per una visione unificata della chimica, ha sempre meno senso tenere distinta la chimica organica dalla chimica inorganica, se si considera che il carbonio è in grado di combinarsi con tutti gli altri elementi del sistema periodico producendo una grandissima varietà di comportamenti che non sono né tipicamente ‛organici' né tipicamente ‛inorganici'. I metodi della chimica fisica servono ugualmente bene tanto al progresso della chimica del carbonio quanto a quello della chimica degli altri elementi. Essi stessi non hanno ragion d'essere se non sono intimamente collegati ai problemi di tutto il ‛materiale' chimico, che è appunto costituito dagli elementi e dai loro composti. Al primo Simposio internazionale di istruzione chimica universitaria, tenutosi a Frascati nel 1969 (v. IUPAC-CTC e CNR, 1970, p. 16) è stata espressa l'opinione che ‟[...] tutte le branche interne della chimica si sono sviluppate a causa di una classificazione artificiale della natura dovuta a un processo di prima approssimazione nella nostra acquisizione della scienza. Obiettivamente non ci sono singole discipline, quali la chimica inorganica, la chimica organica o la chimica fisica, se non nella nostra mente. Classificando la scienza in tal modo, operiamo una distorsione della natura. Più conosciamo la chimica, meno abbiamo bisogno di suddividerla secondo gli schemi tradizionali [...]".
Di conseguenza, si è più disposti a riconoscere che esista il chimico organico, il chimico fisico, ecc., piuttosto che esistano la chimica organica, la chimica fisica, ecc., cioè le discipline che essi professano, in quanto è nell'operatore che sono pensabili dei ragionevoli limiti, non nella scienza. Poiché l'avanzamento delle ricerche in chimica ha prodotto delle brecce profonde nei confini tra le divisioni tradizionali, si pongono oggi problemi di rinnovamento nel modo di presentare la chimica a chi ne intraprende lo studio e nel modo di formare i chimici di domani. Iniziare uno studente a una chimica senza barriere interne, compito che si è rivelato finora molto arduo (v. IUPAC-CTC e CNR, 1970, p. 3), sarebbe un potente catalizzatore per i futuri progressi di questa scienza. Questo è certamente un primo fattore di sviluppo futuro anche per lo studio delle reazioni chimiche.
Ci si può chiedere in quali direzioni si prevede un avanzamento dello studio delle reazioni chimiche negli anni avvenire. La risposta può ragionevolmente venire dai trends attuali della ricerca chimica non solo nell'ambito della chimica in senso accademico, ma anche al di fuori, nella realtà in cui ci muoviamo. Un certo tipo di sviluppo dipende dai progressi delle tecnologie sperimentali. La comparsa di tecniche che permettono di studiare reazioni estremamente rapide dischiude nuovi campi di studio, specialmente tra le reazioni inorganiche. Il ruolo delle interazioni solvente-solvente nelle reazioni chimiche è uno dei problemi di fondo della reattività chimica in soluzione. Esso è meglio compreso se confrontato con il comportamento delle reazioni in fase gassosa. L'attenzione di molti gruppi di ricerca è rivolta verso questi due tipi di problemi ed è facile prevedere che molte reazioni potranno essere intimamente interpretate in seguito a un ulteriore progresso di questi studi. Un'altra direzione molto promettente è lo studio delle reazioni organometalliche e dei meccanismi di catalisi omogenea che passano attraverso intermedi attivi organometallici (v. composti organometallici; v. catalisi omogenea). Molta luce deve ancora essere fatta sulla natura delle reazioni eterogenee che offrono vasti campi di indagine ricchi di conseguenze, dalla catalisi micellare (v. Fendler e Fendler, 1975) alle reazioni che avvengono sulla superficie dei solidi (v. catalisi eterogenea). Molte speranze sono riposte nelle reazioni fotocatalizzate (v. fotochimica) e nelle reazioni chimiche che permettono di immagazzinare energia solare nei sistemi materiali.
Ma se l'integrazione della chimica, con l'abbattimento delle barriere culturali nel suo interno, è un importante fattore di sviluppo, non meno importante è la ricerca nelle aree di interfaccia della chimica con altre discipline: la fisica, la biologia e l'ingegneria. A questo punto è stato dato grande rilievo nel corso dei lavori della Conferenza internazionale di istruzione chimica tenutasi a Snowmass-at-Aspen, Stati Uniti, nel 1970 (v. American Chemical Society, 1971). Occorrerà dare maggior riconoscimento all'importanza del contributo della chimica alla soluzione dei problemi connessi con i sistemi complessi, se si considera che il comportamento di tali sistemi (il motore di un razzo, la cellula vivente, l'atmosfera inquinata) dipende in modo critico dalle proprietà chimiche e, in modo particolare, dalle reazioni chimiche.
bibliografia
American Chemical Society, International conference on education in chemistry, in ‟Journal of chemical education", 1971, XLVIII, pp. 3-13.
Basolo, F., Pearson, R. G., Mechanisms of inorganic reactions, New York 1967.
Chapman, N. B., Shorter, J., Advances in linear free energy relationships, London 1972.
Coetzee, J. F., Ritchie, C.D., Solute-solvent interactions, New York 1969.
Fendler, J. H., Fendler, E. J., Catalysis in micellar and macromolecular systems, New York 1975.
Frost, A. A., Pearson, R. G., Kinetics and mechanism, New York 1961.
Glasstone, S., Thermodynamics for chemists, New York 1947.
Glasstone, S., Laidler, K. J., Eyring, H., The theory of rate processes, New York 1941.
Illuminati, G., Chimica organica, Roma 1971.
Illuminati, G., Solvent effects on selected organic and organometallic reactions. Guidelines to synthetic applications, in Solutions and solubilities (a cura di M. R. J. Dack), parte II, New York 1976, pp. 159-233.
IUPAC-CTC e CNR (Roma), University chemical education, London 1970.
Lewis, G. N., Randall, M., Thermodynamics, New York 1923.
McManus, S. P. (a cura di), Organic reactive intermediates, New York 1973.
March, J., Advanced organic chemistry, New York 1977.
Melander, L., Isotope effects on reaction rates, New York 1960.
Solov'ev, J. I., L'evoluzione del pensiero chimico, Milano 1976.
Tobe, M. L., Reaction mechanisms in inorganic chemistry, London 1972.
Tobe, M. L., Inorganic reaction mechanisms, London-Southampton 1973.