
Catalisi
Catalisi
Catalisi enzimatica, di Herbert Gutfreund
Catalisi eterogenea, di Alessandro Cimino
Catalisi omogenea, di Jack Halpern
Catalisi enzimatica
di Herbert Gutfreund
Lo stato dell'enzimologia nel 1950
Uno dei progressi fondamentali dell'età della biologia molecolare (1950-1970) è stato la descrizione, attraverso i processi chimici, del meccanismo della catalisi enzimatica. Anche se una rassegna della precedente storia dell'enzimologia non sarebbe di alcun aiuto per la comprensione delle teorie attualmente considerate valide, è tuttavia necessario riassumere brevemente le idee correnti nel 1950 per poter spiegare in qual modo le tecniche chimiche, cristallografiche e cinetiche abbiano fornito le informazioni che sono necessarie per descrivere i meccanismi della catalisi enzimatica.
Parecchie migliaia di enzimi, che catalizzavano reazioni chimiche differenti, erano state isolate da piante, animali e microrganismi. In tutti i casi, si era trovato che gli enzimi sono costituiti da una molecola proteica alla quale, in alcuni casi, possono legarsi componenti non proteici, fattori essenziali che contribuiscono all'integrità strutturale o all'attività catalitica.
L'intervallo dei pesi molecolari trovato per le proteine enzimatiche varia entro limiti piuttosto ampi, che vanno da un minimo di 103 a un massimo dell'ordine di 106 Dalton. Studi all'ultracentrifuga lasciavano prevedere che gli enzimi avrebbero potuto essere purificati dando luogo a preparazioni omogenee per dimensione e forma molecolare.
L. Michaelis, sulla base di studi propri e di altri, giunse a due importanti conclusioni sul comportamento delle reazioni enzimatiche: la prima, riguardante la velocità delle reazioni catalizzate da enzimi, a concentrazione di enzima costante, mostra una dipendenza dalla concentrazione di substrato del tutto caratteristica. La fig. 1 illustra come, a bassa concentrazione di substrato, la velocità vari linearmente con questa, mentre a concentrazione più elevata si raggiunga una velocità di saturazione. Questi risultati sono stati interpretati ipotizzando la formazione di un complesso enzima-substrato come prima tappa della reazione catalitica. In condizioni di saturazione tutti i siti catalitici presenti in una soluzione sono occupati da molecole di substrato. Questo risultato suggeriva un'analogia con la teoria dell'adsorbimento isotermico usata nella descrizione della catalisi sulla superficie dei metalli. I parametri di Michaelis di una reazione enzimatica costituiscono importanti caratteristiche di ciascun particolare enzima, anche se il loro significato fisico è risultato successivamente più complesso di quanto indicato in questa introduzione (v. enzimi).
Un secondo punto posto in evidenza da Michaelis riguardava la dipendenza critica dal pH della velocità delle reazioni enzimatiche, che sembrava indicare l'importanza dello stato di protonazione dei gruppi ionizzabili nei residui di amminoacidi dell'enzima. Sia le proprietà ioniche dei residui di amminoacidi, sia gli effetti dell'ambiente chimico e fisico all'interno delle molecole proteiche sono stati ampiamente studiati.
Molti tra gli enzimi più studiati nei primi tempi dell'enzimologia furono quelli che catalizzano reazioni idrolitiche. Ciò diede grande impulso alle ipotesi sui meccanismi enzimatici basati sulla concentrazione locale degli ioni idrogeno in porzioni specifiche della superficie proteica.
L'interesse degli studi sulle reazioni tra gli enzimi e i loro substrati risiede principalmente nella specificità di formazione del complesso enzima-substrato e dei successivi processi catalitici. Il riconoscimento dello specifico substrato da parte dell'enzima rappresenta infatti, dal punto di vista biologico ed enzimatico, la parte concettualmente più importante dello studio degli enzimi. Nelle prime ricerche, le correlazioni quantitative tra le variazioni di struttura del substrato e l'affinità per l'enzima dovevano essere ottenute con metodi indiretti: la costante di Michaelis rappresenta, in molti casi, una misura dell'affinità, ma deve essere interpretata con cautela. Tutti i principi fondamentali della cinetica degli stati stazionari sono stati stabiliti da J. B. S. Haldane (v., 1930). Lo schema originale di Michaelis era basato su questo semplice modello:
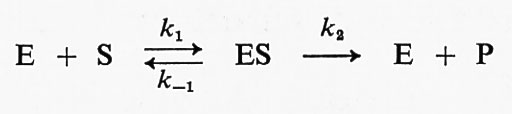
(E rappresenta l'enzima, S il substrato, P il prodotto, ES il complesso enzima-substrato e le k sono le costanti di velocità delle reazioni), con un solo intermedio in rapido equilibrio con l'enzima libero e il substrato. La dissociazione
ES √ E + P
veniva considerata come un processo lento, limitante la velocità della reazione. Quando si verificano queste particolari condizioni, allora
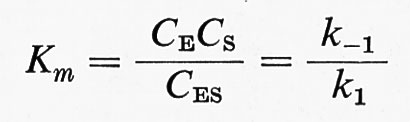
e la velocità di saturazione o velocità massima è
V = k2 C0E ,
in cui le C sono le concentrazioni all'equilibrio delle varie specie e C0E è la concentrazione totale dell'enzima.
G. M. Briggs e J. B. S. Haldane dimostraronò comè fosse possibile derivare espressioni per Km e V senza ricorrere alle ipotesi restrittive di un unico intermedio in equilibrio rapido. Se si misura la velocità iniziale v a una data concentrazione di substrato e se la quantità di substrato consumata durante la misura è trascurabile, si può affermare che la misura della velocità così fatta si riferisce ad una condizione di stato stazionario. Lo stato stazionario di un sistema viene definito tramite gli intermedi di reazione, che possono comprendere più specie della forma ES ed EP:
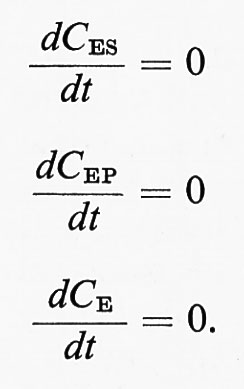
Per ogni meccanismo di reazione, quale, ad esempio,
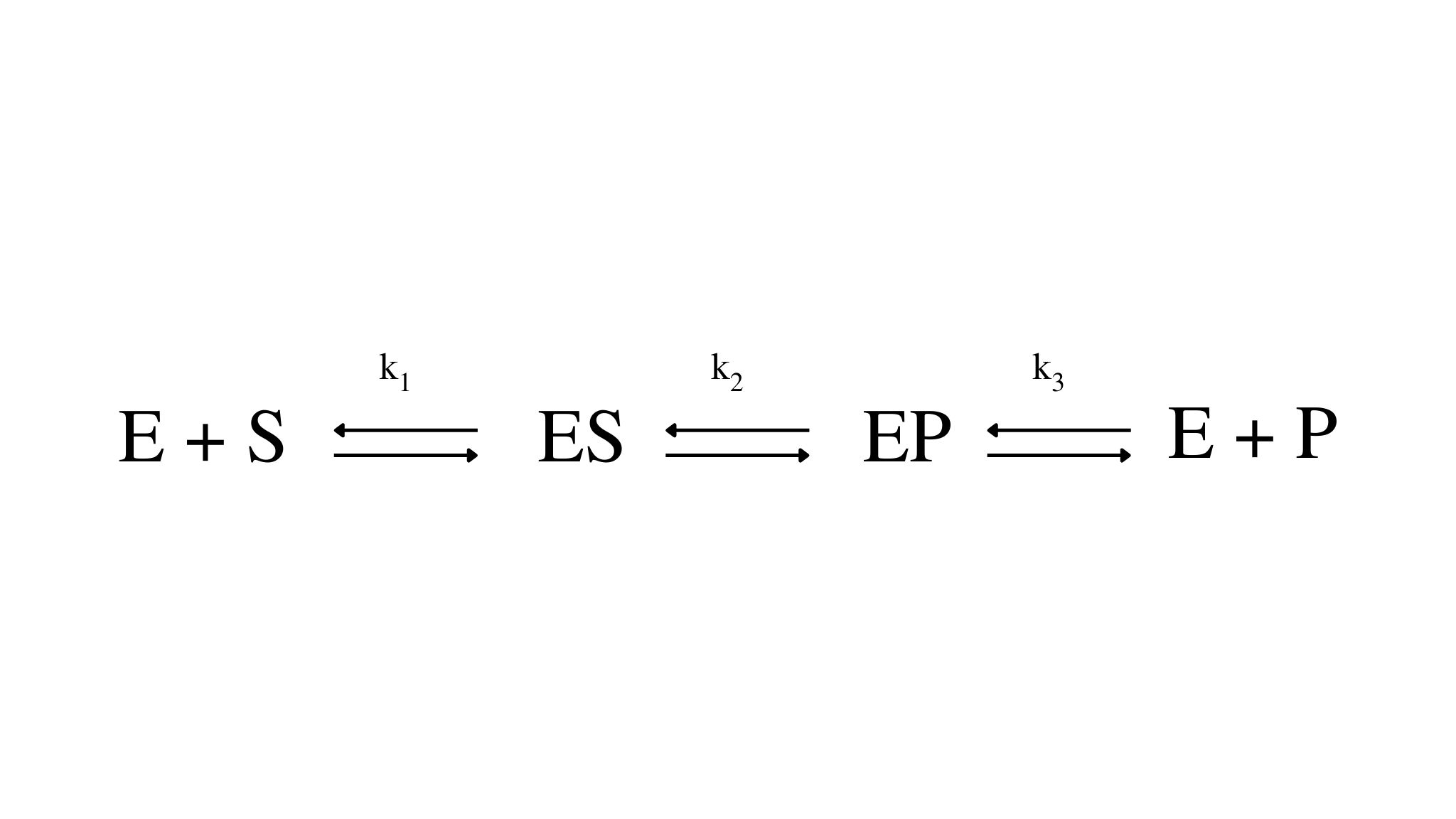
è possibile scrivere un'equazione di stato stazionario per ciascuna forma sotto cui si trova l'enzima. Nell'esempio proposto, l'equazione per la specie ES assume la forma:
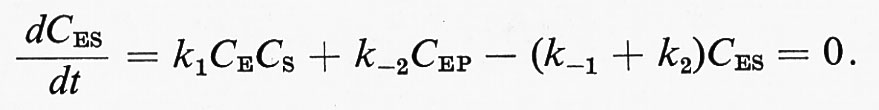
Questo sistema di equazioni, unitamente alla
C0E = CE + CES + CEP,
permette di esprimere la velocità iniziale, definita da
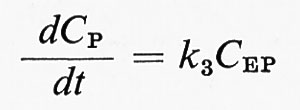
(CP si assume uguale a zero all'inizio della reazione), tramite C0E, CS e due costanti A e B:
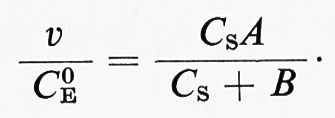
A e B, che sono esprimibili direttamente per mezzo delle costanti di velocità, assumono una forma più complessa via via che aumenta il numero di intermedi previsto dal meccanismo. Per il meccanismo di Michaelis:
A = k2 e B = (k2 + k-1/k1 ;
tuttavia, poiché Michaelis assume k-1 ≪ k2, si potrà assumere B ≅ k-1/k1. B risulta equivalente alla costante di Michaelis e, sebbene la sua espressione risulti spesso molto più complessa di quella data dalla semplice relazione B ≅ k-1/k1, può essere sempre determinata con uno dei metodi rappresentati nella fig. 1.
A partire dal 1912 furono condotti numerosi e approfonditi studi sull'influenza del pH sui parametri cinetici delle reazioni enzimatiche, ma i risultati furono usati a scopo descrittivo piuttosto che nell'intento di trovarne l'interpretazione attraverso opportuni meccanismi. Negli anni cinquanta alcuni autori estesero il metodo degli stati stazionari allo studio di reazioni di enzimi con due substrati e dimostrarono che era possibile stabilire un sistema di equazioni cinetiche adattabile a ogni meccanismo. Ciò risultò assai utile per distinguere tra diversi meccanismi possibili mediante la conoscenza di un appropriato insieme di dati sulle velocità, in funzione delle concentrazioni dei substrati.
Sebbene la maggior parte dei metodi e delle idee che verranno particolarmente discussi più avanti fosse già stata presa in considerazione nei primi studi sugli enzimi, i metodi qui riassunti maturarono soltanto intorno al 1950.
La chimica delle proteine applicata agli enzimi e ai composti enzima-substrato
I primi enzimi a essere studiati in modo approfondito mediante tutte le possibili tecniche moderne furono - per ragioni di carattere pratico, come la facilità di reperirne notevoli quantità - quelli secreti dal pancreas (tripsina, chimotripsina, ribonucleasi). Questi enzimi hanno un peso molecolare relativamente basso (attorno a 20.000) e sono monomerici, si trovano, cioè, in soluzione sotto forma di una singola catena polipeptidica comprendente un certo numero di legami covalenti nella forma di ponti −S−S− che uniscono tra loro le due metà di un residuo di cistina (v. fig. 2).
Il successo ottenuto da F. Sanger nel determinare la sequenza degli amminoacidi nelle catene polipeptidiche dell'insulina ebbe come conseguenza l'applicazione del suo metodo, e di altri simili, per chiarire la struttura primaria di molte altre proteine enzimatiche. La prima conclusione di carattere generale che si poté trarre dall'analisi delle sequenze fu che la sequenza degli amminoacidi di una proteina pura isolata da un animale di una data specie è unica. Paragonando tra di loro le sequenze di una stessa proteina enzimatica isolata da specie differenti si vide che nei diversi preparati vi erano alcune specifiche sostituzioni di residui di amminoacidi. Talvolta, sostituzioni di amminoacidi (mutazioni) si possono trovare anche in campioni isolati da animali diversi, ma appartenenti alla stessa specie. Questi risultati sulle proteine enzimatiche costituiscono oggi uno dei dogmi della genetica molecolare.
Dopo che diversi enzimi delle principali vie metaboliche furono purificati in quantità sufficienti per uno studio particolareggiato, si trovò che essi esistono in soluzione sotto forma di polimeri di catene polipeptidiche tra loro identiche. Le unità polipeptidiche sono tra loro legate da forze non covalenti (ponti salini, legami a idrogeno ed interazioni idrofobiche) e possono essere dissociate reversibilmente in unità individuali mediante vari reagenti. La forma di enzima polimerico che si trova più comunemente è il tetramero, dotato di quattro siti catalitici, ma si trovano anche dimeri, esameri e stati di aggregazione più complessi. Normalmente falliscono i tentativi di ottenere unità monomeriche attive dagli enzimi polimerici. Sembra che la struttura quaternaria (orientamento relativo delle subunità) sia essenziale per mantenere la catena polipeptidica dei monomeri nella corretta configurazione necessaria all'attività enzimatica.
La conoscenza della struttura primaria di una proteina enzimatica costituisce un'informazione indispensabile per tutti gli studi successivi, ma non dà, di per se stessa, alcuna informazione su quali siano i residui di amminoacido che, trovandosi nelle vicinanze del sito attivo, risultano essenziali all'attività catalitica. Osservando, per esempio, la sequenza di amminoacidi illustrata nella fig. 2, sarebbe impossibile prevedere che i residui istidinici 12 e 119 partecipino entrambi alle reazioni del sito catalitico. Uno dei più interessanti problemi chimici sulle reazioni del sito attivo riguarda l'effetto dell'ambiente sulla reattività dei gruppi. La molecola proteica, grazie alla complessa disposizione dei diversi residui di amminoacidi, ha la singolare capacità di inserire i propri gruppi in un dato intorno. Si sono potuti stabilire alcuni principi generali che regolano il modo in cui una catena polipeptidica si avvolge su se stessa assumendo una struttura tridimensionale. I gruppi polari rimangono sulla superficie della proteina, mentre i residui non polari restano vicini fra loro all'interno della molecola. Si osserva frequentemente, però, che proprio nei punti più interessanti, ad esempio nel sito attivo, questa regola non è soddisfatta. L'analisi strutturale e la spettroscopia, di cui discuteremo più avanti, forniscono ragguagli sulla situazione locale in cui si trovano i gruppi di particolare interesse.
Un interessante esempio di reattività selettiva può essere utilizzato per illustrare un ulteriore modo di affrontare lo studio chimico degli enzimi. La tripsina, la chimotripsina e diversi altri enzimi idrolitici vengono inattivati dal diisopropilliuorofosfato (DFP). L'uso di DFP marcato con 32P ha dimostrato che una molecola di inibitore reagisce col gruppo −OH di un residuo di serina dell'enzima:
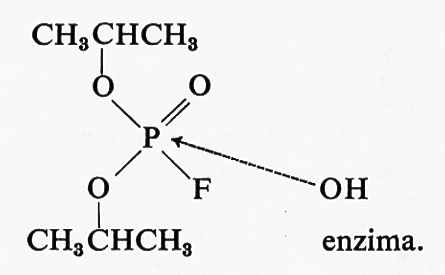
Il risultato più significativo di questi esperimenti sta nella dimostrazione che, pur essendovi altri residui serinici nella molecola di queste proteine, soltanto uno di essi, ben specifico, reagisce col DFP e con reagenti analoghi. Ne segue che questo residuo di serina, dotato di speciale reattività, ha un'importante funzione, potendo dar luogo alla formazione, col substrato, di intermedi chimici transienti.
Molti altri reagenti sono stati utilizzati per lo studio della reattività dei gruppi nucleofili degli enzimi. Talvolta è possibile rendere più selettiva la reazione variando il pH della miscela di reazione. L'alchilazione delle istidine 12 e 119 della ribonucleasi pancreatica mediante iodoacetammide ne costituisce un interessante esempio: trattando l'enzima, intorno a pH 6, con acidi alogeno-sostituiti, si ottengono due frazioni dell'enzima inattivato, che possono essere separate con metodi cromatografici. In una delle frazioni, si trova 1-(1-carbossialchil)istidina nella posizione dell'istidina 119, mentre, nell'altra frazione, si trova 3-(1-carbossialchil)istidina nella posizione dell'istidina 12.
Gli enzimi pancreatici sopra menzionati non possiedono gruppi −SH liberi; tutti i residui di cisteina sono ossidati formando ponti −S−S− intramolecolari. Tuttavia, molti enzimi possiedono un certo numero di gruppi −SH liberi, la cui reattività può assumere valori molto diversi, passando da situazioni in cui essi rappresentano i residui più reattivi dell'intera proteina, ad altre in cui, trovandosi mascherati all'interno della molecola, non sono accessibili ad alcun reagente. Se la proteina viene sgomitolata denaturandola col calore o mediante aggiunta di urea, si può determinare il numero totale di gruppi −SH. In molti enzimi è stato possibile dimostrare che un particolare gruppo −SH, tra i vari presenti nella molecola, mostra una reattività particolarmente elevata ed è implicato nella reazione chimica tra enzima e substrato tutte le volte che si forma un composto dell'enzima in assenza di un secondo substrato necessario per completare la reazione. Per esempio, nel caso della gliceraldeide-3-fosfatodeidrogenasi, che catalizza la reazione reversibile
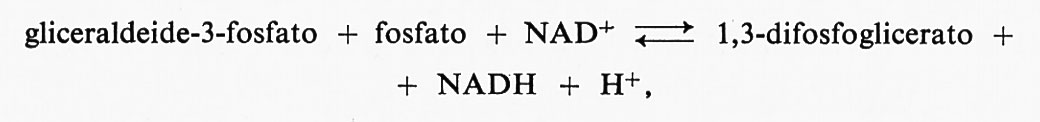
si può dimostrare che si ha formazione del seguente intermedio, che è un acil-enzima:
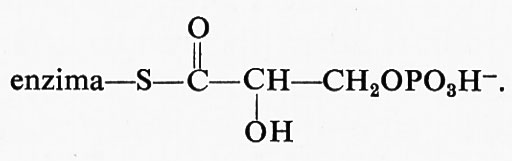
Questo composto può essere ridotto dal NADH per dare l'enzima libero e l'aldeide oppure, mediante addizione di fosfato, può essere fosforilato dando luogo a enzima libero e 1,3-difosfoglicerato.
Anche il ruolo dei gruppi ‛-NH2 dei residui di lisina è stato chiarito mediante lo studio delle reazioni parziali degli enzimi coi loro substrati. Il gruppo funzionale ε-NH2 interviene spesso nella formazione di una base di Schiff, come è illustrato dal seguente esempio in cui l'enzima aldolasi catalizza la reazione reversibile

Se si aggiungono diidrossiacetonfosfato e l'agente riducente NaBH4, si ottengono le seguenti reazioni:
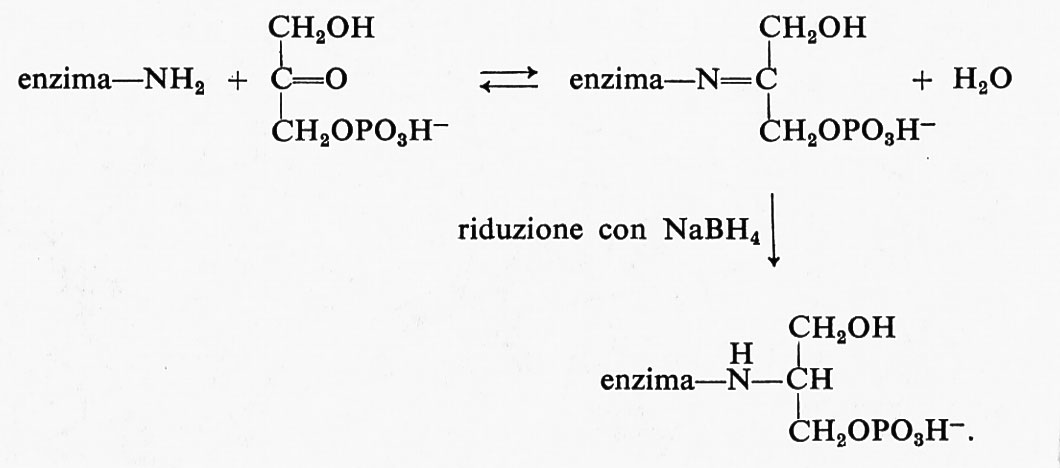
Se in questa reazione viene usato diidrossiacetonfosfato marcato con 14C, su ciascuna subunità dell'aldolasi può essere identificato un particolare residuo di lisina.
Gli esempi sopra riportati rappresentano una ristrettissima selezione dei numerosi studi che hanno permesso l'identificazione dei gruppi funzionali costituenti i siti attivi degli enzimi. Sino a questo punto tutte le funzioni catalitiche dei gruppi su una proteina sono state attribuite a processi del tipo nucleofilo/elettrofilo o acido/base. I processi di ossidoriduzione necessitano di metalli o di cofattori nucleotidici legati alla proteina.
Un particolare importante dell'attività catalitica degli enzimi sta nel fatto che vi sono sempre interessati almeno due gruppi della proteina: nella ribonucleasi due residui istidinici agiscono in armonia; negli enzimi che presenta- no una serina sul sito attivo, un gruppo istidinico è opportunamente collocato nelle vicinanze del gruppo −OH della serina con la funzione generale di catalizzatore basico per il processo transiente di acilazione/deacilazione del residuo serinico.
Le informazioni che si sono potute ottenere, mediante questo tipo di indagine chimica, sulla natura del sito di legame del substrato sono di gran lunga più scarse di quelle ottenute sui residui implicati nel sito catalitico. Mentre i meccanismi catalitici erano stati in molti casi descritti correttamente prima che fosse nota la struttura tridimensionale degli enzimi, il legame col substrato si è potuto descrivere compiutamente soltanto in quei casi in cui è stato possibile determinare la struttura del complesso enzima-substrato.
Studi cristallografici sulle interazioni tra enzima e substrato
Il successo conseguito da M. F. Perutz e J. C. Kendrew nel chiarire la struttura tridimensionale dell'emoglobina e della mioglobina mediante l'analisi strutturale coi raggi X spinse molti studiosi ad applicare questo metodo allo studio degli enzimi. I primi enzimi presi in esame furono enzimi idrolitici monomerici relativamente piccoli, e i primi risultati significativi furono quelli ottenuti sul lisozima dell'uovo di gallina, un enzima che idrolizza i polisaccaridi a livello della membrana della cellula batterica. I risultati di Phillips e dei suoi collaboratori sul lisozima e del gruppo di Lipscomb sulla carbossipeptidasi sono, a tutt'oggi, unici in questo campo. Gli studi cristallografici fornirono la prova dell'esistenza di combinazioni enzima- substrato, prescindendo da ogni informazione di tipo chimico o cinetico, fatta eccezione per la conoscenza della maggior parte della struttura primaria (v. Dickerson e Geis, 1969).
Nel caso del lisozima, numerosi studi furono compiuti per mezzo di una serie di substrati sintetici, costituiti da polimeri dell'N-acetilglucosammina e suoi copolimeri con l'acido N-acetilmurammico. La fig. 3 riporta la struttura tridimensionale del complesso del lisozima con un esamero del copolimero. La determinazione diretta della struttura fu in realtà possibile soltanto sul complesso enzima-trimero; tuttavia, l'insieme dei risultati cristallografici, delle deduzioni tratte dalla costruzione di modelli, degli studi sulle capacità di legame e dell'analisi dei punti di rottura del legame con trimero, tetramero, pentamero ed esamero del substrato, condussero alla conclusione che il substrato si lega entro una crepa che si forma sulla superficie dell'enzima. Durante il processo di formazione del legame si verificano solo lievi spostamenti nella struttura della proteina (0,75 Å), ma vi è una variazione di conformazione (da sedia a mezza sedia) a carico dell'anello appartenente all'unità a livello della quale si ha rottura del legame. È stata avanzata l'ipotesi che questa distorsione a livello del substrato, che porta il C-6 in una posizione più assiale, determini una conformazione più prossima allo stato di transizione. Le posizioni dell'aspartato 52 e del glutammato 35 rispetto al punto di reazione del substrato indicano la partecipazione di questi due residui all'interazione catalitica col substrato.
Mentre per il lisozima ha importanza fondamentale la distorsione a livello del substrato, nel caso della carbossipeptidasi il confronto della struttura dell'enzima con quella dei complessi che forma con i suoi substrati indica notevoli alterazioni a livello della struttura proteica. Il contributo delle variazioni della conformazione proteica durante la formazione del complesso enzima-substrato all'intero meccanismo della catalisi sarà discusso più avanti, quando saranno state menzionate ulteriori dimostrazioni di tali variazioni.
I risultati dell'analisi strutturale completa degli enzimi proteolitici chimotripsina e papaina confermarono i risultati chimici e cinetici sulla composizione del sito catalitico. In entrambi i casi, la conoscenza della struttura tridimensionale, che mette in luce le zone circostanti i gruppi essenziali all'attività catalitica, ha dato delle informazioni che devono essere prese in considerazione nella descrizione particolareggiata di un meccanismo di reazione. Nella fig. 4, per esempio, sono messi in evidenza alcuni particolari delle distanze di interazione, quali nessun altro metodo può fornire. In questo modo lo studio degli enzimi foruisce informazioni, riguardo agli effetti sulla reattività chimica dei gruppi vicinali e dell'ambiente locale, ben più precise di quanto non possa fare lo studio di reazioni più ‛semplici'. Come si vedrà più avanti, le tecniche cinetiche, sviluppate per lo studio di reazioni enzimatiche danno informazioni sulla reattività specifica dei gruppi nel particolare intorno del sito attivo.
Analisi cinetica delle reazioni enzima-substrato
Il numero di turnover degli enzimi, definito come il numero di moli di prodotto che si formano per mole di enzima al secondo, varia entro ampi limiti. Gli enzimi proteolitici idrolizzano il legame peptidico con velocità dell'ordine di 1 s-1 mentre l'enzima anidrasi carbonica catalizza la reazione CO2 + H2O → H2CO3 con velocità dell'ordine di 106 s-1. Un gran numero di enzimi importanti ha, tuttavia, numeri di turnover per sito attivo compresi tra 10 e 100 s-1. È già stato discusso il fatto che la reazione tra enzima e substrato, che produce enzima libero e prodotto, comporta, nella maggioranza dei casi, più trasformazioni del primo ordine, che seguono il processo del secondo ordine consistente nella formazione del complesso enzima-substrato. Il numero di turnover è determinato dalla più lenta di queste reazioni. Lo scopo degli studi cinetici consiste nel chiarimento del meccanismo di reazione mediante l'identificazione esatta dei diversi intermedi e nella determinazione delle costanti cinetiche per ogni singolo passaggio (v. cinetica chimica).
In condizioni di stato stazionario, la cinetica può dare informazioni soltanto su quegli intermedi che, in tali condizioni, sono presenti in quantità significative. Un maggior numero di informazioni si può ottenere mediante metodi che permettano di osservare le velocità di formazione e decomposizione di stati intermedi transienti, durante il breve periodo di tempo compreso tra l'istante in cui l'enzima ed il substrato vengono posti a reagire e l'istante in cui si stabiliscono le condizioni di stato stazionario. In molte reazioni enzimatiche, questo periodo transiente, compreso tra l'inizio della reazione e il primo ciclo enzimatico completo, può essere convenientemente studiato in tempi dell'ordine dei millisecondi. Le tecniche che permettono di osservare variazioni nello spettro di assorbimento 0 nella fluorescenza, su questa scala di tempi, sono state sviluppate da Hartridge, Roughton, Chance e, Gibson durante gli ultimi cinquant'anni. L'applicazione su larga scala di tali tecniche allo studio degli enzimi è tuttavia di data più recente (v. Gutfreund, 1965). Nella fig. 5 è riportata una rappresentazione schematica dell'apparato di scansione rapida col metodo dello stopped flow (flusso intermittente), che permette di osservare variazioni spettrali che avvengono molto rapidamente.
Esistono essenzialmente due modi di affrontare teoricamente l'analisi dei fenomeni cinetici transienti nelle reazioni enzimatiche. Se si considera lo schema
E + S ⇄ ES ...... ES ⇄ E + P.
è possibile seguire la cinetica transiente per il processo di formazione del prodotto, oppure si può seguire la cinetica transiente per il processo di avvicinamento degli intermedi ES, ... EP alle rispettive concentrazioni di stato stazionario. Consideriamo dapprima un metodo che ci permetta di osservare la somma delle concentrazioni di tutte le forme di prodotto (legato all'enzima e libero).
La fig. 6 riporta la rappresentazione, elaborata da un calcolatore, di un meccanismo che prevede quattro intermedi di reazione, due dei quali nella forma di complesso enzima-substrato e due nella forma di complesso enzima-prodotto. La curva rappresenta, in funzione del tempo, la somma delle varie forme di prodotto. In essa si distinguono tre fasi: una prima fase, in cui si nota un aumento nella velocità di formazione degli intermedi enzima-substrato, è seguita da una seconda fase, in cui si ha la formazione transiente degli intermedi enzima-prodotto. Nella terza fase, la velocità di formazione del prodotto di reazione è giunta allo stato stazionario.
La reazione della fosfatasi di Eschenchia coli, che catalizza la reazione di idrolisi degli esteri fosforici, può essere rappresentata come segue:

dove A rappresenta il gruppo fosforico che forma con l'enzima un complesso transiente. Se il gruppo uscente B è un cromoforo (come, ad es., nel caso che il substrato sia il nitrofenilfosfato), la velocità con cui B viene prodotto può essere usata come un indicatore per la produzione del fosfato, legato all'enzima e libero in soluzione. La fig. 7 riporta la registrazione di un esperimento di questo tipo, nelle condizioni in cui si osservano soltanto le fasi che rappresentano la produzione della specie EA e la velocità di produzione del prodotto A in condizioni di stato stazionario. L'aumento di velocità iniziale non è osservabile perché avviene durante il tempo morto dello strumento. I risuitati più interessanti che si sono ottenuti da esperimenti di questo tipo eseguiti sulla fosfatasi di Escherichia coli si traggono dal confronto delle velocità di produzione di EA con substrati aventi diversi gruppi uscenti B. Da alcuni risultati chimici si sa che la specie EA rappresenta un complesso enzima-fosfato, il che significa che durante la formazione di EA si ha un trasferimento di fosfato dal substrato all'enzima. Se fosse possibile osservare direttamente lo stadio chimico di trasferimento del gruppo fosforico, ci si dovrebbe aspettare che la sua velocità dipenda sensibilmente dalla natura del gruppo uscente. Ciò che effettivamente si osserva, mediante esperimenti del tipo di quello illustrato nella fig. 7, è che la velocità di formazione di EA è del tutto indipendente dal substrato. Il 4-nitrofenilfosfato ed il 2,4-dinitrofenilfosfato, ad esempio, reagiscono con l'enzima, dando luogo alla formazione dell'intermedio enzima-fosfato, con velocità identiche.
I risultati degli esperimenti descritti dimostrano che il processo che determina la velocità di formazione del complesso intermedio enzima-fosfato non è la reazione chimica di trasferimento del gruppo fosforico, ma qualche processo di modificazione dell'enzima stesso. È stata pertanto avanzata l'ipotesi che l'enzima possa esistere sotto due forme, E ed E*, e che il processo che limita la velocità consista nelle trasformazioni che avvengono tra l'una e l'altra di queste due forme. In seguito ad esperimenti paralleli di cinetica e di equilibrio di combinazione eseguiti mediante l'uso di substrati contenenti un gruppo cromoforo (Halford, Trentham e Gutfreund, riportati in Gutfreund, v., 1971), si è giunti alla formulazione di uno schema di reazione del tipo:
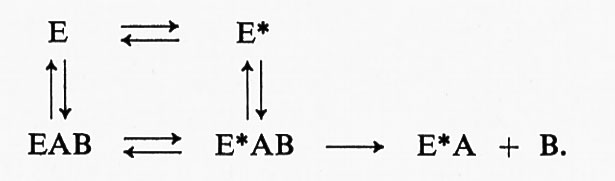
Tale processo prevede una variazione di conformazione della proteina collegata col substrato e corrisponde al modello di D. E. Koshland jr. dell'adattamento indotto. Koshland ha infatti dimostrato che la specificità di un enzima non è dovuta all'esistenza di un rigido sito di legame per il substrato, ma che, al contrario, un particolare importante della specificità enzimatica consiste nella risposta di un sito di legame flessibile alla presenza di un substrato; soltanto quando quest'ultimo è presente, il sito di legame assume la struttura adatta alla formazione di un complesso reattivo enzima-substrato. Torneremo più avanti sullo studio della formazione del complesso enzima-substrato.
Un altro esempio di studio sulla formazione di prodotti transienti, che ha dato luogo a interessanti risultati, è quello che riguarda l'enzima alcooldeidrogenasi del fegato, la cui reazione con i substrati può essere descritta come segue:
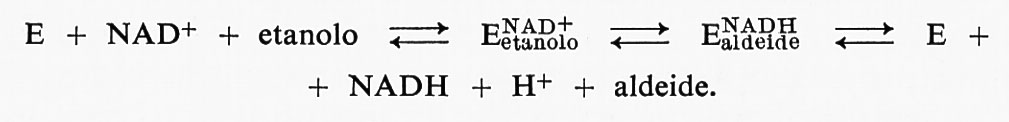
Nella trattazione presente, possiamo trascurare le reazioni del second'ordine fra l'enzima e i substrati, in quanto questi passaggi possono essere resi più veloci di ogni altra trasformazione del primo ordine aumentando la concentrazione del substrato. La registrazione di un esperimento in cui l'alcooldeidrogenasi di fegato di cavallo viene posta a reagire con NAD+ ed etanolo in uno spettrofotometro a scansione rapida col metodo dello stopped flow, e in cui viene registrata la velocità di formazione di NADH legato all'enzima e libero, è formalmente identica a quella riportata nella fig. 7. La rapida formazione di una mole di NADH per mole di sito attivo dell'enzima è seguita da lenta produzione di NADH in condizioni di stato stazionario. La fase rapida di formazione di NADH legato all'enzima rappresenta la velocità di conversione del primo complesso enzima-substrato nel complesso enzima-prodotto. Ancora una volta è necessario progettare un esperimento da cui si possa vedere se è lo stadio chimico a limitare la velocità del processo. In questo caso è stato usato un metodo che dimostra l'utilità di un'altra tecnica: l'effetto delle sostituzioni isotopiche sulle velocità di reazione.
Se si ripete la misura della velocità di formazione del NADH facendo uso, come substrato, di etanolo deuterato, il processo rapido di formazione del complesso transiente ENalAdDeiHde avviene con una velocità sei volte minore: ciò dimostra chiaramente che è il processo chimico di trasferimento dell'idruro quello che limita la velocità. È stato appurato che, nel caso dell'alcooldeidrogenasi e di altre deidrogenasi coniugate col NAD+, la formazione di ENetAaDn+olo è seguita da un processo di isomerizzazione del complesso enzima-substrato, ma, nelle condizioni in cui è stato condotto l'esperimento descritto, questa trasformazione è veloce rispetto al processo chimico. Questo fatto apre la via all'interessante possibilità di studiare la dipendenza della velocità di trasferimento dell'idruro dalla struttura e dalla reattività di substrati diversi. L'alcooldeidrogenasi del fegato reagisce con un'ampia gamma di alcoli differenti; lo studio di tali reazioni potrà essere reso anche più interessante dal fatto che sarà presto nota la struttura tridimensionale completa dell'enzima.
In molti casi, intermedi di reazione diversi (complessi ES ed EP) possiedono proprietà spettrali nettamente distinte. Ciò può essere imputato alla perturbazione di un cromoforo della proteina enzimatica, di un gruppo prostetico o, infine, di un substrato. In questi casi, l'osservazione delle rapide variazioni spettrali che avvengono nell'interconversione di intermedi transienti fornisce un maggior numero di informazioni di quante non se ne possano trarre da risultati puramente cinetico, poiché, in primo luogo, vi è una dimostrazione diretta dell'esistenza di certi intermedi che possono essere realmente osservati e, in secondo luogo, è spesso possibile dare un'interpretazione degli spettri e delle loro variazioni in funzione delle proprietà chimiche o strutturali dei composti intermedi. Ulteriori importanti indicazioni di eventi specifici si possono trarre dalle variazioni di fluorescenza. L'interpretazione di tali segnali, unitamente ai quali conviene menzionare quelli di risonanza di spin elettronico e nucleare, come indici di variazioni conformazionali che avvengono durante una reazione enzimatica, richiede una procedura complessa che trascende il proposito di questo articolo. Tali studi forniscono informazioni essenziali sulla mobilità dei complessi enzimatici e i loro risultati, unitamente all'informazione diretta ma statica derivante dalla cristallografia, hanno contribuito a fornire le conclusioni che saranno discusse nel prossimo capitolo.
Delle variazioni negli spettri di assorbimento e nella fluorescenza è stato fatto largo uso per seguire la formazione di complessi enzima-substrato. In tali ricerche, oltre alle tecniche di stopped flow discusse in precedenza, sono state impiegate le tecniche di ‛rilassamento' sviluppate da Eigen e de Maeyer per lo studio generale delle reazioni di combinazione con leganti, che si sono dimostrate di grande utilità nello studio delle reazioni enzimatiche. Il principio che sta alla base dei metodi di rilassamento consiste semplicemente in una variazione rapida della temperatura di un sistema che si trova in condizioni di equilibrio a una certa temperatura (la temperatura è soltanto uno dei parametri che si possono utilizzare per variare l'equilibrio di un sistema). In un tempo di circa 5 μs si può ottenere un aumento di temperatura di oltre 10 °C e si può osservare la conseguente reazione chimica con cui il sistema si porta alla nuova condizione di equilibrio.
L'analisi cinetica delle misure effettuate col metodo dello stopped flow o con quello di rilassamento permette di distinguere i processi a un solo stadio, del tipo E + L ⇄ EL (dove L può essere un substrato o un opportuno analogo), dai processi più complessi che implicano interconversioni successive del primo ordine, del tipo EL ⇄ E*L. Nella precedente discussione sulla fosfatasi, si era avanzata l'ipotesi che si potessero formare successivamente più complessi enzima-substrato. Mediante l'uso di un composto analogo al substrato, capace di legarsi all'enzima, ma non di venire idrolizzato (il nitrobenzilfosfonato), è stato possibile studiare il processo
E + L ⇄ EL ⇄ E*L.
Fortunatamente le velocità delle variazioni conformazionali misurate in presenza dell'analogo del substrato risultarono in completo accordo con quelle previste dalla cinetica dei transienti per la reazione col substrato.
Nel caso di enzimi con due substrati, è possibile studiare la combinazione di un substrato alla volta. Alcune regole generali possono essere enunciate sulla base dei risultati delle ricerche sui diversi tipi di reazione enzima-substrato. Il processo iniziale del secondo ordine, rappresentato dalla combinazione col substrato, non comporta specificità, se non in minima parte. Ciò porta al fatto che, in tutti i casi studiati, le costanti di velocità per questa reazione risultano, a meno di un fattore 10, eguali a 3•102 M-1s-1. Se, d'altra parte, si determina la frequenza di collisione tra molecole delle dimensioni degli enzimi e dei substrati, si ottiene un valore approssimativamente eguale a 5•109 M-1s-1. Tenendo conto del fatto che l'area della superficie di una molecola proteica è circa 100 volte maggiore di quella di un substrato di media grandezza, se ne deduce che la frequenza di collisione del substrato con il sito di legame sulla proteina corrisponde alla costante cinetica della reazione di combinazione dell'enzima col substrato. La specificità del sistema deve pertanto dipendere dal contatto iniziale.
In tutte le reazioni tra enzima e substrato che si sono studiate in modo approfondito, si è visto che dopo l'aggiunta di substrato (o substrati) avvengono delle modifiche strutturali. Mcuni studi hanno dimostrato l'esistenza di un numero di possibili conformazioni per i complessi enzima-substrato o per l'enzima libero: in questi casi, gli studi cinetici devono poter determinare gli stadi necessari nella via principale di reazione e gli eventuali equilibri collaterali. L'equilibrio delle forze responsabili del mantenimento della struttura tridimensionale di una proteina in soluzione acquosa è estremamente critico. La configurazione spaziale delle catene polipeptidiche è determinata da legami a idrogeno, ponti salamini e interazioni idrofobiche. Le energie di tali interazioni, perché i legami si possano mantenere nell'acqua, dipendono tutte dalla struttura dielettrica locale. Poiché il processo di combinazione con una molecola di substrato determina certamente un'alterazione dell'intorno locale, esso può dare il via ad una variazione conformazionale. Tale variazione conduce alla formazione di un complesso, la cui orientazione è particolarmente favorevole alla catalisi, nel caso in cui il substrato sia quello specifico per l'enzima.
Alcuni principi generali della catalisi enzimatica
La teoria delle velocità assolute di reazione tratta delle velocità di reazione in funzione dell'energia libera di attivazione G≠. Tale entità rappresenta la differenza tra l'energia libera dello stato fondamentale e quella dello stato di transizione (complesso attivato). Lo stato di transizione si definisce come quello stato in cui una molecola è in condizioni tali da dare luogo alla reazione entro un periodo di tempo corrispondente all'inverso della frequenza di vibrazione della molecola stessa (v. fig. 8).
In presenza del catalizzatore (enzima), gli stati iniziale e finale della reazione sono gli stessi; ciò significa che, mentre l'equilibrio del sistema non viene modificato, l'energia libera di attivazione si è abbassata. L'abbassamento di tale energia può essere dovuto sia a una diminuzione della differenza di energia libera tra lo stato fondamentale e lo stato di transizione di un particolare meccanismo di reazione, sia a una modificazione della chimica della reazione. Si possono distinguere essenzialmente tre fattori che contribuiscono ad aumentare le velocità di reazione secondo l'uno o l'altro dei modi di catalisi sopra menzionati: a) variazioni conformazionali del substrato; b) relazioni spaziali e di ambiente locale nel complesso enzima-substrato; c) intermedi covalenti enzima-substrato.
È stato detto in precedenza che nella reazione del lisozima coi suoi substrati ha luogo una variazione conformazionale che modifica la geometria del substrato, avvicinandola a quella caratteristica dello stato di transizione. Misure del contributo all'energia di legame apportato dai sei anelli glucidici che si sistemano nel sito attivo dell'enzima hanno stabilito che cinque di questi danno luogo a un ΔG compreso tra −2 e −5 kcal ciascuno, mentre il sesto anello, che subisce una distorsione, si lega con un ΔG ≅ +5 kcal. Il processo di combinazione complessivo è pertanto energeticamente favorevole nonostante il lavoro necessario per il processo di distorsione. Questo tipo di bilancio energetico può essere espresso in modo generale dicendo che la formazione del complesso enzima-substrato può determinare una struttura in cui alcuni dei livelli energetici del sistema si portano più in alto. Una situazione simile si riscontra nei casi in cui gruppi elettricamente carichi sono portati a contatto o sono trasferiti da un ambiente acquoso a uno apolare.
Un altro fattore che, nella formazione del complesso enzima-substrato, contribuisce ad accelerare la reazione è costituito dalle relazioni spaziali. Questo fenomeno è stato studiato a fondo per mezzo di modelli nei quali i due gruppi reagenti vengono mantenuti a distanza ravvicinata. Raffrontando le velocità di reazione tra due gruppi reagenti uniformemente distribuiti e mantenuti vicini, sì trova che la velocità di reazione è, al massimo, accelerata di un fattore eguale a circa 100. È difficile fare un raffronto diretto tra la velocità, ad esempio, dell'imidazolo libero in soluzione che reagisce con un estere e quella dell'imidazolo di un residuo istidinico che costituisce un sito attivo che reagisce con un substrato. È tuttavia opportuno dire che si cerca ancora una spiegazione per aumenti di velocità dell'ordine di 108-1010.
La precisa struttura del complesso enzima-substrato può contribuire molto di più che non l'avvicinamento dei due gruppi reagenti a una certa distanza opportuna. È stato visto che una particolare caratteristica dei meccanismi enzimatici consiste nel fatto che almeno due gruppi della proteina cooperano nell'interazione con il substrato. Anche altre interazioni tra i gruppi collocati sul sito attivo possono avere, come è illustrato ad esempio nella fig. 4, notevoli effetti sulla reattività. Nell'ambito di un complesso enzima-substrato, i fattori che sembrano contribuire agli effetti di prossimità possono essere enumerati come segue: a) prossimità di un certo numero di gruppi; b) corretta e precisa orientazione dei gruppi; c) effetto dell'ambiente sulla reattività dei gruppi.
Un'interessante indicazione sugli effetti ambientali si può trarre dal confronto tra le proprietà di una molecola libera in soluzione e quelle di una molecola legata a un sito attivo. I nucleotidi flavinici, che si combinano con un certo numero di proteine per formare sistemi di trasferimento di idrogeno, possono variare il loro potenziale di ossidoriduzione di una quantità equivalente a circa 5 kcal, quando si leghino al sito specifico dell'enzima. Tale variazione, espressa mediante l'energia di attivazione di uno degli stadi di reazione, corrisponde a una variazione di velocità di un fattore 104.
È a tutt'oggi difficile esprimere in termini quantitativi il contributo degli effetti ambientali e sterici all'efficienza catalitica di un enzima. Studi più precisi sulla struttura e sulla reattività dei siti attivi di un numero sempre maggiore di enzimi potranno fornire in futuro i dati necessari per la loro correlazione quantitativa.
La formazione di composti covalenti tra enzimi e substrati, di cui si è detto sopra, costituisce la chiave di volta del meccanismo enzimatico. Quale contributo possono portare questi fenomeni all'accelerazione enzimatica di una reazione? Prendiamo in considerazione la reazione della chimotripsina col suo substrato. La reazione completa consiste nell'idrolisi dei legami peptidici. L'idrolisi non è altro che un caso speciale di trasferimento di un gruppo acile, in cui una molecola di acqua funge da accettore. Le reazioni di trasferimento del gruppo acilico si possono rappresentare con lo schema generale:
AB + C → AC + B.
dove A rappresenta il gruppo acilico, B e C rispettivamente il gruppo uscente e quello entrante (nucleofilo). La velocità del processo dipenderà dall'efficienza del gruppo B come gruppo uscente e da quella del gruppo C come nucleofilo. La sequenza di reazioni schematizzata nella fig. 9 costituisce una possibile interpretazione dei dati a disposizione. Per l'argomento che stiamo trattando il nostro interesse sarà rivolto soltanto al ruolo del gruppo −OH della serina in posizione 195. Dagli esperimenti con inibitori, sui quali si è già riferito in precedenza, e da esperimenti con substrati in condizioni speciali, si sa che si viene a formare un complesso serina-O-estere come intermedio transiente tra la senna 195 della chimotripsina e il gruppo acilico del substrato. Il gruppo −OH della serina è un nucleofilo migliore dell'acqua, e reagirà quindi, a parità di condizioni, più velocemente di questa. Una volta formatosi, l'intermedio dovrà subire l'idrolisi, cioè reagire con l'acqua, per dar luogo ai prodotti finali. Il particolare importante, per questo secondo stadio, consiste nel fatto che l'−OH della serina è un gruppo uscente più efficiente del gruppo α-amminico del substrato. Queste sono, ancora una volta, argomentazioni puramente qualitative: considerazioni quantitative dovrebbero essere basate sulla conoscenza dell'efficienza, come nucleofilo e come gruppo uscente, della funzione −OH della serina, in questo particolare ambiente.
Controllo e altre funzioni degli enzimi
Un aspetto importante della catalisi enzimatica riguarda la capacità di regolare i processi biologici. Gli enzimi non sono responsabili soltanto delle complesse sequenze di quelle reazioni chimiche che portano alla biosintesi e alla rottura dei costituenti cellulari, ma partecipano anche al controllo di molte funzioni fisiche. La contrazione muscolare, gli impulsi nervosi e la visione sono soltanto alcuni dei fenomeni fisiologici più strettamente correlati con l'azione enzimatica. Bisogna tener presente che l'evoluzione che ha subito un particolare enzima non è stata quella che lo ha reso il catalizzatore più efficiente di una certa reazione: è verosimile che un buon chimico organico potrebbe sintetizzare un enzima più efficiente del catalizzatore naturale per quella particolare reazione. Tuttavia, l'enzima sintetico potrebbe non soddisfare molte delle necessità insorgenti in un sistema biologico integrato. In questo capitolo saranno discusse le seguenti funzioni degli enzimi: a) meccanismi di controllo per la regolazione delle velocità; b) funzioni dei composti enzima-substrato. Lo studio di entrambi questi aspetti è strettamente connesso con lo studio del meccanismo catalitico.
In questa trattazione ci limiteremo ad un particolare tipo di fenomeno di controllo: la cooperatività (per una trattazione particolareggiata può essere vantaggiosamente consultato l'articolo di Koshland jr., v., 19703). Oggi si ha per certo che molte molecole enzimatiche sono costituite da un certo numero di subunità: l'essenza del concetto di cooperatività consiste nel fatto che una certa informazione viene trasmessa da una subunità a un'altra. In questo contesto, l'informazione più importante riguarda lo stato di occupazione dei siti di combinazione del legante. Vi possono essere più siti di combinazione per leganti diversi per ogni subunità: siti per i substrati, per ioni attivatori o inibitori e altri metaboliti attivatori o inibitori. In certi casi la struttura in subunità è più complessa, e per funzioni di legame diverse si trovano unità polipeptidiche differenti. Bisogna poi ricordare che non è affatto vero che tutti gli enzimi polimerici presentano fenomeni cooperativi. Vi sono altri vantaggi nell'avere enzimi costituiti di unità multiple e l'interazione tra i siti è frutto dell'evoluzione di quei sistemi per i quali tale situazione è risultata vantaggiosa.
Il più semplice esempio di fenomeno cooperativo nell'azione enzimatica è illustrato dalla combinazione coi substrati. Consideriamo un tetramero con quattro siti di legame per un substrato, intrinsecamente identici. Se non ci fosse interazione (trasferimento di informazione) tra i siti, le curve di saturazione (v. fig. 1), o di legame col substrato, sarebbero, nel tetramero, indistinguibili da quelle di un sistema che avesse lo stesso numero di siti su monomeri liberi in soluzione. Se, tuttavia, come conseguenza del primo legame di una molecola di substrato con uno qualsiasi dei quattro siti, si avesse un cambiamento di conformazione che aumentasse l'affinità per il substrato, la variazione di conformazione potrebbe essere considerata come l'informazione scambiata tra le subunità (v. fig. 10). J. Monod e F. Jacob hanno proposto un meccanismo del ‛tutto o nulla' in cui tutte le subunità devono trovarsi contemporaneamente o nella forma ad alta affinità o in quella a bassa affinità. Sia che si possa applicare il meccanismo del ‛tutto o nulla', sia che l'affinità aumenti progressivamente a mano a mano che procede l'occupazione dei siti, in ogni caso la curva di saturazione o combinazione col substrato assume una forma sigmoide, del tipo rappresentato nella fig. 11.
Una conseguenza della forma della curva di legame cooperativo sta nel fatto che, nella zona di mezzo (CES/CE0 ≅ 0,5), la risposta a variazioni di concentrazione del substrato è molto maggiore di quanto non sia nel caso di siti a legame indipendente: per questo motivo, si potrebbe dire che la cooperatività agisce come un interruttore dissipativo.
Un fenomeno che ha polarizzato l'attenzione sull'interazione cooperativa tra enzimi e leganti è l'inibizione per retroazione feedback). In una reazione enzimatica isolata A →B, la velocità netta della reazione nella direzione da A a B diminuirà all'aumentare di B perché la reazione inversa diventa significativa e perché B può agire come inibitore competitivo per A al sito attivo. Gli inibitori competitivi per lo stesso sito sono in genere strutturalmente correlati col substrato. Sono stati trovati sistemi di vie metaboliche in cui un prodotto finale della via agisce da inibitore competitivo per un enzima che catalizza uno dei primi stadi, come è indicato nello schema seguente:
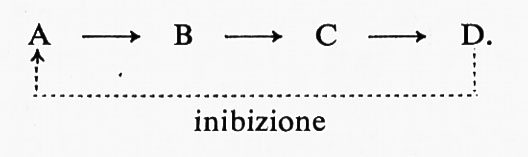
Si è trovato che l'inibizione competitiva da parte di un prodotto distante, che non sia più strutturalmente correlato col substrato, non ha luogo nel sito di legame del substrato. Tali inibitori allosterici si legano su siti diversi (allosterici) e inducono variazioni conformazionali che vengono trasmesse al sito di legame del substrato. Si trova in genere che queste variazioni di conformazione modificano l'affinità per il substrato piuttosto che la velocità di catalisi. Nella combinazione cooperativa col substrato, le variazioni conformazionali indotte dal substrato aumentano l'affinità per questo ultimo, mentre le variazioni conformazionali indotte dall'inibitore producono l'effetto opposto.
Fino ad ora non è stato possibile ottenere raffigurazioni della struttura tridimensionale degli enzimi che presentano queste variazioni di conformazione di carattere regolativo. Risultati spettroscopici e cinetici portano alla conclusione che tali enzimi possono esistere sotto due forme distinte, R (alta affinità per il substrato) e T (bassa affinità per il substrato), in equilibrio dinamico tra di loro. Come conseguenza delle relazioni termodinamiche per gli equilibri multipli, la presenza di un substrato, che manifesta affinità maggiore per la forma R, sposterà l'equilibrio R ⇄ T verso sinistra. Inversamente, l'inibitore, che ha maggior affinità per la forma T, sposterà l'equilibrio verso destra.
Occorre ricordare espressamente che in certi enzimi l'inibitore non soltanto si lega in siti separati, ma possiede talvolta una speciale subunità per il suo sito di legame. È stato studiato un altro fenomeno che può essere importante per.la regolazione metabolica: esso consiste nella cooperatività negativa, il cui risultato è quello di diminuire l'affinità per il substrato a mano a mano che aumenta il numero dei siti occupati dell'enzima.
Questi fenomeni devono essere menzionati in una discussione moderna sui meccanismi enzimatici. Tuttavia, poiché per nessuno di essi è ancora stato descritto in modo compiuto il meccanismo, portarne degli esempi costituirebbe una digressione troppo lunga. La descrizione di un certo numero di sistemi e i riferimenti bibliografici relativi ad altri si possono trovare altrove (v. Koshland jr., 19703).
Un altro argomento che mette in relazione i meccanismi enzimatici con la regolazione metabolica è connesso col fatto che molti enzimi si trovano in concentrazioni paragonabili a quelle dei loro substrati. Nella classica definizione di catalizzatore, si afferma che la concentrazione del catalizzatore è trascurabile rispetto a quella dei reagenti. In alcune vie metaboliche, come ad esempio nella glicolisi, si trova che una parte significativa degli intermedi di reazione esiste sotto forma di composti con l'enzima e che la loro utilizzazione dipende dalla velocità con cui vengono rilasciati dal sito di legame. Ha assunto anche un certo interesse il fatto che si possano ottenere informazioni sulle proprietà dei substrati quando sono legati all'enzima. Alcuni di questi fenomeni saranno illustrati con esempi.
È stata più volte avanzata l'ipotesi che i prodotti legati all'enzima possano reagire con l'enzima successivo nella sequenza metabolica, senza che si debbano prima dissociare allo stato libero in soluzione. Tale ipotesi sembra particolarmente interessante per il controllo della disponibilità di prodotti come il NADH, che sono substrati per un notevole numero di reazioni enzimatiche. Fino ad ora, tuttavia, tutti gli studi condotti in proposito, sebbene siano in numero limitato, hanno indicato che, tra due reazioni enzimatiche consecutive, i substrati devono passare attraverso lo stato libero.
I prodotti di molte reazioni enzimatiche reagiscono ulteriormente con un'altra molecola di substrato, dando luogo ad un processo in cui l'enzima esplica un ruolo molto speciale e non strettamente catalitico. Per esempio, il primo stadio nella sintesi delle proteine dagli amminoacidi comporta la formazione di adenilati degli amminoacidi e di pirofosfato libero. Lo stadio successivo nella sintesi proteica comporta la formazione di un composto tra il gruppo acilico dell'amminoacido e uno specifico acido nucleico; anche in questo caso, esiste un particolare acido nucleico per la reazione con ciascun amminoacido. Anidridi miste, come gli adenilati degli amminoacidi, si idrolizzano molto rapidamente quando sono poste in acqua e sono molto reattive con tutti gli agenti nucleofili. La seconda funzione dell'enzima che catalizza la formazione degli adenilati degli amminoacidi sta nella preservazione di questo prodotto per la reazione con l'apposito gruppo accettore dell'acido nucleico specifico. L'importanza dell'enzima in questa reazione non consiste nell'accelerare la velocità di reazione, ma piuttosto nella sua funzione di ‛binario' per la reazione stessa.
Esempi analoghi di siffatte funzioni ‛binario' si possono trovare anche in certe sequenze di processi ossidoriduttivi. I nucleotidi flavinici e i gruppi eme vengono rapidamente ossidati dall'ossigeno molecolare quando si trovano allo stato libero, mentre quando sono combinati con una proteina specifica le loro forme ridotte non reagiscono con l'ossigeno in soluzione. Il sito attivo delle proteine preserva i gruppi per la reazione con gli accettori specifici e li protegge da reazioni con gli abbondanti reagenti non specifici.
Bibliografia
Dickerson, R. E., Geis, I., The structure and function of proteins, New York 1969.
Gutfreund, H., An introduction to the study of enzymes, Oxford 1965.
Gutfreund, H., Transient and relaxation kinetics of enzyme reactions, in ‟Annual review of biochemistry", 1971, XL, pp. 315-344.
Gutfreund, H., Enzymes. Physical principles, London-New York 1972.
Haldane, J. B. S., Enzymes, London 1930.
Koshland, D. E. Jr., The molecular basis for enzyme regulation, in The enzymes (a cura di P. Boyer), vol. I, Enzyme regulation and control, London-New York 19703, pp. 342-397.
Catalisi eterogenea
di Alessandro Cimino
Introduzione
Definizione e cenno storico
Il termine ‛catalisi', introdotto nel 1836 da J. J. Berzelius, designa il fenomeno, causato da una sostanza chiamata ‛catalizzatore', di un aumento della velocità di raggiungimento dell'equilibrio chimico tra le sostanze reagenti, senza che il catalizzatore partecipi alla stechiometria della reazione. Per esempio, la reazione H2 + ½O2 = H2O avviene a velocità trascurabile a temperatura ambiente, ma avviene istantaneamente in presenza di platino finemente suddiviso: il platino è un catalizzatore per la reazione. Se reagenti e catalizzatore appartengono alla stessa fase si parla di ‛catalisi omogenea', se essi appartengono a fasi diverse, di ‛catalisi eterogenea'. L'esempio prima citato è di catalisi eterogenea, dato che i reagenti (H2 e O2) sono gas, e il catalizzatore (Pt) è un solido. Tra i diversi casi di catalisi eterogenea distinti a seconda delle fasi presenti (per es. gas-solido, gas-liquido, liquido-solido), i più importanti sono quelli che vedono la partecipazione di un catalizzatore solido, dato che per essi si hanno sia maggiori applicazioni industriali sia problemi concettualmente più complessi.
La catalisi, che costituiva all'inizio una curiosità scientifica, ha assunto importanza industriale crescente a partire dal 1868 ‛reparazione catalitica del cloro). In un primo periodo essa ha principalmente interessato l'industria inorganica pesante (cloro, ammoniaca, acido nitrico, ecc.); si è estesa poi in questo secolo all'industria dei prodotti organici. Dopo la seconda guerra mondiale, l'industria petrolifera ha portato un nuovo grandissimo impulso all'applicazione della catalisi. È stato stimato che il 90% dei nuovi processi chimici (che rappresentano più della metà di tutti i processi chimici oggi in operazione) si basano su catalisi eterogenea.
L'enorme importanza applicativa ha spinto a cercare una razionalizzazione dei fenomeni catalitici, tale da permettere un miglioramento dei processi conosciuti, o la scoperta di nuovi. Si sono quindi sviluppati gli studi sull'argomento e, in particolare, a partire dal 1920 circa, sono stati oggetto di attenzione sia la definizione cinetica dei processi catalitici, sia lo studio dei processi all'interfase e quindi delle interazioni tra superficie del solido e reagenti. Nel corso di un decennio circa, a cavallo del 1930, sono state poste le basi per la descrizione fenomenologica dei processi che avvengono sulla superficie (I. Langmuir, H. 5. S. Taylor, E. Rideal, A. Balandin, G. M. Schwab) ed è anche di quegli anni (1938) l'introduzione dell'equazione di Brunauer, Emmett e Teller (BET), che ha permesso la misura dell'area superficiale di un solido disperso.
Natura e problemi del fenomeno della catalisi eterogenea
La ricerca nel campo della catalisi eterogenea tende oggi ad avere carattere interdisciplinare. Essa richiede infatti conoscenze che inizialmente appartenevano a campi diversi della chimica (termodinamica, cinetica, chimica inorganica) e che in tempi più recenti si sono estese verso la fisica dei solidi, la chimica teorica, l'ingegneria. Per formarsi un quadro logico della grande varietà di studi oggi condotti sotto l'etichetta di ‛catalisi eterogenea' e per delineare i problemi attuali e le prospettive di sviluppo, è opportuno premettere alcune considerazioni sulla natura della catalisi eterogenea.
Abbandonata l'idea iniziale che il catalizzatore avesse proprietà misteriose atte a far unire tra loro i corpi in modo specifico e affermata già nel secolo scorso la validità dei principi della termodinamica, per cui la reazione può essere catalizzata solo se termodinamicamente realizzabile, è apparso chiaro che la presenza di un catalizzatore offre una via diversa all'attuazione di un processo che anche in assenza del catalizzatore avverrebbe, se pure a velocità ridotta. Quando sono possibili diverse reazioni, il catalizzatore può favorirne preferenzialmente una, permettendo così la selezione del prodotto finale tra i molti termodinamicamente possibili. A ciò è dovuta l'azione selettiva, che ha tanta importanza pratica, e che è simile in principio all'azione degli enzimi (v. catalisi enzimatica).
Il fenomeno della catalisi va quindi inquadrato nei problemi di reattività chimica. Problemi di reattività si pongono anche per reazioni non catalitiche. Per esempio, nel caso di due molecole semplici A e B la teoria mostra (v. legame chimico) che le strutture elettroniche di A e B devono fornire la spiegazione sia per la possibilità termodinamica della formazione di un legame tra A e B, sia per il meccanismo e quindi per la cinetica del processo. Se A e B sono molecole semplici, si può sperare di descrivere la loro reazione in termini di base, cioè di meccanica quantistica. Se A e B sono molecole complesse e la trattazione quanto-meccanica è impossibile, si può, rinunciando al rigore, ricorrere a parametri empirici e persino accontentarsi di una descrizione fenomenologica. Nel caso di una reazione catalitica tra A e B in presenza del catalizzatore X, il meccanismo di reazione, e cioè la successione dei ‛passi elementari' che conducono alla reazione, è modificato dal catalizzatore, che entra in uno o più passi elementari. Quanto detto vale per la catalisi sia omogenea (v. catalisi omogenea) sia eterogenea. Se il catalizzatore X è un solido, A o B ovvero entrambi devono formare un vero e proprio legame con il solido, cioè con gli atomi superficiali di questo. Nella catalisi eterogenea, quindi, vi sono specie intermedie che intervengono nella reazione, caratterizzate dal fatto che la loro esistenza è limitata alla superficie (‛complessi superficiali').
Sulla superficie di un solido può formarsi una grande varietà di specie. Per esempio, anche una molecola semplice come O2 può dar luogo sulla superficie a diverse specie, quali O2, O2-, O22-, O, O-, O2-. Quando si esamini una molecola più complessa, il numero delle specie è molto ampio, anche se alcune di esse sono meno probabili di altre. Idealmente, quindi, si dovrebbe poter rispondere alle seguenti domande: quali specie si formano sulla superficie? da quali parametri dipendono le loro concentrazioni? qual è il loro ruolo nel processo globale? In pratica, si è ben lontani dal poter rispondere esaurientemente a tali domande. Appare chiaro, infatti, che la catalisi eterogenea, pur obbedendo agli stessi principî di ogni reazione chimica, presenta la peculiarità che almeno una delle sostanze reagenti, e cioè il catalizzatore solido X, è necessariamente complessa, anche se A e B sono molecole semplici. È pertanto impossibile pensare ad una trattazione rigorosa quanto-meccanica. Le conoscenze sulla struttura elettronica dei solidi si sono sviluppate in tempi relativamente recenti e quelle sulla superficie dei solidi reali sono tuttora suscettibili di rapido avanzamento. Vi è quindi un settore della ricerca teso a correlare la struttura dei solidi con l'attività catalitica e ciò può essere conseguito mediante metodi e impostazioni diversi. D'altra parte, proprio come nel caso di reazioni tra molecole complesse si è fatto ricorso a una razionalizzazione su base empirica, così nel campo della catalisi eterogenea troviamo numerosi studi che si ispirano a questo criterio. Infine, di importanza pratica notevole sono gli studi che attraverso un esame di parametri cinetici, pur non mirando necessariamente a chiarire la natura delle interazioni molecole-superficie, tendono a ottenere una definizione cinetica del fenomeno tale da poter prevedere le condizioni atte a produrre un composto voluto alla velocità più conveniente. Per questo ultimo tipo di studi sono essenziali conoscenze che appartengono anche al campo dell'ingegneria chimica, in particolare quelle che si ottengono attraverso l'esame dei fenomeni di trasporto di massa e di calore.
In quanto segue si esamineranno gli aspetti più significativi del fenomeno catalitico, così come esso appare da questi cenni introduttivi. Si parlerà cioè dell'interazione tra una molecola e una superficie, con l'avvertenza che per molecola si intenderà una specie generica che potrà essere molecola, radicale, ione o atomo in senso tradizionale. Si illustreranno i tipi di trattazione con cui si è tentata una descrizione della formazione del complesso superficiale in termini di base. Si potranno quindi esaminare le vie intese a dare una razionalizzazione del vero e proprio fenomeno catalitico.
Interazione tra una molecola e una superficie
Adsorbimento chimico
Un solido, per poter agire da catalizzatore, deve adsorbire almeno uno dei reagenti, deve cioè legarlo sulla superficie. Il fenomeno della catalisi eterogenea è pertanto intimamente connesso a quello dell'adsorbimento. La natura delle forze agenti nell'interazione solido-molecola dipende sia dalla struttura elettronica della molecola sia da quella del solido e sarà riconducibile ai medesimi schemi validi per il caso di interazione tra due atomi o molecole. In particolare, si possono distinguere: a) interazioni molecola-superficie nelle quali non si ha né sovrapposizione di orbitali né trasferimento di elettroni tra atomi del solido e molecola adsorbita; b) interazioni nelle quali si ha sovrapposizione di orbitali o trasferimento di elettroni, ovvero le due condizioni simultaneamente. Nel caso a) viene quindi mantenuta l'attribuzione degli elettroni alle specie reagenti prima dell'adsorbimento. Le forze, che sono simili a quelle responsabili della liquefazione di un gas, sono del tipo detto di ‛dispersione' o di London e non si formano legami dei tipi incontrati all'interno delle molecole. Si classifica questo caso come ‛adsorbimento fisico'. Il caso b) conduce alla formazione di legami chimici veri e propri e viene distinto come ‛adsorbimento chimico'. La distinzione tra i due casi è utile in quanto le energie di interazione del primo tipo sono più piccole di quelle del secondo. Per esempio, il calore di adsorbimento fisico di H2 è circa 1 kcal/mole contro un calore di diverse decine di kcal/mole misurato per l'adsorbimento chimico. In genere il calore di adsorbimento fisico non è superiore a 10 kcal/mole. La distinzione però non è netta, in quanto anche nel primo caso (adsorbimento fisico) gli elettroni della molecola e del solido dovranno trovare nuove distribuzioni e, in ultima analisi, si può immaginare un passaggio continuo tra il caso a) e il caso b).
L'adsorbimento fisico, oltre a presentare un interesse rilevante per sé, inquadrandosi nella problematica generale dello studio delle interazioni tra corpi, ha un interesse per la catalisi in quanto, come si vedrà, esso può essere considerato come lo stadio precursore dell'adsorbimento chimico. Le limitazioni di spazio non consentono qui un esame del fenomeno e delle teorie dell'adsorbimento fisico.
L'adsorbimento chimico porta alla formazione di un legame tra atomo o molecola adsorbiti e atomi superficiali analogo ai legami che portano alla formazione delle molecole o dei cristalli. Questa asserzione trova la dimostrazione sperimentale nella corrispondenza, per esempio, tra calori di adsorbimento di vari gas (O, H, N) e calori di formazione del composto stechiometrico in massa (ossidi, idruri, nitruri) (v. fig. 1).
La descrizione della formazione del legame tra molecola e superficie dovrebbe tener conto, come detto, delle strutture elettroniche sia della molecola adsorbita sia del solido. Sorgono però difficoltà pratiche non sormontabili se si tenta una descrizione dettagliata, anche nel caso in cui l'adsorbato sia un atomo singolo. Pertanto sono stati sviluppati diversi approcci, che sottolineano aspetti particolari del fenomeno. Per esempio, si è tentato di descrivere il fenomeno mediante parametri generali e macroscopici del solido e, all'altro estremo, si è descritto il fenomeno come un'interazione tra l'atomo adsorbito e un atomo del solido quasi fosse isolato, pur tentando di valutare l'effetto collettivo della presenza dell'insieme di atomi. Esempi dell'uno e dell'altro approccio saranno dati in seguito. È importante peraltro notare che ciascun tipo di approccio tende a valutare e a evidenziare aspetti particolari del fenomeno. Pertanto, ciascun tipo ha maggiore o minore successo nella descrizione dettagliata dei diversi aspetti del fenomeno (energetici, strutturali, correlativi con il tipo di solido o con il tipo di atomo o molecola adsorbiti).
Quanto detto vale, come principio, anche per il fenomeno della catalisi, strettamente connesso con quello dell'adsorbimento; infatti, tutte le trattazioni che tendono a una razionalizzazione del fenomeno catalitico rispecchiano le diverse tfilosofie' che possono essere adottate per lo studio dell'interazione tra superficie e molecola.
È bene sottolineare che ogni tentativo di razionalizzazione presuppone una conoscenza fenomenologica precisa, che a sua volta presenta difficoltà di varia natura. Da un lato vi è la difficoltà sperimentale di misurare le grandezze ritenute significative, dall'altro la difficoltà di chiarire proprio quali grandezze abbiano rilevanza diretta. Spesso, a causa delle difficoltà sperimentali, ci si deve limitare a misurare grandezze solo indirettamente collegate all'intima natura del fenomeno dell'adsorbimento e della catalisi. Lo spazio non consente qui un esame anche sommario della fenomenologia dell'adsorbimento, per la quale si rinvia alla bibliografia.
Curve di energia potenziale
La descrizione dei fenomeni di adsorbimento mediante curve di energia potenziale (cioè energia E in funzione della distanza r dalla superficie) permette di rappresentare ciò che può avvenire all'avvicinarsi di una molecola alla superficie, chiarisce il ruolo dell'adsorbimento fisico e illustra i problemi che si incontrano nella descrizione del processo in termini di base. In quanto segue, i calori di adsorbimento sono positivi se il processo è esotermico (il segno è quindi contrario a quello del ΔH).
Se si considera una molecola H2 che si avvicina a una superficie (per es., di un metallo) e si prende come riferimento (E = 0) lo stato con r uguale a infinito, l'adsorbimento fisico dà luogo a una curva quale la a della fig. 2. Il minimo P corrisponde all'energia di adsorbimento fisico QP della molecola H2 sul metallo. La curva b rappresenta l'adsorbimento degli atomi H. Per r → ∞ essa è al di sopra dello zero della quantità ΔHd uguale al calore di dissociazione della molecola (H2) in atomi (2H). Al diminuire di r la curva b passa per il minimo C, che rappresenta l'adsorbimento chimico degli atomi sul metallo. Si notino sia la maggiore profondità del minimo (calore di adsorbimento chimico QC più elevato), sia il minor valore di r corrispondente al minimo, se paragonato alla curva a.
Le due curve, a e b insieme, illustrano quindi il processo che avviene quando una molecola H2 si avvicina alla superficie. La molecola, senza energia di attivazione, viene prima adsorbita fisicamente (P), ma se essa aveva o se acquista l'energia EB (rispetto allo zero) è anche in grado di passare la barriera di potenziale B e di venire chimicamente adsorbita come atomi. Dalla figura appare chiaro che quando la molecola si trova nella buca P, è necessaria una energia QP + EB per l'adsorbimento chimico, superiore all'energia QP richiesta per desorbirsi di nuovo. Il desorbimento è quindi più probabile. Solo poche molecole passeranno la barriera B rispetto a quelle che sono sottoposte al processo di adsorbimento-desorbimento in P. Il processo di adsorbimento chimico si applica quindi a poche molecole dotate di particolare energia. Si può però dare il caso che la particolare situazione relativa delle curve a e b sposti il punto B al di sotto dello zero. Ciò, per esempio, può avvenire se b dà un minimo pronunciato e/o se a presenta valori elevati di QP, cioè minimi pronunciati. In tal caso, una molecola adsorbita in P verrà più facilmente chemiadsorbita che desorbita. Si può concludere che lo stato di adsorbimento fisico, pur dotato di bassa energia, è un precursore essenziale dell'adsorbimento chimico. Inoltre, gli esempi mostrano come sia desiderabile calcolare con sufficiente precisione le curve a e b per poter predire teoricamente il fenomeno dell'adsorbimento. La definizione delle curve a e b, come già detto, presenta oggi notevoli difficoltà e costituisce un terreno d'indagine aperto per il futuro.
Le curve di energia potenziale consentono anche di descrivere in modo conveniente ciò che si verifica al crescere della copertura ϑ, dove per ‛copertura' si intende la frazione di superficie coperta dalle molecole adsorbite, e quindi 0 ≤ ϑ ≤ 1. Infatti, la fig. 2 si riferisce a un particolare valore della copertura, per esempio ϑ = 0. Vi sono diversi aspetti del fenomeno dell'adsorbimento che giustificano l'esistenza di una variazione di Q con ϑ . Nell'adsorbimento fisico, dove il fenomeno è poco rilevante, si può pensare essenzialmente a interazioni tra molecole adsorbite. Nell'adsorbimento chimico, dove si osservano variazioni di Q assai notevoli, si può pensare a diverse cause: a) non uniformità della superficie, o ‛eterogeneità a priori'; b) variazione della forza di legame dovuta a una variazione nella struttura elettronica indotta dall'adsorbimento, o ‛eterogeneità indotta'; c) esistenza di diverse forme di adsorbimento con calori distinti, in rapporti variabili con la copertura; d) interazioni tra molecole adsorbite. Se il calore di adsorbimento QC varia con il variare di ϑ, la curva b si sposterà al variare di ϑ. In modo schematico ciò è rappresentato nella stessa fig. 2 dove, oltre alla curva b, che si riferisce a ϑ = 0, è riportata la curva b′ che si riferisce a ϑ ≠ 0. Si vede che all'aumentare di ϑ diminuisce la velocità di adsorbimento, poiché aumenta il valore della barriera B. Inoltre, l'incremento ΔΕ tra b e b′ è inferiore al decremento ΔQ tra i valori QC (relativo a b) e QC′ (relativo a b′). La barriera energetica che ora si oppone al desorbimento QC′ + EB′ è inferiore quindi a QC + EB′, e pertanto il desorbimento sarà più veloce. Si raggiunge quindi uno stato stazionario sulla superficie per un dato valore di ϑ: valori elevati di ϑ verrebbero rapidamente diminuiti dal rapido processo di desorbimento, mentre valori piccoli verrebbero aumentati da un rapido processo di adsorbimento.
Il quadro illustrato viene ulteriormente complicato dall'esistenza di diverse forme di adsorbimento. Si può mostrare che al variare di ϑ si tende a favorire l'una o l'altra delle diverse forme possibili.
La fig. 2 mostra anche che tra ΔΕ e ΔQ vi è una relazione, in dipendenza dalle curve di potenziale. La relazione può essere approssimata da ΔΕ = −f ΔQ, con f 〈 1. Essa mette in relazione una grandezza tipica termodinamica, come il calore (o entalpia) di adsorbimento molare, con una grandezza cinetica, l'energia di attivazione. Essa, cioè, tende a correlare costanti di velocità con costanti di equilibrio. Più in generale, correlazioni tra costanti di velocità e costanti di equilibrio (o calori di reazione) sono state introdotte in catalisi, sia omogenea sia eterogenea. Di esse si parlerà ancora quando si tratteranno le correlazioni.
È logico che la comprensione delle interazioni molecola-superficie sia anche alla base della descrizione cinetica del processo di adsorbimento. Non è qui possibile soffermarsi su tale descrizione che ha notevole importanza nelle trattazioni cinetiche dei processi catalitici e per la quale rinviamo ai testi citati in bibliografia.
Aspetti elettronici del fenomeno di adsorbimento
Si è sottolineato all'inizio di questo articolo che il catalizzatore interviene in una reazione grazie alla formazione di veri e propri legami chimici tra molecola adsorbita e superficie, la natura del legame dipendendo sia dalla struttura elettronica della molecola adsorbita, sia da quella del solido.
La struttura elettronica dei solidi è stata ampiamente studiata negli ultimi quarant'anni. I risultati fornivano una base teorica sufficientemente valida per tentare di utilizzare i concetti in essa contenuti per lo studio del fenomeno della catalisi. Ciò è stato svolto in alcune trattazioni a partire dal 1950 circa. Sarà perciò utile richiamare alcuni concetti e la terminologia usata nella fisica dei solidi.
Nella ‛teoria delle bande' (v. solidi, fisica dei) si distinguono i casi: a) del metallo; b) del semiconduttore; c) dell'isolante. La distribuzione dei livelli di energia che possono accomodare un elettrone è caratteristica nei tre casi.
Un parametro caratteristico della struttura elettronica di un solido è il tlivello di Fermi' EF. Esso è correlato alla distribuzione degli elettroni: per E = EF la probabilità di occupazione è uguale a ½ valore di EF caratterizza anche l'energia libera di un elettrone nel solido; esso, cioè, è l'analogo del potenziale chimico di una specie in una miscela. L'energia richiesta per allontanare un elettrone dal solido, o energia di estrazione (workfunction) ϕ è data dalla differenza tra il valore dell'energia del sistema con l'elettrone fermo a distanza infinita (presa come E = 0) e l'energia con l'elettrone nel solido, EF. Inversamente, l'aggiunta di un elettrone al solido porrà l'elettrone al livello di energia EF.
Un altro importante aspetto preso in esame dalla teoria delle bande è l'influenza esercitata sulla struttura elettronica da un numero piccolo di difetti. Si può così mostrare che atomi di impurezze possono creare nuovi livelli, entro la zona proibita, che possono donare elettroni (livelli ‛donatori'), in modo che la conduzione sia dovuta a elettroni carichi negativamente (‛semiconduttore tipo n'), o accettare elettroni (livelli ‛accettori'), in modo che la conduzione sia dovuta ai buchi positivi lasciati dagli elettroni (‛semiconduttore tipo p').
In un esame delle proprietà adsorbenti di una superficie, in relazione a fenomeni di catalisi, è necessario anche richiamare una differente trattazione del legame nei metalli, dovuta a L. Pauling, che si basa sul metodo del ‛legame di valenza' (valence bond) e che si applica in particolare ai metalli di transizione. La teoria suppone che non tutti gli elettroni d entrino negli orbitali ibridi che intervengono nei legami interatomici e che si possa pertanto caratterizzare il metallo con la percentuale di carattere d (designata con δ) che entra nel legame. Valori elevati di δ corrispondono a legami più forti e a raggi atomici più piccoli. Si può pensare che se il legame atomo (o molecola) adsorbito-superficie è formato con rottura dei legami ibridi metallici e utilizzazione degli orbitali resi disponibili, si avrà una correlazione tra forza di legame adsorbato-superficie e δ. Peraltro, anche se sono utilizzati glì orbitali atomici, si avrà una correlazione con δ, perché il valore di δ fissa anche la frazione di elettroni non impegnati in detti orbitali. I tentativi di correlazione tra calori di adsorbimento e δ debbono quindi essere interpretati come tentativi di una razionalizzazione della fenomenologia e non come una vera teoria dell'adsorbimento. Si vedranno in seguito applicazioni degli stessi concetti a problemi catalitici.
Adsorbimento sui metalli
Una trattazione semplificata si può ottenere caratterizzando il metallo solo con alcuni parametri, quali l'energia di estrazione, e non occupandosi quindi della distribuzione dei livelli entro le bande di energia. In particolare, come nel caso del legame chimico nelle molecole semplici, si può schematizzare il sorgere o di un legame essenzialmente ionico o di un legame essenzialmente covalente.
Il primo caso potrà aversi per adsorbati che abbiano bassa energia di ionizzazione, dato che il metallo è in genere un buon accettore di elettroni, a causa della elevata densità di livelli energetici vuoti disponibili. Il calcolo basato su un trasferimento elettronico dell'atomo adsorbato al metallo dà per Q un corretto ordine di grandezza, ma non sempre un buon accordo: per esempio, si calcola per Na su W: Q = 30,5 kcal/mole e per Cs su W: Q = 45,7 kcal/mole. I valori osservati sono rispettivamente 32 e 64 kcal/mole.
La formazione di un legame essenzialmente covalente nel caso semplice dell'idrogeno H su un metallo M è descritta secondo il metodo di Pauling. I calcoli differiscono secondo la scelta dei valori di elettronegatività. Come esempio si riportano, in kcal/mole, i valori di Q per l'adsorbimento di idrogeno su diversi metalli.
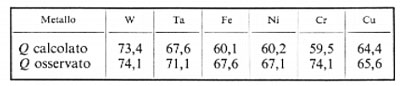
Mentre l'ordine di grandezza dei valori calcolati appare corretto, l'accordo non è particolarmente buono per alcuni metalli. Oltre alla difficoltà di misurare con precisione Q, si hanno evidentemente delle lacune nelle ipotesi. Per esempio, non si prende in esame la rottura dei legami M−M durante l'adsorbimento e le variazioni delle proprietà metalliche che ne derivano.
Recentemente, anziché partire dalla struttura a bande dei metalli per descrivere i processi di trasferimento di carica e di formazione di legami (di tipo ionico, oppure di tipo covalente o metallico), si è posto l'accento sulla utilizzazione degli orbitali disponibili. Questo approccio non si limita al caso delle superfici metalliche. Peraltro, anche sui metalli si può immaginare che vi siano degli orbitali o delle valenze non sature in superficie, responsabili dell'adsorbimento. La simmetria e l'energia degli orbitali deve condizionare la natura del legame formato. Questo tipo di trattazione tende a prendere in esame non solo molecole semplici, ma anche molecole più complesse quali ossigeno, azoto, etilene, ecc. Per esempio (v. fig. 3), dalla faccia (100) del nichel (cubico a facce centrate) emergono due orbitali, dyz e dxz, a 45° sul piano, e un orbitale, dz2, a 90° sul piano. Gli altri orbitali restano paralleli al piano e non emergono. Un atomo quale H, avente un elettrone s può utilizzare l'orbitale dz2. Una molecola quale l'etilene, avente un doppio legame, può essere adsorbita usando sia l'orbitale dz2, sia i due orbitali dyz e dxz. L'esempio è indicativo del tipo di trattazione, simile a quelle incontrate nei composti di coordinazione.
Adsorbimento su semiconduttori
Molti catalizzatori solidi hanno caratteristiche di semiconduttori, come ossidi, solfuri, ossidi misti. Anche i catalizzatori cosiddetti metallici, impiegati nell'industria o in laboratorio in condizioni in cui la superficie esposta non rimanga in realtà una superficie metallica pulita, dovrebbero essere considerati come ossidi (o solfuri) piuttosto che come metalli. Si comprende quindi l'interesse a trattare questi solidi, semiconduttori, come una classe a sé stante e ad utilizzare per essi concetti e conoscenze sviluppati negli ultimi trent'anni.
In un primo approccio al problema si è presa per base la struttura elettronica del solido quale essa appare dalla teoria dello stato solido, nella sua forma schematica del modello a bande. Appartengono a questo approccio le tre trattazioni svolte indipendentemente da Aigrain e collaboratori (Francia), Hauffe e collaboratori (Germania) e Weisz (S.U.A.). Esse hanno condotto alla formazione della teoria detta dello ‛strato esterno' (boundary layer o Randschicht). T. W. Wolkenstein (URSS) ha sviluppato una trattazione più generale, sempre basata sulla struttura elettronica del solido, descritta da un metallo a bande (v. Wolkenstein, 1963). Queste trattazioni, e particolarmente l'ultima, sono state considerate come ‛teorie elettroniche della catalisi'. Con il procedere degli anni e con l'approfondimento delle conoscenze della fisica delle superfici da un lato, e delle conoscenze sperimentali dall'altro, le teorie elettroniche hanno incontrato limitazioni concettuali e hanno subito trasformazioni tali da rendere più dubbia la loro capacità reale di inquadramento semplice del fenomeno catalitico in un contesto fisico che permettesse la previsione delle caratteristiche del fenomeno su superfici diverse. La teoria elettronica ha però indubbiamente il merito di aver attirato l'attenzione su alcuni aspetti della fisica dei solidi e di aver evidenziato il carattere interdisciplinare della catalisi. Essa inoltre fornisce per alcuni processi un linguaggio descrittivo e logico; traccia della sua influenza si ritrova anche in trattazioni non impostate sulla teoria elettronica. È ragionevole aspettarsi nella evoluzione delle trattazioni teoriche dei fenomeni di adsorbimento e di catalisi un più accentuato superamento di queste teorie, ma non la negazione di certi aspetti e di alcuni principî. È per questo che si ritiene opportuno richiamare alcuni concetti, ad illustrazione della via seguita.
È stato sperimentalmente osservato che spesso l'adsorbimento di un gas su un semiconduttore provoca una variazione della conducibilità elettrica del solido. Ciò indica una variazione della concentrazione di trasportatori di corrente, elettroni o buchi secondo che si tratti di tipo n o p. Si può quindi assumere per semplicità un modello che ipotizzi un trasferimento elettronico all'atto dell'adsorbimento. Per esempio, se si adsorbe ossigeno su ZnO (tipo n) la conducibilità diminuisce; ciò può essere spiegato con la formazione di ioni O- sulla superficie, ottenuta per trasferimento di elettroni dal semiconduttore sugli atomi adsorbiti. Si forma così uno strato esterno (da cui il nome della trattazione) di cariche negative O-. Dato che diminuisce la concentrazione dei trasportatori, si parla di edsorbimento (o strato) con impoverimento' (depletive chemisorption o Verarmungsrandschicht). Analogamente, si ha uno ‛strato di arricchimento' (cumulative chemisorption o Anreicherungsrandschicht). La conseguenza importante di questi fenomeni è che si ha un incurvamento delle bande vicino alla superficie, come nel caso di un contatto metallo-semiconduttore. Il trasferimento elettronico è possibile se l'energia di estrazione spesa per allontanare un elettrone è inferiore all'elettroaffinità A dell'atomo adsorbito (v. fig. 4). Con il procedere dell'adsorbimento, si crea una barriera di altezza V e l'energia di estrazione diventa ϕ + V, dove ϕ è l'energia di estrazione dalla superficie nuda. Conseguentemente, si arriverà al caso in cui A = ϕ + V: oltre tale valore l'adsorbimento anionico di O non sarà più possibile. Fatte opportune ipotesi sul parametro V, l'applicazione dell'equazione di Poisson permette di valutare il numero massimo degli atomi adsorbiti e mostra che l'adsorbimento è inferiore all'l% del totale dei siti. Quanto detto vale anche per l'adsorbimento cationico su semiconduttori p. La trattazione per il caso di impoverimento dà quindi una spiegazione fisica razionale dell'osservazione che le coperture sono in genere molto ridotte, pur ottenendosi un sensibile effetto sulla conducibilità. Il caso di adsorbimento con arricchimento (per es. H su ZnO, che fornisce elettroni al solido) incontra delle difficoltà di principio per la sua applicazione. In genere l'adsorbimento è più esteso, come è da attendersi, data la disponibilità di livelli accettori.
Una delle limitazioni del modello è il requisito del trasferimento di carica. La formazione di legami covalenti, infatti, non può essere prevista e trattata nella stessa forma semplice e suggestiva. Un notevole contributo allo sforzo di razionalizzazione del fenomeno dell'adsorbimento su semiconduttori è stato dato da Wolkenstein. Due aspetti fondamentali sono presi in esame dalla teoria: la possibilità che esistano diverse forme di adsorbimento e il tentativo di considerare in dettaglio il tipo di legame tra atomi e superficie, riconoscendo a quest'ultima il carattere non già di superficie ideale geometrica, bensì di insieme di atomi con loro proprie caratteristiche.
Sulla base di considerazioni teoriche, Wolkenstein mostra che devono essere presi in esame due tipi di legame tra atomi o molecole adsorbiti e superficie, uno definito ‛debole' e uno ‛forte'. I due termini sono usati in senso relativo l'uno rispetto all'altro e non assoluto; inoltre, il legame debole non deve essere confuso con quello di van der Waals. È solo nel legame forte che vengono coinvolti elettroni e buchi liberi del semiconduttore, che agiscono da valenze libere del solido. L'adsorbimento di tipo forte avviene quindi grazie all'esistenza degli elettroni o dei buchi liberi: questi costituiscono pertanto i centri attivi che rendono possibile l'adsorbimento. Il legame debole è un tipo di legame monoelettronico, come quello presente nella molecola ionizzata H2+. Il legame che comunemente viene preso in esame nell'adsorbimento è però quello forte, come appare, per esempio, dalla teoria dello strato esterno. Per illustrare i diversi casi possibili si veda la fig. 5, dove sono mostrati i legami deboli e forti su un solido ionico binario (NaCl) formati da atomi Na e Cl. I casi A e D mostrano il legame debole su catione e anione rispettivamente (non intervengono né elettroni e né buchi p del solido). I casi B ed E fanno intervenire un elettrone, C e F fanno intervenire un buco elettronico p. Viene messo in luce che l'adsorbimento forte ha come casi limite possibili il legame ionico da un lato e quello covalente dall'altro.
I risultati più importanti tratti da questa teoria possono essere riassunti nel modo seguente: 1) esiste la possibilità di forme differenti di chemiadsorbimento, con buchi o elettroni, che corrispondono ai casi limite di legami ionici o covalenti; 2) durante il processo di adsorbimento possono essere generati elettroni o buchi liberi: l'adsorbimento, pertanto, non richiede la distruzione delle valenze libere del solido. Questo punto è di particolare importanza e corrisponde, nella terminologia usuale, a dire che i centri attivi per l'adsorbimento possono essere creati dal processo stesso; 3) le forme differenti sono a volte saturate nelle loro valenze e a volte no (forme radicaliche). Le diverse forme saranno quindi caratterizzate da grande differenza di reagibilità; 4) è possibile la trasformazione di una forma nell'altra, come è mostrato per esempio nella fig. 5. È chiaro che la determinazione delle concentrazioni d'equilibrio delle varie forme è di primaria importanza ai fini dell'attività catalitica. L'autore dimostra che l'equilibrio delle varie forme è dettato dal livello di Fermi. I risultati sono mostrati nella fig. 6. In questa figura sono rappresentate le concentrazioni d'equilibrio di tre forme per ciascun atomo (o molecola) adsorbito. Le concentrazioni portano l'apice °, +, - rispettivamente per le forme deboli, donatrici o accettrici di elettroni. Si noti in particolare che a seconda della posizione del livello di Fermi si può avere prevalenza di una delle tre forme. Ogni causa che faccia modificare la posizione del livello di Fermi provocherà una variazione delle concentrazioni relative alle diverse forme. Tra le cause sono da ricordare la presenza di piccole quantità di sostanze estranee, come i cosiddetti e noti ‛promotori' e ‛veleni'. Un'altra possibile causa di variazione è l'irraggiamento della superficie.
È da notare che, anche se la situazione reale e la posizione del livello di Fermi in superficie non sono note, una modificazione all'interno del solido si riflette in uno spostamento di EF che, sia in superficie sia all'interno, è sempre nella stessa direzione. Quindi, almeno qualitativamente, gli spostamenti provocati all'interno mediante aggiunte chimiche si riflettono in modo qualitativamente analogo in superficie. È infine da notare che una superficie reale non è praticamente mai omogenea. Se si ricorda che la posizione relativa del livello di Fermi determina le concentrazioni delle varie forme, si vede che su una superficie reale possono essere presenti ‛isole' con differenti caratteristiche nei riguardi delle specie adsorbite. Nell'esame dei fenomeni catalitici si tornerà su questa teoria.
Struttura e chimica delle superfici dei solidi
Anche una trattazione che esamini superfici ideali deve riconoscere l'esistenza di configurazioni locali che variano da sito a sito. La situazione è ulteriormente complicata quando si prendono in esame superfici reali, dato che l'esistenza di difetti di diversa natura e l'esposizione di piani preferenziali fanno variare le configurazioni locali e le specie dei siti. A ciò aggiungasi che proprio l'atto dell'adsorbimento (in presenza o in assenza di reazione catalitica) può modificare la configurazione locale del sito di adsorbimento. Si può quindi parlare di una chimica delle superfici, in modo analogo alla chimica in tre dimensioni. Essa tende a fornire informazioni dettagliate sulla natura e sulle concentrazioni dei diversi arrangiamenti superficiali, sia in assenza delle molecole adsorbite, sia in presenza di queste.
La chimica delle superfici è una branca relativamente nuova. La difficoltà principale risiede nell'osservazione sperimentale; solo tecniche recenti hanno permesso un avanzamento sensibile, per cui si può oggi asserire che questo ramo subirà un'evoluzione importante nei prossimi decenni. A tale branca è interessata non solo la catalisi, ma anche tutta una serie di campi nei quali abbia importanza una interfase che coinvolga un solido (solido-solido, solido-liquido, solido-gas). Lo studio della superficie è intrinsecamente diretto a esaminare quantità di materia estremamente più piccole di quelle esaminate negli studi riguardanti la massa del materiale, dato lo sfavorevole rapporto (atomi di superficie)/ (atomi nel volume). Si può ovviare in parte a tale inconveniente con lo studio di materiali finemente suddivisi. Solo in casi estremi (centinaia di m2/g), il rapporto (atomi di superficie)/(atomi totale) è notevole (0,1 o superiore). Però la dispersione altera la chimica delle superfici, e quindi non sempre è estrapolabile a un solido disperso ciò che si deduce per la superficie di un solido compatto per es. un cristallo) o viceversa.
Diverse tecniche sono correntemente impiegate per lo studio delle superfici, ma l'importanza dello studio delle superfici come problematica a sé stante e le difficoltà insite in ciascuna tecnica limitano spesso lo studio delle superfici alle particolari condizioni imposte da ciascuna tecnica. Non sempre, cioè, vi è la possibilità di estendere lo studio del solido nelle condizioni in cui esso viene impiegato nella reazione catalitica.
Per quanto riguarda più propriamente la catalisi, è opportuno notare che l'esistenza sulla superficie di diverse specie, in concentrazioni assai differenti tra loro e di diversa reattività, introduce complicazioni notevoli non solo per l'osservazione sperimentale, ma anche per la valutazione del loro ruolo nel meccanismo catalitico. Una specie presente in concentrazioni ridotte e la cui osservazione può sfuggire alle tecniche impiegate, può fornire una via energeticamente più facile ed essere quindi responsabile della reazione globale. Vi è analogia, in questo, con la cinetica delle reazioni omogenee (v. cinetica chimica), dove specie cineticamente importanti (quali atomi o radicali) sono in concentrazioni che non risultano rilevabili con i mezzi ordinari.
Le tecniche sperimentali di studio delle superfici oggi a disposizione tendono a dare informazioni prevalentemente su uno dei due aspetti, strutturale o dinamico, che possono essere riconosciuti in una prima suddivisione. Le informazioni di tipo dinamico si ottengono con la scelta appropriata del processo da eseguire e spesso con l'ausilio di isotopi. Studi concernenti la mobilità di specie superficiali, l'inserzione di atomi (appartenenti al solido o solo allo strato adsorbito) nei prodotti finali, il meccanismo di avvelenamento, rappresentano alcuni esempi. È auspicabile e probabile un'espansione di questi studi, sia in condizioni vicine allo stato stazionario, sia lontane da esso (‛metodi di rilassamento'). Le indagini di tipo strutturale possono essere rivolte alla superficie del solido o alla specie adsorbita e ciò in dipendenza dalle condizioni richieste dalla tecnica in uso, anche se si tende a sviluppare mezzi di informazione che esaminino entrambi i problemi.
Un esame anche sommario delle tecniche richiederebbe una trattazione a parte. Tutto il campo dell'osservazione sperimentale dimostra nella varietà dei mezzi l'interdisciplinarità oggi richiesta dallo studio dei fenomeni catalitici. È ovvio che accanto a tecniche più raffinate sono sempre impiegati metodi che possono essere definiti tradizionali, in quanto mirano all'ottenimento di dati termodinamici (calori ed entropie di reazione, isoterme di adsorbimento, ecc.) o morfologici (area superficiale, porosità). Queste indagini sono sempre indispensabili per una corretta valutazione di altre informazioni e hanno registrato un notevole avanzamento tecnico, per es. attraverso l'impiego di tecniche microgravimetriche e microcalorimetriche.
Dal punto di vista strutturale, un notevole contributo allo studio delle superfici è stato dato dalle tecniche di diffrazione di elettroni a bassa energia (Low Energy Electron Diffraction o LEED) e dalla microscopia a emissione di campo, elettronica (FEM) o ionica (FIM), quest'ultima di particolare interesse per l'estensione a problemi di reattività. Interessante per i possibili sviluppi è la spettroscopia di fotoelettroni, variamente designata (ESCA, PES, XPS, IEE).
Alcune tecniche sono impiegabili per ottenere informazioni sia sul solido (e sulla sua superficie), sia sulle specie adsorbite. Esse comprendono tecniche spettroscopiche e di altra natura (misurà di proprietà elettriche o magnetiche). Le tecniche spettroscopiche hanno potenzialmente un grande interesse e, anche se esistono dei limiti nella loro sensibilità, si avvantaggiano continuamente di ogni ritrovato che aumenti sensibilità e risoluzione. È quindi plausibile attendersi un forte sviluppo delle tecniche spettroscopiche, anche perché esse si prestano spesso a essere impiegate in condizioni simili a quelle impiegate in catalisi, e congiuntamente ad altre misure. In particolare, è da ricordare la spettroscopia all'infrarosso di molecole adsorbite. Essa ha mostrato direttamente l'esistenza di complessi superficiali (per es. carbonili, in adsorbimento di CO, e di alcolati nell'adsorbimento di alcoli) e quindi di specie molecolari simili, ma non necessariamente uguali a quelle note nella chimica. Essa è però poco sensibile a particolari raggruppamenti e non rivela complessi superficiali presenti in bassa concentrazione, che potrebbero essere responsabili del meccanismo della reazione. È anche da menzionare la tecnica ESR (da Electron Spin Resonance: risonanza di spin elettronica) o EPR (da Electron Paramagnetic Resonance: risonanza paramagnetica elettronica), molto sensibile purché siano soddisfatti certi requisiti, e che ha fornito indicazioni preziose sia sull'esistenza di specie particolari nel solido (stati di ossidazione insoliti, quali Cr5+, Mo5+, Ni3+), sia, recentemente, di specie superficiali, quali O2-.
Cinetica delle reazioni catalitiche eterogenee
All'inizio di questo articolo si è sottolineato il fatto che il fenomeno della catalisi eterogenea rientra nella categoria dei problemi di reattività chimica. Gli aspetti cinetici del fenomeno sono quindi riconducibili agli stessi principi generali che governano ogni reazione, ma la presenza di un solido introduce complicazioni tali da rendere la cinetica delle reazioni eterogenee oggetto di considerazione come problema a sé stante. È chiaramente fuori degli scopi di questo articolo dare una trattazione anche sommaria dei diversi casi incontrati in pratica, ma è opportuno richiamare alcuni aspetti di particolare rilievo per la problematica generale.
La trasformazione dei reagenti nei prodotti avviene attraverso passi successivi che possono essere raggruppati nei seguenti stadi (o regimi):
a) diffusione dei reagenti verso la superficie del solido;
b) diffusione dei reagenti dalla zona della superficie esterna verso la superficie interna (pori) del catalizzatore;
c) adsorbimento di uno o più reagenti; d) reazioni sulla superficie, che coinvolgono una o più specie adsorbite;
e) desorbimento delle specie formate nel processo
d); f) diffusione dei prodotti nei pori verso l'esterno (inverso di b);
g) diffusione dei prodotti lontano dal catalizzatore nella massa fluida (inverso di a).
Gli stadi a) e g) (regime di ‛diffusione esterna') sono governati dai principî che sovrintendono al trasporto di massa in fluido. Gli stadi b) e f) (regime di ‛diffusione interna') sono dipendenti dai fattori che governano il trasporto di massa entro i pori. Quindi essi dipendono dalle caratteristiche geometriche dei pori e, in generale, dalla ‛tessitura' del catalizzatore. Gli stadi c), d) ed e) (regime cinetico chimico) riassumono tutti i fenomeni di formazione e rottura di legami, e di riarrangiamento molecolari, con partecipazione della superficie. Di questi si è parlato nei capitoli precedenti a proposito dei fenomeni di adsorbimento e di formazione di complessi superficiali.
La reazione globale procede a una velocità che è dettata dallo stadio più lento esistente nella successione dei diversi stadi. Per esempio, gli stadi di diffusione esterna sono dominanti nell'ossidazione dell'ammoniaca durante il processo di sintesi dell'acido nitrico, con un catalizzatore Pt-Rh particolarmente attivo. Le caratteristiche fluidodinamiche del sistema sono di importanza essenziale. Gli stadi di diffusione interna sono importanti quando si tratti con catalizzatori dispersi molto porosi. In tali casi sia l'attività catalitica sia la selettività sono influenzabili dalla tessitura del solido. Sono state sviluppate trattazioni teoriche in base ai vari parametri che influenzano il fenomeno in presenza di regimi diffusionali. C'è da notare che le ricerche cinetiche che mirano a chiarire gli stadi più propriamente chimici (c, d, e) devono garantirsi dell'assenza di interferenza della regione diffusionale (b, f; g). È però difficile alterare la tessitura di un catalizzatore senza alterare le proprietà chimiche della superficie. Questo accoppiamento costituisce una delle difficoltà che si incontrano quando si voglia trasportare al campo dei catalizzatori industriali ciò che si può chiarire sugli stadi chimici con catalizzatori di caratteristiche morfologiche particolari atte ad essere esenti da interferenze con regimi diffusionali (film o fili metallici, materiali molto sinterizzati).
Un breve commento merita il problema del regime cinetico chimico. La reazione chimica di trasformazione dei reagenti in prodotti sulla superficie avviene tramite una successione di passi elementari, cioè non riducibili. In generale, viene indicato come ‛meccanismo di reazione' l'insieme dei passi elementari che trasforma i reagenti in prodotti. In alcune reazioni dette ‛singole' o ‛semplici' il numero dei prodotti è limitato per es. CO + ½O2 → CO2), ma spesso si hanno diversi prodotti e diverse reazioni, consecutive e parallele, che trasformano le sostanze reagenti (‛schema di reazione'). Per esempio, la trasformazione dell'n-butano in butadiene,
CH3−CH2−CH2−CH3 → CH2=CH−CH2 + 2H2
può essere rappresentata dallo schema (semplificato per l'esclusione di reazione di cracking),
Grafico
dove B = butano, c2E = cis-2-butene, t2E = trans-2-butene, 1E = 1-butene, D = butadiene. Si hanno in totale nove reazioni. Ciascuna di queste nove reazioni singole può comprendere diversi passi elementari. Un primo obiettivo è quello di avere informazioni cinetiche su ciascuna delle nove reazioni, senza di che si avrebbe solo un'informazione globale di scarsa utilità ai fini della comprensione dei meccanismi di reazione, e quindi del ruolo del catalizzatore. Già l'ottenimento dei singoli dati cinetici è un'impresa non sempre possibile e spesso tutt'altro che semplice. Come accennato nell'introduzione, la conoscenza dei dati cinetici permette di condurre una reazione con la scelta dei parametri più appropriati per l'ottenimento di un particolare prodotto. Lo studio della cinetica delle reazioni eterogenee è quindi di interesse applicativo assai notevole. La definizione cinetica, inoltre, permette di ricondurre la reazione a un insieme di reazioni singole o semplici, per ciascuna delle quali è sperabile poter dedurre sia un meccanismo di reazione sia il ruolo del catalizzatore.
Si è detto che una reazione, anche singola, procede attraverso passi elementari. Questi coinvolgono specie transienti, a vita più o meno breve, alcune delle quali sono necessariamente presenti solo sulla superficie del catalizzatore. Può un'analisi cinetica condurre a riconoscere i passi elementari e la natura delle specie transienti? Non si può dare una risposta valida per tutti i casi. Si può a volte dedurre la presenza di alcune specie, ma spesso la risposta sulla successione dei passi elementari è ambigua, anche se limitata a un numero non grande di possibilità. Si richiede, in genere, non solo un'analisi accurata dei possibili passi, ma anche la formulazione di ipotesi, fisicamente plausibili, sui singoli passi e sulle caratteristiche delle specie superficiali, quali la reversibilità o l'irreversibilità del passo, la distribuzione (uniforme o no) dei siti di adsorbimento e l'equilibrio tra forme particolari delle specie adsorbite. Le trattazioni date da diversi autori tendono a fornire una base solida della formulazione fisico-matematica, sì da ricondurre il processo a un modello matematicamente affrontabile. Ciò è stato fatto, per esempio, per la sintesi dell'ammoniaca e per le reazioni di idrogenolisi di idrocarburi semplici. Inoltre, per alcune reazioni semplici quali le reazioni di scambio isotopico tra molecole di ossigeno, la decomposizione di N2O, lo scambio tra idrogeno e deuterio, ecc., è possibile avere una descrizione tale da collegare il dato cinetico a un meccanismo particolare. Sotto questo aspetto la trattazione cinetica non è fine a se stessa, ma costituisce uno dei mezzi impiegati per approfondire lo studio della natura del processo catalitico.
Correlazioni in catalisi
Nei capitoli precedenti si sono illustrati problemi che si trovano alla base del fenomeno della catalisi eterogenea e si sono descritti i tentativi di valutazione dei parametri caratteristici del solido che determinano in definitiva il suo comportamento come adsorbente. Tuttavia, appare chiaro come non sia semplice pervenire ad una razionalizzazione del ruolo di ciascun parametro e quindi alla scelta di un certo catalizzatore per una particolare reazione. Nella ricerca di una risposta empirica ma razionale si è quindi portati a valutare il ruolo di ciascun parametro caratterizzante il solido mediante correlazioni, cioè mediante l'esame di classi di solidi (o di classi di reazioni), che permettano una correlazione tra attività catalitica da un lato e un particolare fattore che distingua ciascun solido (o ciascuna reazione) nella classe considerata. Il fattore che rappresenta la variabile può essere scelto in modo diverso a seconda dell'enfasi che viene data a quel particolare aspetto: può essere scelto, per esempio, un parametro che rifletta la struttura elettronica del solido o un parametro che dipenda da una proprietà chimica di superficie (quale l'acidità superficiale). Tale scelta dipenderà sia dalla scuola di pensiero che viene sentita come più congeniale, sia dal tipo stesso di reazione. Si ritrovano quindi nella descrizione del fenomeno catalitico i punti di vista diversi ma non necessariamente contrastanti già menzionati a proposito dell'adsorbimento. In particolare, si potrà mettere l'accento sulle proprietà elettroniche collettive del solido o sulle proprietà di complessi superficiali formati durante la reazione e visti come specie reattive intermedie.
Ora, dato il carattere di questo articolo, si cercherà di dare uno sguardo a questi tentativi di razionalizzazione, piuttosto che di riassumere le conoscenze specifiche già raggiunte nei diversi settori in cui la catalisi è già applicata.
Parametri termodinamici dell'adsorbimento e catalisi
L'adsorbimento chimico è un processo essenziale per la catalisi. Si può quindi pensare che, affinché una superficie sia cataliticamente attiva, essa debba adsorbire i reagenti. Un primo esame dell'abilità di adsorbimento può orientare verso la scelta di un materiale per classi di reazione. Un esempio tipico è lo studio dell'adsorbimento di molecole diverse su vari metalli. Si è trovato che la forza di adsorbimento con cui molti metalli legano alcune tipiche molecole (con l'eccezione dell'oro che non adsorbe ossigeno) varia in genere nell'ordine:
O2 > C2H2 > C2H4 > CO > H2 > CO2 > N2.
Si può allora fare una classificazione dei metalli sulla base delle specie (tra quelle elencate) che il metallo è in grado di adsorbire (v. Bond, 1962). Si può così stabilire che i metalli dei gruppi che vanno dal IV fino all'VIII-1 del sistema periodico adsorbono N2, mentre Co (VIII-2) e Ni (VIII-3) non l'adsorbono. Nella sintesi di NH3, la molecola di N2 deve essere attivata, e ciò può avvenire per adsorbimento di N2 (anche se attualmente si pensa che forme di adsorbimento particolari in presenza di idrogeno possano essere responsabili dell'attivazione). La classificazione suggerisce allora che solo il Fe, e non il Ni e il Co, sia attivo tra i metalli dell'VIII gruppo, e ciò è quanto si osserva in realtà. Un altro esempio è fornito da reazioni di idrogenazione. Metalli quali Al e Mg non adsorbono idrogeno e non sono attivi.
Questo primo criterio qualitativo non esaurisce però il problema. Tra i metalli che adsorbono una data specie molecolare, si hanno notevoli variazioni di attività catalitica. L'adsorbimento è caratterizzato dal calore di adsorbimento, che è in relazione alla forza di legame stabilito con la superficie. Se è vero che un legame deve essere formato, e affinché ciò avvenga (v. fig. 2) il calore di adsorbimento deve essere apprezzabile, è logico aspettarsi che qualora il legame molecola-superficie sia troppo forte la specie adsorbita non sarà più incline a entrare in passi elementari successivi e quindi non vi sarà attività catalitica malgrado il verificarsi dell'adsorbimento. Quindi l'attività catalitica presenterà un massimo in funzione del calore di adsorbimento. Questa tipica correlazione, detta dall'aspetto curva ‛a vulcano', è rappresentata schematicamente nella fig. 7 e mostra come esista una relazione tra una grandezza termodinamica e l'attività catalitica. Ciò si riscontra in pratica in molti processi. Per esempio, la sintesi dell'ammoniaca prima citata non solo non procede su Ni (adsorbimento nullo di N2) ma procede male anche sui metalli alla sinistra del ferro, che hanno calori di adsorbimento di N2 più elevati di quelli del Fe. Un altro esempio è offerto dalla decomposizione dell'acido formico. In questo caso, per avere una misura dell'energia di interazione, si è preso il calore di formazione del formiato metallico, mostrando così un modo di utilizzazione di dati termodinamici indiretti (v. fig. 8), di cui si è discusso a proposito dell'adsorbimento.
Può essere interessante notare che una correlazione tra attività catalitica (grandezza cinetica) e energia di legame (grandezza termodinamica) può essere dedotta da considerazioni di base. Si è già notato a proposito delle curve di potenziale nell'adsorbimento che può essere scritta una relazione tra la variazione ΔE dell'energia di attivazione e la variazione del calore di reazione ΔQ (Polanyi) (si ricorda che ΔQ è positivo per un processo esotermico):
ΔΕ = − f ΔQ.
La relazione è generale e ha notevole importanza. Nel campo omogeneo, per esempio, la relazione di Brönsted per catalisi acido-base può essere derivata dalla relazione di Polanyi. La relazione di Polanyi trova applicazione anche in catalisi eterogenea non solo per reazioni catalitiche acido-base, ma anche quando si desideri introdurre nella trattazione cinetica la non uniformità dei siti e la variazione dei calori di adsorbimento con la copertura. Essa conduce a esprimere la costante di velocità k di ciascun passo della reazione come
k = costante × Kf
dove K è la costante di equilibrio e f deriva dalla relazione di Polanyi. L'utilità di questa correlazione risiede nella possibilità di esprimere le equazioni cinetiche in funzione di un numero ridotto di parametri cinetici, dato che per alcuni passi vengono sostituite costanti di equilibrio, cioè grandezze termodinamiche. Si può anche dimostrare, come corollario, che deve valere una correlazione del tipo ‛a vulcano' tra la velocità globale della reazione e i calori di adsorbimento (v. Boudart, 1968). L'esistenza della correlazione del tipo ‛a vulcano' è logica ma non sempre evidente, in quanto può avvenire che uno dei due rami sia sperimentalmente non accessibile. Una situazione che si verifica spesso è quella del ramo discendente, cioè si trova spesso che l'attività catalitica varia in senso inverso all'energia di legame. Ciò è stato riscontrato, per esempio, nella idrogenazione dell'etilene su metalli di transizione. Chiaramente, tale comportamento non è estrapolabile ai limiti estremi dei valori di adsorbimento, né è estendibile alla generalità delle reazioni. Questi esempi ripropongono l'importanza della comprensione del fenomeno dell'adsorbimento, onde poter razionalizzare l'energetica del processo. Infatti, il calore di adsorbimento dovrebbe essere desunto da proprietà di base della molecola adsorbita e del solido adsorbente: sorgeranno quindi correlazioni tra parametri tipici del solido ed energia di adsorbimento e in definitiva tra detti parametri e attività catalitica. Per esempio, si è visto come il calore di adsorbimento possa dipendere da δ (percentuale di carattere d del metallo): si avrà quindi una correlazione tra attività catalitica e δ. Correlazioni di questo tipo sono state mostrate per l'idrogenazione dell'etilene (Beek) e per l'idrogenolisi di idrocarburi semplici quali l'etano (Sinfelt). La correlazione non risolve il problema di base di poter definire a priori il ruolo di ciascun fattore nell'attività catalitica, ma consente un orientamento razionale nella scelta del metallo adatto.
Infine, un'interessante correlazione tra dati cinetici e termodinamici è stata suggerita recentemente e dovrà nel futuro essere più estesamente esplorata, a proposito del problema della selettività di un catalizzatore. In breve, l'idea, introdotta da Sachtler e collaboratori nel caso di un catalizzatore per ossidazione, consiste nel riconoscere che il catalizzatore tenderà a promuovere l'ossidazione totale (a CO2 e H2O) di un composto organico (per es. C6H5CHO) se sarà facile agli intermedi di reazione riuscire a strappare l'ossigeno superficiale. In caso contrario, l'intermedio avrà maggior probabilità di non subire ulteriore ossidazione, e quindi di essere desorbito, dando così luogo a ossidazione selettiva (per es. C6H5COOH). La facilità con cui l'ossigeno viene rimosso durante l'ossidazione della molecola, cioè mentre la superficie tende a impoverirsi di ossigeno, sarà misurata dal gradiente ∂(ΔH)/∂/x, dove ΔH è l'entalpia di legame ossigeno-superficie e x è il contenuto di ossigeno. Un elevato gradiente implica una difficoltà energetica alla riduzione superficiale del contenuto di ossigeno e favorisce l'ossidazione parziale (selettiva).
Il fattore elettronico in catalisi: metalli e leghe
Si è già visto a proposito dell'adsorbimento che vi sono giustificazioni di principio in base alle quali si può collegare la struttura elettronica di un solido con la sua capacità di formare specie superficiali particolari, e quindi con la sua attività catalitica.
Una correlazione si ritrova a proposito dell'energia di legame e del carattere d. I metalli di transizione con banda d incompleta sono in genere molto più attivi in diverse classi di reazioni. È stato osservato (Dowden) che non è sufficiente prendere in esame i livelli disponibili entro le bande, ma la variazione della loro concentrazione con il variare dell'energia ∂n(E)/∂Ε. Non sempre le informazioni sulla struttura elettronica dei metalli sono sufficientemente dettagliate. Ma, anche nei casi favorevoli, una relazione quantitativa è complicata dalla necessità di specificare la struttura elettronica superficiale. Quest'ultima riflette quella di massa, ma non coincide con essa. Si verificano sperimentalmente e si giustificano così in linea di principio variazioni dovute alla faccia cristallina esposta. Inoltre è da considerare che l'attività catalitica, per atomo metallico ‛esposto', dovrebbe variare quando il cristallo diventa molto piccolo, dato che la struttura elettronica, al di sotto di una certa grandezza del cristallo, dipende dalla dimensione. Ciò non è sperimentalmente facile da provare. Vi sono anzi esempi che tendono a mostrare il contrario. Sembrerebbe quindi che vi siano reazioni nelle quali è più importante la natura atomica del metallo che non le sue proprietà elettroniche collettive o la particolare configurazione geometrica superficiale presentata dagli atomi metallici. È opportuno notare la complessità del problema dal punto di vista sperimentale. L'intervallo di dimensioni in cui gli effetti strutturali possono assumere un peso considerevole è limitato; ciò pone il problema della dispersione del metallo. Se il metallo è disperso su un supporto può verificarsi un'interazione con il supporto, che simula un effetto di dimensione. Infine, la possibilità di misurare il numero degli atomi ‛esposti' - misura necessaria per poter definire un'attività catalitica per atomo - non è sempre attuabile e dipende dalla natura del metallo.
L'impiego di leghe può offrire in linea di principio un mezzo per far variare in modo continuo le proprietà elettroniche. Sono però da tener presenti ancora una volta le difficoltà sperimentali, che smentiscono a volte l'ottimismo con cui sono state considerate certe correlazioni. In mancanza di informazioni precise sull'omogeneità della lega, è azzardato ritenere che discontinuità nel comportamento catalitico siano senz'altro da attribuire al riempimento di bande e non piuttosto a formazione di fasi superficiali particolari, come avviene, ad esempio, nel caso della lega CuNi. Inoltre, l'attività catalitica è di difficile misurazione, data l'area piccola dei catalizzatori formati da leghe, che abbiano composizione possibilmente omogenea.
In definitiva, mentre l'esistenza qualitativa di una correlazione appare provata e razionalizzabile in linea di principio, una sua formulazione quantitativa è ancora lontana. Il campo è indubbiamente suggestivo ed è da attendersi un progresso nella misura in cui sarà possibile ottenere informazioni più precise sulla struttura e sulla composizione della superficie, sulla struttura elettronica in superficie e su metodi sensibili di definizione dell'attività catalitica.
Il fattore elettronico in catalisi: semiconduttori
Per motivi storici, si tende a impiegare il termine ‛fattore elettronico' quando questo è riferito a un modello a bande, cioè a un modello collettivo, non localizzato. In accordo con la consuetudine, si parlerà quindi di modelli collettivi.
Nella teoria dello strato esterno, l'adsorbimento è legato a un trasferimento elettronico o verso il solido o verso l'adsorbato. Il passo lento della reazione può essere legato a un trasferimento di elettroni verso l'adsorbato (reazione ‛accettrice') o verso il solido (reazione ‛donatrice'); quest'ultima può essere anche vista come trasferimento di una buca elettronica (positiva) verso l'adsorbato. Una variazione della concentrazione elettronica, o dei buchi positivi nel solido, influenzerà allora la reazione. L'esame di una stessa reazione condotta su solidi diversi, caratterizzati da concentrazioni diverse di elettroni o di buchi positivi, permetterà allora di dedurre se la reazione è di classe accettrice o donatrice. Per esempio, la reazione di scambio tra H2 e D2 è catalizzata da diversi ossidi: si trova che trattamenti che aumentano la concentrazione elettronica, per esempio con gas o con aggiunte di ioni eterovalenti, tendono ad aumentare la velocità di scambio, e viceversa. Si deduce allora che la reazione è accettrice.
Un altro esempio è costituito dall'ossidazione di NH3 a N2O, studiata (Krauss) su MnO, Fe2O3, CoO, NiO. Il carattere p è qui legato al contenuto di ossigeno in eccesso. Si trova che l'attività catalitica (percentuale di N2O formata) è proporzionale al contenuto di O in eccesso; ciò prova che si è di fronte a una reazione donatrice, vale a dire favorita dal carattere p.
Infine, un esempio classico è quello della reazione di decomposizione di N2O. Scrivendo diversi ossidi in ordine decrescente di attività era stato osservato che i più attivi erano di tipo p (Cu2O, CoO, NiO) e i meno attivi di tipo n (ZnO, Fe2O3). Ciò era stato interpretato come indice che la reazione, i cui passi elementari possono essere scritti
N2Ogas + e → N2O−ads,
N2O−adsN2 gas + O-
2O- → O2 gas + 2e
è di tipo donatrice, cioè il passo lento sarebbe dovuto alla terza reazione. In realtà, tale deduzione non è senza ambiguità. Infatti anche l'adsorbimento di una specie accettrice (per es. O2) è favorito dal carattere p; quindi detto carattere potrebbe intervenire nella prima reazione, anziché nella terza.
La correlazione tra attività catalitica e fattore elettronico non deve essere interpretata necessariamente come un rapporto di causa ed effetto. Il fattore elettronico, inteso come risultato di interazioni cooperative, dipende a sua volta dalle configurazioni dei singoli atomi costituenti e l'aspetto cooperativo può non essere fondamentale (v sotto). Inoltre, quando si confrontano solidi diversi non è detto che il passo lento rimanga sempre lo stesso. Un esempio di mancanza di univocità è dato dall'ossidazione di CO, studiata da diversi autori. La fig. 9 riporta la variazione dell'energia di attivazione apparente E e la conducibilità elettrica (misurata a 400 °C e a 25 °C) per NiO puro, con aggiunte di Li2O (aumenta il carattere p) e con aggiunte di Cr2O3 (aumenta il carattere n). La correlazione appare suggestiva, ma a bassa temperatura essa è inversa, cioè l'aggiunta di Li2O innalza, anziché diminuire, il valore di E. Questi e altri esempi di ambiguità se da un lato possono essere spiegati con una variazione del meccanismo o del passo lento, mostrano l'impossibilità di prevedere a priori in quale direzione debbano essere modificate le proprietà di un solido per favorire l'attività catalitica.
Gli esempi di correlazione per reazioni che assumono il trasferimento elettronico presentano un punto debole nella rigidità dei modelli ionici delle specie adsorbite. Particolare attenzione merita quindi la trattazione di Wolkenstein, che rimuove in linea di principio questa limitazione.
Si è già illustrato a proposito dell'adsorbimento come la trattazione di Wolkenstein conduca a considerare diverse forme di adsorbimento, le cui concentrazioni dipendono dal livello di Fermi. La presenza di forme diverse dà maggiore flessibilità alla teoria. Si consideri una sostanza che può reagire in due modi differenti, per esempio CH3CH2OH
in cui a) è la reazione di disidratazione e b) la reazione di deidrogenazione. L'esempio illustra un caso molto comune in catalisi e riguarda il problema della ‛selettività' di un catalizzatore. Schematicamente, Wolkenstein suppone che a) e b) possano procedere con meccanismi tali che implicano: in a) una donazione di e al reticolo, cioè una reazione donatrice; in b) una donazione di p al reticolo, cioè una reazione accettrice. La trattazione matematica dei due casi a) e b) conduce a concludere che la disidratazione a) è favorita da un abbassamento del livello di Fermi, mentre la deidrogenazione b) è favorita da un innalzamento del livello stesso, ciò che appare confermato sperimentalmente. Si può quindi influire sulla selettività con aggiunte opportune al catalizzatore. È poi da ricordare che non solo reazioni differenti, ma anche passi elementari di una stessa reazione possono appartenere a classi differenti (accettrici o donatrici), come ricordato per la decomposizione di N2O, o come può essere mostrato per l'ossidazione di CO. A seconda che il passo lento sia l'uno o l'altro, lo spostamento del livello di Fermi può favorire od ostacolare la reazione. Differenze di temperatura e di preparazione possono spostare il passo lento dall'uno all'altro; si spiegano così le discordanze riscontrate in alcuni casi, come quello citato nell'ossidazione di CO.
La teoria di Wolkenstein non è esente da critiche, sia per quanto riguarda la scelta del parametro (livello di Fermi) da cui dipendono tutte le grandezze significative, sia per l'aggiustabilità dei diversi parametri, cosicché è possibile in genere giustificare a posteriori un fenomeno, ma non prevederlo. Essa ha però meriti concettuali indubbi, e l'importanza di aver data attenzione a certi parametri del solido si riflette sia nelle teorie derivanti dalla trattazione di Wolkenstein, sia in teorie che cercano vie diverse. Ciò appare, per esempio, nella trattazione di fenomeni di elettrocatalisi o ancora nella trattazione di Krylov (v., 1970) che, mediante considerazioni sempre basate sul modello a bande dei solidi, mostra la possibilità di correlare l'attività catalitica con l'ampiezza della zona proibita.
Il ruolo della configurazione elettronica localizzata
Le difficoltà presentate dai modelli collettivi del solido e l'indagine sperimentale diretta, specie con la spettroscopia infrarossa, di molecole adsorbite, hanno riproposto all'attenzione degli studiosi di adsorbimento e catalisi il problema di un legame chimico localizzato. Le specie esistenti sulla superficie di un solido possono essere infatti trattate come ‛complessi superficiali', concentrando l'attenzione sulla struttura elettronica del sito coordinante. Si considera valido, cioè, in prima approssimazione il modello di un legame localizzato e appartenente a un complesso superficiale a cui il resto del solido apporta solo una perturbazione.
I progressi realizzati nella chimica dei composti di coordinazione hanno dato notevole impulso a una trattazione del tipo ‛localizzato'. Si avevano, infatti, sia modelli di diverse specie superficiali sia strumenti teorici per la loro trattazione sia, infine, una vasta casistica che forniva una base per correlazioni empiriche ma razionali.
In questa cornice vanno inquadrati diversi lavori apparsi a partire dal 1960. La prima trattazione esplicita lungo queste linee è dovuta a Dowden, il quale pone particolare attenzione agli ioni dei metalli di transizione, che costituiscono la classe di maggior interesse in reazioni catalitiche ossidoriduttive. Questo approccio è in continua evoluzione e si va arricchendo di nuovi spunti e di nuove osservazioni. Tra questi, possono essere citati non solo gli studi su ossidi diversi, ma anche quelli aventi per oggetto o soluzioni solide di solidi, o zeoliti con ioni di metalli di transizione. In entrambi i casi è difficile descrivere il solido alla stregua di un modello a bande, mentre il mantenimento dell'attività catalitica, nonché delle caratteristiche riscontrate per ossidi puri anche in sistemi aventi ioni isolati, tende a suggerire la validità di un modello localizzato.
La teoria del campo cristallino (v. stereochimica) e la successiva elaborazione della teoria del campo dei leganti avevano mostrato come la reattività dei complessi dipendesse dalla configurazione elettronica e dalla simmetria del complesso. Anche nei fenomeni di superficie può essere individuato l'aspetto di simmetria. Infatti l'adsorbimento su un atomo superficiale completa la sua sfera di coordinazione e, per esempio, per un atomo posto sul piano (100) si passa da una configurazione di piramide a base quadrata a una di tipo ottaedrico. Le distorsioni esistenti nella realtà rispetto alle simmetrie complete (rilassamenti, non equivalenza dei legami) non comportano variazioni tanto grandi da non tentare anche nel caso delle superfici una correlazione con la struttura elettronica. L'energia del complesso superficiale dipende sia dal campo creato dai leganti (quindi dalla natura degli ioni vicini allo ione metallico e dalla molecola adsorbita), sia dal numero di elettroni posseduti dallo ione metallico. Il campo cristallino, pertanto, introduce una modulazione nell'energia, cioè, al variare del numero nd di elettroni d, si ottengono dei minimi di energia di stabilizzazione per particolari valori di nd Poiché i fenomeni dinamici sono molto sensibili a variazioni anche piccole di energia e tenuto conto (v. curve ‛a vulcano', fig. 7) che l'intervallo di energia interessante i fenomeni di superficie è in realtà ristretto, si può capire come possa verificarsi un'analoga modulazione dell'attività catalitica con nd, cioè una correlazione con nd.
Vi sono ormai diverse classi di reazioni, già studiate per serie di ossidi, che presentano questa correlazione. Alcuni esempi sono riportati nella fig. 10. Non sempre la correlazione è evidente, né d'altra parte essa deve essere sempre esistente per poter affermare che i meccanismi di reazione sulla superficie possono essere trattati come quelli dei complessi molecolari. Infatti è da ricordare che vi sono complicazioni proprie della superficie e che la presenza di difetti di varia natura può offuscare o alterare il quadro deducibile da una trattazione basata su una superficie ideale. Così, per esempio, esistono stati di valenza anomali, corrispondenti a livelli accettori-donatori, e vi sono anche configurazioni non prevedibili da una struttura ideale a causa di vacanze atomiche o di ioni interstiziali. Inoltre, il passo lento può essere di natura diversa (adsorbimento, desorbimento o altri processi confinati sulla superficie) e ciò può portare a trovare massimi di attività catalitica per una particolare configurazione elettronica solo per certe reazioni. Per diverse reazioni, peraltro, è possibile poi mettere in evidenza la presenza di un passo che coinvolge la rottura (o la formazione) di uno stesso legame. Per esempio, l'ossidazione di H2, di CO, di CH4, di NH3, lo scambio isotopico di O2 e la decomposizione di N2O coinvolgono tutte reazioni con O-s, dove O-s rappresenta un ossigeno di superficie. Se questo passo governa o fa sentire il suo peso nella reazione globale si avranno correlazioni con l'energia di legame (e quindi con nd) per tutte le dette reazioni. Ciò è quanto si osserva sperimentalmente.
La correlazione tra nd e attività catalitica rappresenta, analogamente a quella con il carattere di semiconduttore, un utile punto di partenza, ma non può soddisfare pienamente i quesiti e i problemi incontrati in catalisi. Essa rappresenta un vivido esempio di confluenza di idee da campi diversi (stato solido, spettroscopia, chimica inorganica) e mostra la stretta correlazione esistente tra catalisi eterogenea e catalisi omogenea. Un esempio di questa correlazione è offerto dall'esame di una stessa reazione (ossidazione del cicloesene) condotta su ossidi o su complessi (in soluzione) di metalli di transizione. La fig. 11 mostra il parallelismo di attività catalitica per i due prodotti idroperossido e chetone.
Infine, nell'esporre il punto di vista della trattazione localizzata, è opportuno accennare, sia pure brevemente, ad altri punti di contatto tra complessi superficiali e composti di coordinazione. Si può così notare che per i complessi superficiali può avvenire un riarrangiamento elettronico tramite orbitali σ e π analogo a quanto avviene per un complesso molecolare. Questi riarrangiamenti portano a variazioni delle geometrie molecolari ed a stati elettronici assimilabili a stati eccitati. Pertanto, le caratteristiche della molecola adsorbita possono essere differenti da quelle normalmente conosciute, ma simili a quelle di una molecola eccitata. Ciò può avere molta importanza sia per l'attivazione di molecole semplici (quali O2), sia per l'attivazione di legami in molecole più complesse, quali olefine (per es. in reazioni di dismutazione).
Catalisi stereospecifica
Tra gli esempi di trattazione localizzata merita un cenno particolare il caso della catalisi stereospecifica, che implica sia concetti riguardanti la struttura elettronica, sia aspetti geometrici del sito attivo e della molecola reagente.
Dopo la scoperta dell'azione catalitica per la polimerizzazione dell'etilene mostrata da composti di metalli di transizione insieme a un alchile metallico (Ziegler), l'estensione a sistemi di α-olefine (Natta) ha portato alla scoperta di polimeri stereoregolari, cioè con gruppi sostituenti orientati in modo definito, non casuale (v. polimeri). Per esempio, il propilene può dar luogo a due polimeri stereoregolari (‛isotattico' e ‛sindiotattico') oltre a quello non regolare (‛atattico'), come schematizzato nella fig. 12. Oltre ad applicazioni pratiche assai notevoli, date le caratteristiche fisiche, pregevoli dei polimeri stereoregolari, la scoperta dei catalizzatori e delle reazioni Ziegler-Natta ha focalizzato l'attenzione sulla necessità di un intermedio che deve tendere a riprodurre come in uno stampo una sola configurazione tra le molte possibili. È opportuno ricordare brevemente alcune caratteristiche della reazione e degli schemi proposti, dato che esse riassumono diversi concetti espressi in catalisi e data l'importanza che essi possono avere non solo per queste reazioni, ma anche per reazioni complesse quali quelle enzimatiche. Si riconosce che deve esistere un centro sulla superficie, identificato nello ione del metallo di transizione, che coordina l'olefina. L'agente alchilante produce un legame attivato metallo-carbonio. Se si prende come esempio TiCl3 e AlR3 quali catalizzatori e C2H4 quale alchene, si può scrivere
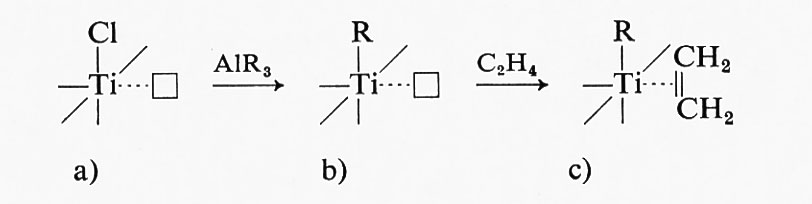
dove in c) si realizza un legame π con l'olefina. L'intermedio c) si trasforma facilmente:
Si può notare che il sito vacante viene alternativamente a trovarsi in due posizioni (b = f, d). Inoltre la fissazione del legante al sito vacante e la reazione tra i due leganti coordinati (c, e) hanno luogo facilmente solo se i reagenti hanno una determinata conformazione. Ciò conduce sia a un meccanismo di accrescimento indefinito, sia al fatto che l'accrescimento della catena, avviene con ‛pezzi' obbedienti a uno ‛stampo', cioè è stereoregolare. Per questo schema è stata anche fatta una dettagliata descrizione degli orbitali molecolari in gioco e della stereochimica di superficie e si è potuto così giustificare il fatto che l'azione è svolta da alcuni ioni e non da altri. In particolare, si è potuto giustificare il fatto che siano attivi solo gli ioni con configurazioni da d0 a d3 e non altri con orbitali d più riempiti.
Catalisi e centri acidi superficiali
Alcune reazioni sono catalizzate da acidi, sia in sistemi omogenei sia in sistemi eterogenei. È logico pensare che il meccanismo sia fondamentalmente lo stesso e che coinvolga gli stessi tipi di specie intermedie. Reazioni catalizzate da acidi sono, per esempio, le seguenti: isomerizzazioni, polimerizzazioni, alchilazioni e cracking. Si può scrivere un meccanismo che coinvolge uno ione carbonio favorito da acido di Brönsted (donatore di protoni) o di Lewis (accettore di coppia elettronica). La superficie di un solido, come già menzionato, può presentare gruppi e configurazioni diverse e quindi possono essere presenti siti acidi di tipo Brönsted e ‛i tipo Lewis. Data la natura ‛composita' della superficie, la presenza di siti acidi di un tipo non esclude la presenza di siti acidi dell'altro tipo; un esempio tipico di superficie con siti acidi di entrambi i tipi è la silice-allumina. È possibile correlare l'attività catalitica e la selettività per alcune classi di reazioni con l'acidità della superficie e avere quindi una guida alle modificazioni da apportare per poter influire sulla reazione nel senso voluto. Un esempio di correlazione è mostrato nella fig. 13.
Catalizzatori polifunzionali
Di particolare importanza sono i catalizzatori che, oltre a mostrare una funzione acida, presentino anche capacità ossidoriduttiva. Essi rientrano, e ne sono un tipico esempio, nei catalizzatori che posseggono due o più funzioni e che sono pertanto detti ‛bifunzionali' o in genere ‛polifunzionali'. Un esempio tipico è costituito dal Pt su silice-allumina. Oltre all'azione deidrogenante-idrogenante del Pt, essi posseggono quella di isomerizzazione e di cracking. Così, per esempio, il C16H34 (esadecano) viene convertito in ottani C8H18 perché, grazie all'azione del Pt, viene formata prima l'olefina (deidrogenazione) e da questa mediante cracking si forma l'ottene, grazie all'azione di SiO2-Al2O3; infine, si formano gli ottani per idrogenazione. Il bilanciamento tra le diverse funzioni e l'influenza di fattori diversi sulla selettività rendono possibile, in linea di principio, ottenere su un unico tipo di catalizzatore distribuzioni di prodotti diversi e ciò ha un'importanza pratica assai grande. L'obiettivo del disegno di un catalizzatore industriale che effettui le operazioni volute implica in genere l'appropriato uso di più funzioni presenti su un catalizzatore appositamente studiato.
Nota conclusiva
Si può notare come, pur essendo partiti da un esame dei problemi di adsorbimento che ambiva trattare con una certa profondità e un certo rigore il fenomeno dell'adsorbimento, quale passo essenziale per la comprensione del fenomeno della catalisi, a mano a mano che si riconosceva l'intervento di aspetti diversi e complessi si sia abbandonata l'idea di una trattazione a priori del fenomeno catalitico nel suo complesso. Ciò è oggi caratteristico della catalisi eterogenea. Essa presenta un lungo fronte il cui inizio ha basi solide nella conoscenza dei fenomeni chimici elementari, ma che si sviluppa riprendendo spesso il carattere di empirismo razionale proprio della chimica. La catalisi eterogenea è simile in ciò ad altri campi della chimica, al confine con discipline diverse, che stanno lentamente evolvendosi con la ricerca innanzi tutto della valutazione dei fattori realmente importanti, il che implica problemi sperimentali non semplici e in continua evoluzione. Si può anche dire che già appare la impossibilità, dato il carattere stesso del fenomeno, di dare una risposta quantitativa univoca al quesito su quali siano i fattori che determinano l'attività catalitica.
Bibliografia
Advances in catalysis, voll. I ss., New York-London 1948 ss.
Bond, G. C., Catalysis by metals, London 1962.
Boudart, M., Kinetics of chemical processes, Englewood Cliffs, N. J., 1968.
‟Catalysis reviews" (a cura di H. Heinemann), New York, anni 1967 ss.
Clark, A., The theory of adsorption and catalysis, London 1970.
Flood, E. A. (a cura di), The solid-gas interface, New York 1967.
Fripiat, J., Chaussidou, J., Jelli, A., Chimie-physique des phénomènes de surface, Paris 1971.
Krylov, O. V., Catalysis by non-metals, London 1970.
Roberts, M. V., Thomas, J. M. (a cura di), Surface and defect properties of solids, London 1972.
Schwab, G. M., Catalysis, New York 1937 (traduzione dall'originale tedesco con aggiunte di H. S. Taylor e R. Spence).
Somorjai, G. A., Principles of surface chemistry, Englewood Cliffs, N. J., 1972.
Thomas, J. M., Thomas, W. J., Introduction to the principle of heterogeneous catalysis, London 1967.
Thomson, S. J., Webb, G., Heterogeneous catalysis, Edinburgh 1968.
Wolkenstein, T. W., The electronic theory of catalysis on semiconductors, Oxford 1963.
Catalisi omogenea
di Jack Halpern
Introduzione
La definizione generalmente accettata di catalisi comprende quei fenomeni che si manifestano con l'aumento della velocità di una reazione chimica mediante una sostanza ('catalizzatore'), che non viene consumata durante la reazione. L'indagine empirica di tali fenomeni risale perlomeno al 1836, allorché J. J. Berzelius introdusse il termine ‛catalisi' per indicare l'accelerazione, mediante platino e altre sostanze, di reazioni quali la decomposizione del perossido d'idrogeno in ossigeno e acqua, e l'ossidazione dell'alcool ad acido acetico. Esempi di catalisi erano conosciuti in realtà molto prima di allora. Un esempio è il processo, importante dal punto di vista industriale, delle camere di piombo per la produzione di acido solforico, introdotto nel 1746, nel quale NO è aggiunto come catalizzatore per accelerare l'ossidazione di SO2 a SO3. I fenomeni catalitici hanno perciò costituito dei temi importanti nella scienza chimica, cosi come nella tecnologia chimica, per almeno 200 anni.
L'argomento di questa voce è limitato alla ‛catalisi omogenea', a reazioni, cioè, nelle quali il catalizzatore è disperso molecolarmente nella stessa fase (di solito gassosa o liquida) dei reagenti. Queste reazioni catalitiche presentano una grande diversità nei meccanismi particolareggiati. Si può tuttavia asserire, generalmente, che il catalizzatore partecipa come reagente e viene esso stesso trasformato chimicamente nel corso della reazione, per essere infine rigenerato, cosicché la sua concentrazione non diminuisce. La ossidazione di SO2 catalizzata da NO, quindi, riferendoci a quanto sopra, può essere interpretata mediante un meccanismo del tipo:
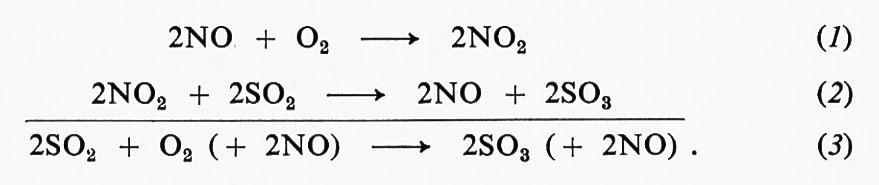
Le tecniche usuali per l'indagine dei meccanismi delle reazioni chimiche per es., misure cinetiche, isotopi traccianti, criteri stereochimici, effetti di sostituzione, rivelazione fisica e chimica dei prodotti intermedi) si possono anche applicare, e sono state in effetti ampiamente applicate, alle reazioni catalitiche. Alcune delle classi più familiari di reazioni catalitiche omogenee, oggi riconosciute come tali dopo essere state a lungo studiate e i cui meccanismi sono stati compresi almeno parzialmente, vengono illustrate dagli esempi seguenti.
Catalisi acido-base
La catalisi mediante acidi e basi è ampiamente diffusa, particolarmente per reazioni organiche, come l'idrolisi degli esteri e delle ammidi, l'idratazione delle olefine e delle aldeidi, l'alogenazione dei chetoni, la polimerizzazione e diverse trasposizioni molecolari. La catalisi mediante acidi deriva comunemente dall'aumento della reattività tramite aggiunta di un protone al reagente, come esemplificato dall'idrolisi degli esteri catalizzata da un acido secondo il meccanismo seguente:
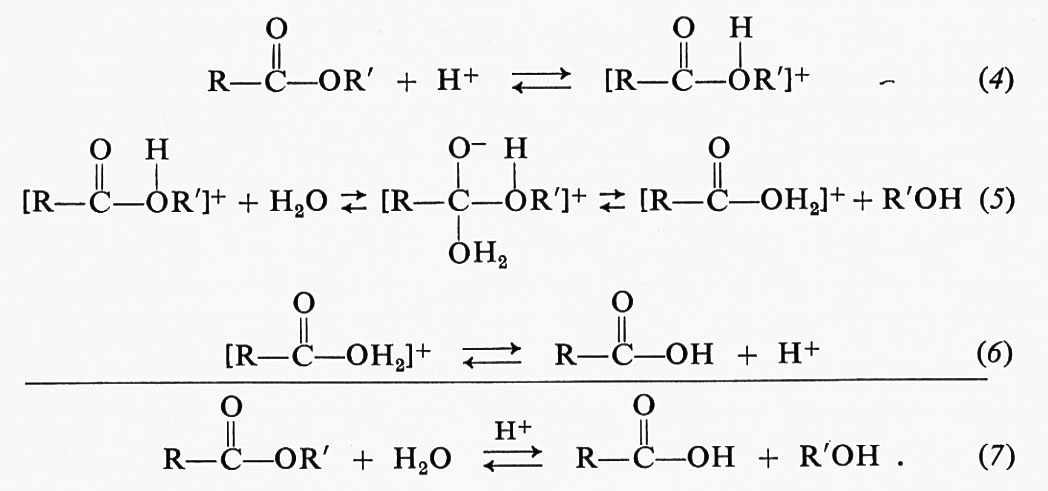
La catalisi mediante basi può derivare o dall'aumento di reattività per aggiunta di una base al reagente o, alternativamente, per sottrazione di un protone al reagente. Questi due tipi di meccanismo sono illustrati dagli esempi che seguono:

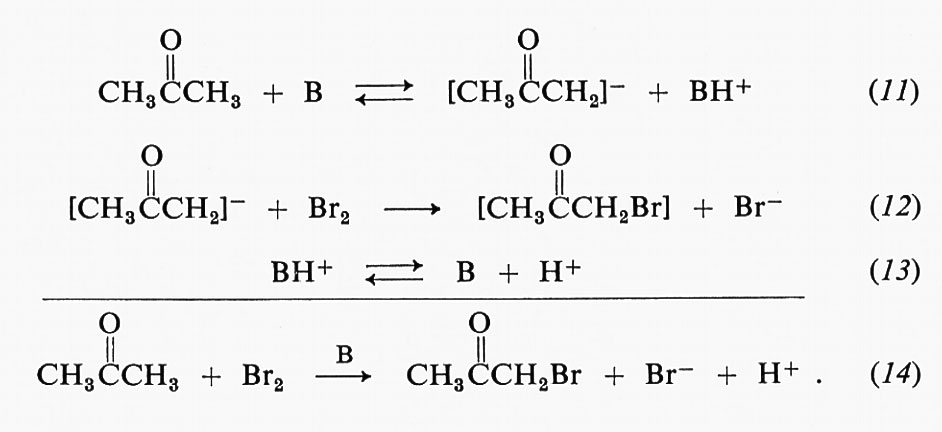
Tali reazioni catalizzate da un acido o da una base presentano un diverso comportamento particolareggiato (per es., catalisi generale acida o basica rispetto alla catalisi specifica da H3O+ o OH-), dipendente dalla circostanza che la prima fase determini la velocità o sia uno stato di preequilibrio, dalla natura della partecipazione del solvente, ecc. Di solito, più di uno di questi processi dettagliati contribuisce alla catalisi mediante acido o base complessivamente osservata per una data reazione (v. Bell, 1941 e 1959; v. Bender, 1971; v. Jencks, 1969).
Reazioni a catena di un atomo e di un radicale libero
Questa vasta classe di reazioni è esemplificata dalla combinazione di idrogeno e cloro catalizzata da NO, la cui cinetica corrisponde al seguente meccanismo:
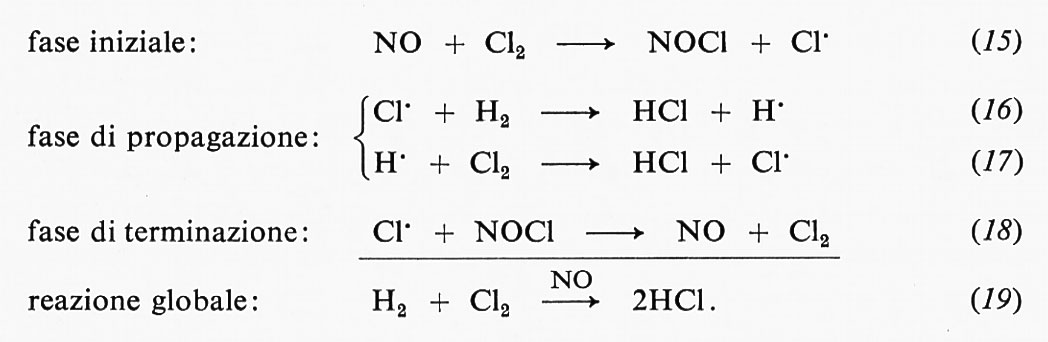
Poiché la catalisi della fase iniziale è ‛amplificata' dalla successiva fase di propagazione, nella quale si possono generare molte molecole del prodotto in ogni ciclo catalitico (prima cioè della fase di terminazione), queste reazioni a catena sono particolarmente sensibili alle influenze catalitiche (v. Dainton, 19662).
Dagli esempi precedenti dovrebbe essere chiaro che la catalisi omogenea non può essere interpretata solo attraverso un singolo schema meccanicistico unificato. Il meccanismo di ogni processo catalitico è infatti specificatamente legato sia alla particolare reazione chimica, sia al particolare catalizzatore. L'argomento della catalisi omogenea è, di conseguenza, assai ampio e vario e si può ritenere che comprenda l'intero campo dei meccanismi e dei tipi di reattività delle reazioni omogenee.
Nello stesso contesto, si dovrebbe anche notare che la designazione di una particolare specie come catalizzatrice di una reazione chimica è in un certo senso arbitraria e senza significato fondamentale. Riferendoci allo schema di reazione illustrato dalle equazioni 1-3, si nota che la specie catalitica NO entra nella reazione ed è trasformata chimicamente nella prima fase, nella quale essa ha essenzialmente funzione di reagente. Il fatto che il suo ruolo sia o non sia ‛anche' quello di catalizzatore dipende dalla circostanza che la stessa specie venga rigenerata in una reazione successiva (per es. l'equazione 2 del caso in esame).
La definizione generalmente accettata di catalisi, da noi usata, non preclude la possibilità che più di una molecola di una specie catalitica sia prodotta in ogni ciclo di una reazione catalitica. Un esempio di ciò è l'ossidazione di H2 con Cu2+, catalizzata da Cu+, secondo il meccanismo descritto dalle equazioni (20-22):
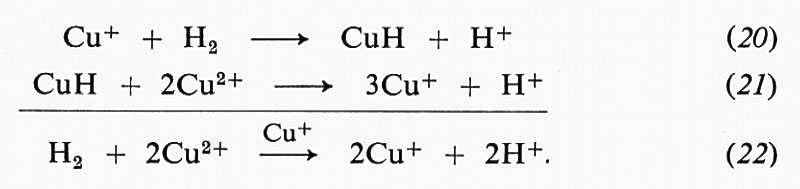
Una caratteristica di queste reazioni, che sono dette ‛autocatalitiche' o reazioni a ‛catena ramificata', è che la velocità può aumentare col procedere della reazione (tendendo in taluni casi al comportamento esplosivo), a causa dell'aumento della concentrazione del catalizzatore.
Gli esempi sopra discussi sono intesi a illustrare alcuni aspetti generali e taluni principî fondamentali della catalisi omogenea. Essi sono illustrativi di quei tipi di sistemi (specialmente quelli che comportano la catalisi acido-base e reazioni a catena di un radicale libero, quali i processi di ossidazione e di polimerizzazione) che hanno dominato il campo della catalisi omogenea sino a tempi relativamente recenti. Questi sistemi sono stati studiati e documentati ampiamente e si è già progredito notevolmente nella loro comprensione (v. Ashmore, 1963).
A partire dal 1950, la catalisi omogenea è stata sempre più dominata da nuovi temi, specialmente da quelli connessi con la catalisi di reazioni in soluzione tramite composti di coordinazione e composti organometallici, in modo particolare dei metalli di transizione (v. Halpern, 1969; v. Jones, 1968; v. composti organometallici). Stimolato dalla rinascita della chimica inorganica e dallo spettacolare sviluppo della chimica dei composti organometallici, questo campo di ricerca è stato contrassegnato da un'intensa attività e da numerose scoperte di processi catalitici di grande attualità e di considerevole importanza scientifica e tecnologica. Tra questi sono l'idrogenazione catalitica omogenea delle olefine e di altri composti non saturi; l'idroformilazione delle olefine in aldeidi (processo oxo); la carbonilazione degli alcoli e degli alogenuri organici ad acidi e acil-alogenuri; l'ossidazione delle olefine ad aldeidi, chetoni ed esteri vinilici, catalizzata da sali di palladio (processo Wacker) e una gran varietà di reazioni di polimerizzazione e di ciclooligomerizzazione di olefine, dieni e alchini (idrocarburi acetilenici). Di questi temi e di quelli a essi collegati tratteranno ampiamente le sezioni successive di questa voce.
Stabilizzazione dei prodotti intermedi di una reazione mediante coordinazione
I catalizzatori agiscono fornendo nuove vie di sviluppo alle reazioni chimiche e i loro contributi si riflettono in un aumento delle velocità di reazione. Talvolta tali andamenti catalitici sono profondamente collegati a quelli attraverso i quali la reazione procede in assenza di un catalizzatore; più in generale, come già indicato, quello catalitico è un processo caratteristico nel quale il catalizzatore entra come un reagente, subisce una reazione chimica, ma è alla fine rigenerato, cosicché la sua concentrazione rimane inalterata.
Per ogni procedimento catalitico, se ne può di solito costruire uno corrispondente non catalizzato, che può o no fornire un importante contributo alla reazione effettiva in assenza di catalizzatore. In questo senso, il ruolo del catalizzatore può essere interpretato come quello di stabilizzatore dei prodotti intermedi della corrispondente reazione non catalizzata, essendo questa stabilizzazione necessariamente maggiore di quella dei reagenti.
Un'illustrazione di questo tema è fornita dalla catalisi con rame(II), in soluzione acquosa, dell'ossidazione dell'idrogeno molecolare da parte di vari ossidanti, quali il cromo(VI), il ferro(III) e l'ossigeno. Questa catalisi avviene secondo il meccanismo descritto dalle equazioni(23-25) ove la reazione che determina la velocità è quella della scissione eterolitica dell'H2 con Cu2+:
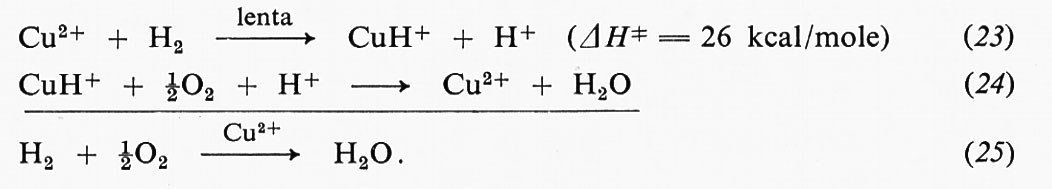
La corrispondente reazione non catalizzata, descritta dalle equazioni (26-28):
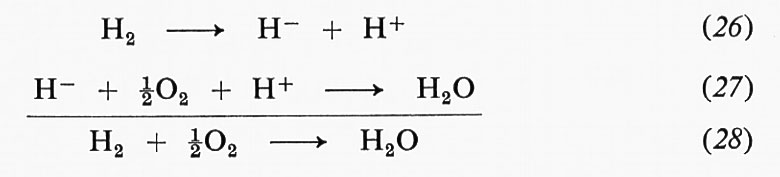
è molto meno favorevole, a causa dell'endotermicità della reazione (26). Si può calcolare che la corrispondente energia di attivazione (ΔΗ≠) superi le 35 kcal/mole, mentre per la reazione catalizzata da Cu2+, ΔΗ≠ è solamente di 26 kcal/mole. Questa diminuzione dell'energia di attivazione può essere attribuita alla stabilizzazione del prodotto intermedio H- dovuta al catalizzatore Cu2+. Altri esempi di catalisi possono essere interpretati in modo analogo (v. Halpern, 1959 e 1965).
Nel contesto di questa interpretazione della catalisi, la notevole versatilità dei metalli di transizione e dei loro composti nel catalizzare una così grande varietà di reazioni può essere compresa se si tiene conto della capacità di questi metalli di stabilizzare una varietà corrispondentemente grande di leganti per mezzo della coordinazione. Questi leganti comprendono specie a legame σ, come H, gruppi alchilici e arilici, come pure una varietà di specie a legame π, come le olefine, gli alchini, i polieni, i gruppi allilici, ecc. Nella tab. I sono riportati alcuni esempi di leganti, la stabilizzazione dei quali mediante coordinazione è importante dal punto di vista della catalisi, e di vari tipi di reazioni nelle quali compaiono come prodotti intermedi (v. Bird, 1967) dei complessi contenenti tali leganti.
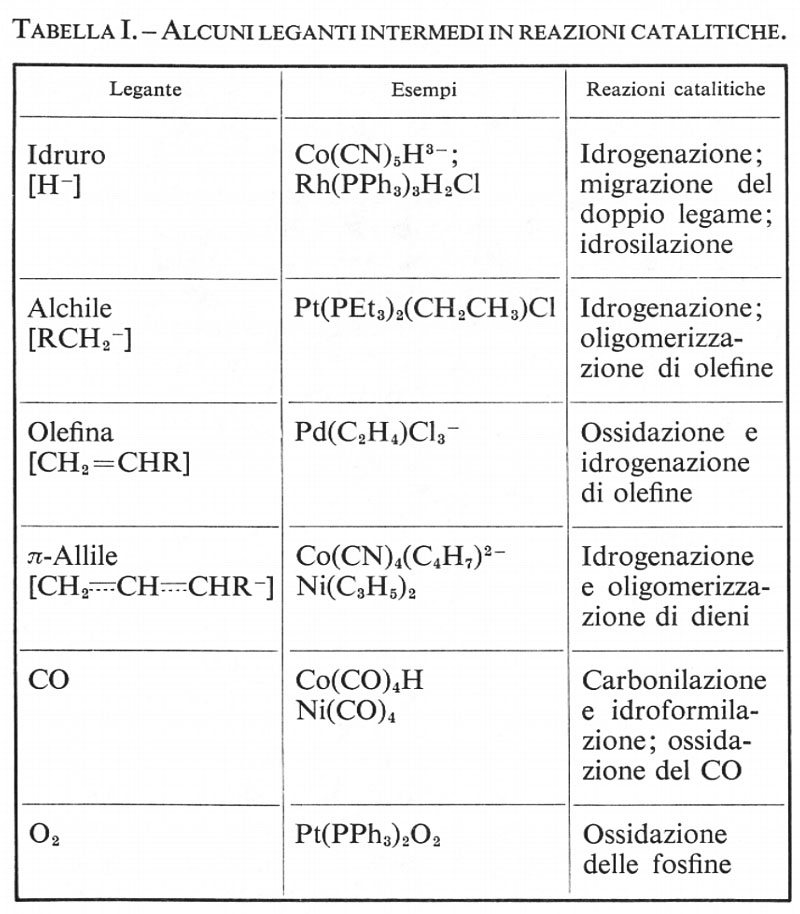
Occorre far notare che, mentre alcuni tipi di complessi elencati nella tab. I, quali i complessi di carbonili metallici e di olefine, sono conosciuti da tempo, molti altri (per es., quelli contenenti un idruro, un alchile, un allile π, diossigeno e diidrogeno coordinati con metalli di transizione) sono stati scoperti e caratterizzati soltanto di recente (dopo il 1950 circa). I complessi dell'ultimo tipo, specialmente i complessi organometallici dei metalli di transizione, sono particolarmente importanti come catalizzatori e come prodotti catalitici intermedi. Il numero di tali composti, che possiedono caratteristiche chimiche nuove e distintive, tra cui insolite proprietà catalitiche, è ancora in via di rapida espansione e allo stato attuale sembra che le possibilità in questo campo siano lungi dall'essere compiutamente realizzate.
Modelli di reattività collegati con la catalisi omogenea
Addizione ossidativa e reazioni connesse
Un'altra via per comprendere la notevole efficienza catalitica e la versatilità dei composti dei metalli di transizione è quella di riguardare la frequenza con cui si incontrano complessi relativamente stabili, quindi accessibili, ma altamente reattivi, i quali, in virtù delle loro proprietà elettroniche e strutturali, presentano alti gradi di reattività che assomigliano profondamente a quelli dei prodotti intermedi reattivi della chimica organica, come i radicali liberi, i carbanioni, i carbeni, gli ioni carbonio, ecc. Le reattività di queste ultime specie sono connesse con la grande stabilità della configurazione a guscio chiuso del carbonio saturo (4 elettroni di coordinazione, 8 di valenza). In conseguenza di questa stabilità, le specie con configurazione a guscio aperto, come i radicali liberi e i carbeni, tendono a presentare comportamenti reattivi caratteristici, comprendenti reazioni le quali ripristinano la configurazione a guscio chiuso del carbonio saturo (per es., sottrazione di un atomo da parte di radicali liberi, reazioni di inserimento di carbeni, spostamento nucleofilico da parte di carbanioni, ecc.).
Tra i complessi dei metalli di transizione a basso spin, quali, ad esempio, i carbonili, i cianuri, le fosfine e i composti organometallici, la configurazione a guscio chiuso con 6 elettroni di coordinazione d6 (18 di valenza) è di solito molto stabile. I complessi la cui configurazione differisce da questa presentano quindi elevate reattività e danno facilmente reazioni che tendono a ripristinare la configurazione stabile a guscio elettronico chiuso. Le caratteristiche di reattività risultanti, illustrate nella tab. II, presentano straordinarie analogie con quelle delle specie organiche corrispondenti, come ad esempio quelle esistenti tra complessi d7 5-coordinati e radicali liberi, tra complessi d8 4-coordinati e carbeni, tra complessi d8 5-coordinati e carbanioni, ecc. (v. Halpern, 1968).

Tra le reazioni più importanti comprese in questi modelli di reattività, vi sono quelle che comportano l'ossidazione di taluni complessi di metalli di transizione da parte di molecole sature, con incorporamento dei frammenti della scissione riduttiva nei gusci di coordinazione, ad esempio:
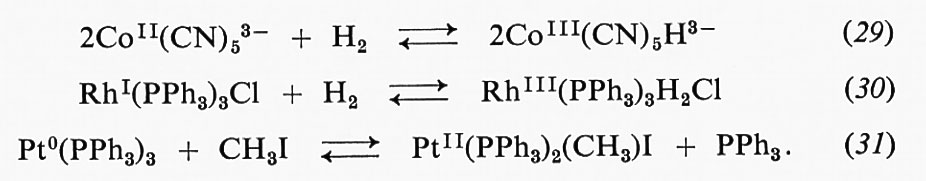
Tali reazioni, caratteristiche dei complessi d7 5-coordinati, d8 5-coordinati e d8 4-coordinati (e anche taluni d10), sono solitamente indicate come reazioni di ‛addizione ossidativa', ciò che riflette l'aumento del numero di ossidazione del metallo (v. Halpern, 1970). Queste reazioni di addizione ossidativa sono comuni e spesso forniscono facili vie alla dissociazione di molecole stabili come quella dell'idrogeno e di alogenuri organici, unita alla formazione, rispettivamente, di complessi idruro- e organometallici di metalli di transizione. Questo ha ovviamente importanti implicazioni per la catalisi, per esempio nelle reazioni di idrogenazione catalitica, che di solito procedono secondo meccanismi caratterizzati da uno stadio nel quale la dissociazione di H2 avviene per mezzo di ‛addizione ossidativa' al catalizzatore. Anche le reazioni inverse a quelle di addizione ossidativa (e cioè di eliminazione riduttiva), che forniscono un meccanismo efficace per la formazione di nuovi legami covalenti, per esempio H−H, C−H, C−C e C−X (X=Cl, Br, I, S ecc.), costituiscono stadi importanti in molti dei processi catalitici che saranno descritti in seguito.
Mentre il parallelismo che abbiamo menzionato tra i modelli di reattività di taluni complessi di metalli di transizione e i corrispondenti analoghi organici ha una portata assai vasta, non del tutto inaspettate si presentano anche delle importanti differenze. Una di queste è dovuta al fatto che le specie organiche reattive, come i radicali liberi, i carbeni e i carbanioni, sono tipicamente instabili, talvolta intrinsecamente, talaltra rispetto a reazioni con il mezzo circostante; esse si presentano quindi come prodotti intermedi di reazioni transitorie a breve vita. D'altro canto i composti di coordinazione ‛analoghi', come Co(CN)53-, Rh(PPh3)3Cl e Mn(CO)−5, sebbene altamente reattivi, sono di solito piuttosto stabili sia in soluzione sia come reagenti allo stato solido e possono quindi essere preparati direttamente per essere utilizzati come catalizzatori.
Reazioni di inserimento migratorio
Un'altra classe di reazioni di grande importanza in molti processi catalitici, in cui sono implicati complessi di un metallo di transizione, è quella delle reazioni definite di ‛inserimento' o di ‛inserimento migratorio'. Tali reazioni comportano l'inserimento di una molecola non satura X (per es., CO, olefina, acetilene, SO2, ecc.) in un legame ‛ metallo-legante M-L (ove L può essere H, alchile, arile, un altro atomo metallico, ecc.) cioè,
M−L + X → M−X−L (32)
oppure
Alcuni esempi specifici ditali reazioni sono:
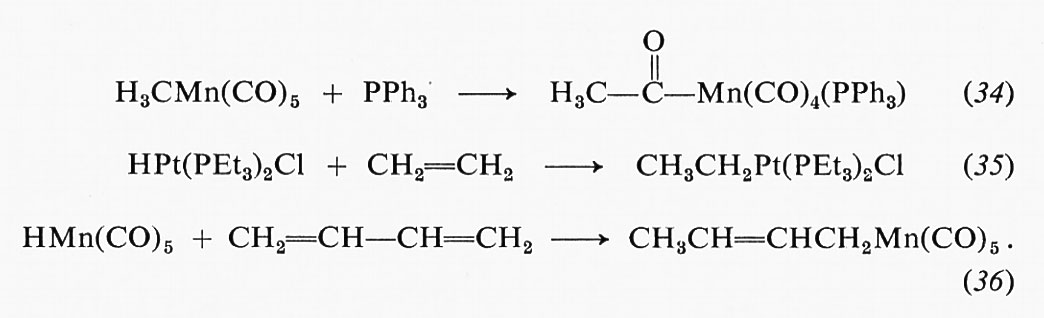
Tali reazioni costituiscono degli stadi importanti in molti processi catalitici, come nella carbonilazione e nelle reazioni di addizione alle olefine, quali l'idrogenazione, la polimerizzazione, ecc. Nonostante siano assai frequenti (v. Heck, 1965; v. Lappert e Prakai, 1967), l'identificazione di tali reazioni e la valutazione della loro importanza nella catalisi sono di data relativamente recente. I loro meccanismi non sono ancora stati compresi del tutto e il loro ulteriore studio costituisce un tema di fondamentale importanza nel campo della catalisi omogenea. In accordo con il meccanismo chiaramente implicato nell'equazione (34), sembra che molte, ma non la totalità, di tali reazioni di inserimento procedano per trasposizioni migratorie all'interno del guscio di coordinazione, essendo necessaria la coordinazione della molecola non satura X al metallo prima dell'inserimento. Prodotti simili a quelli delle reazioni di inserimento possono anche derivare dall'attacco di un reagente esterno nucleofilico, o elettrofilico, a un legante coordinato non saturo, come nell'esempio seguente che è profondamente legato a una delle fasi caratteristiche dell'ossidazione catalitica di CO da parte di taluni ioni metallici:
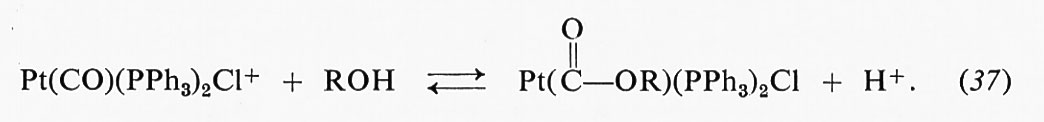
Descrizione e meccanismi di tipi specifici di reazioni catalitiche omogenee
Catalisi di reazioni nucleofiliche mediante cattura di elettroni dai reagenti tramite coordinazione
La coordinazione a uno ione metallico carico positivamente si presume che aumenti la densità della carica positiva sul substrato, rendendolo di conseguenza più sensibile ad un attacco nucleofilico. Il ruolo di uno ione metallico catalizzatore è, in tali casi, essenzialmente lo stesso di quello di un acido di Lewis o di un ‛superacido' e l'origine di questa attività catalitica è intimamente collegata con quella che sta alla base di molti dei comuni effetti catalitici degli acidi di Brönsted (per es., equazioni 4-7).
Nonostante possano portare cariche positive più e!evate, gli ioni metallici non sono di solito tanto efficaci nel polarizzare i substrati coordinati o nel trasferirvi cariche positive a causa del loro legame maggiormente ionico (specialmente quando siano coordinati a piccoli atomi elettronegativi, come ossigeno o azoto). Ciò si riflette, ad esempio, nelle acidità relative degli acidi protonati e coordinati, alcuni valori dei quali sono elencati nella tab. III.
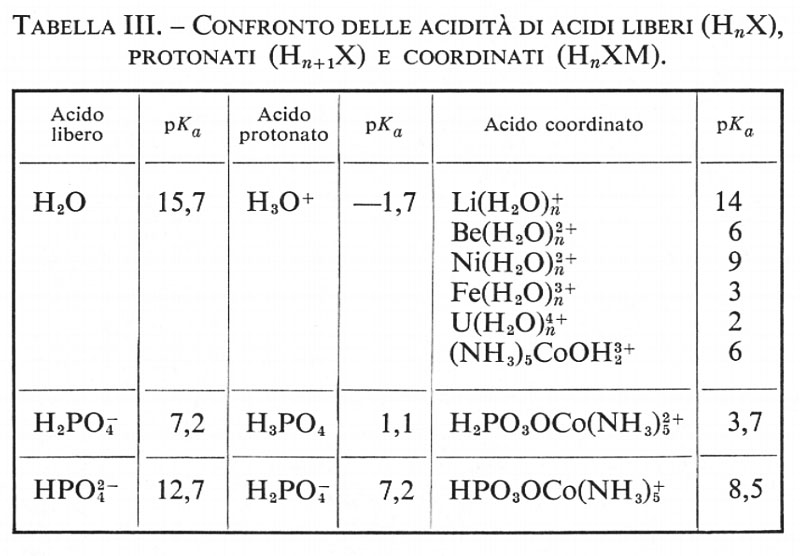
Vi sono, tuttavia, alcune circostanze nelle quali gli ioni metallici possono essere più efficaci dei protoni nel catalizzare le reazioni nucleofiliche (v. Halpern, 1969). Tra queste ricordiamo le seguenti.
1. Con specie (come basi soft o di ‛classe B'), quali gli ioni alogenuri, che presentano scarsa basicità verso i protoni, ma elevata basicità verso taluni ioni metallici. Lo spostamento degli ioni da entrambi gli alogenuri, organico e inorganico, promosso da ioni metallici (una classe di reazioni che generalmente non è suscettibile della catalisi acida di Brönsted), serve a illustrare questo effetto:
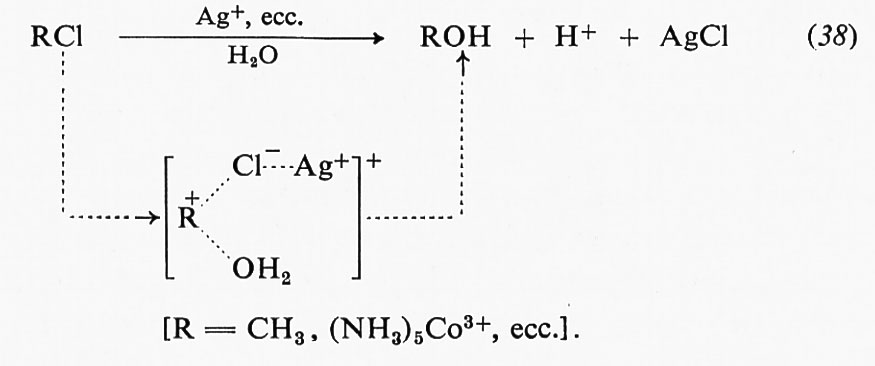
2. Con specie quali olefine, CO, alchini, ecc. la cui coordinazione a ioni metallici dipende dal legame π tramite la donazione di elettroni dπ da parte del metallo al legante. La catalisi mediante ioni metallici di reazioni del tipo dell'ossidazione di CO o di olefine e dell'idratazione degli alchini può essere interpretata sulla base di meccanismi che comportano l'attacco nucleofilico dell'acqua al substrato coordinato (equazione 37). Alcuni esempi relativi a essa saranno discussi in seguito (v. sotto, § d).
3. Quando la chelazione contribuisce al legame del substrato con lo ione metallico, ovvero alla stabilizzazione del prodotto con questo ione, come negli esempi seguenti:
a) catalisi mediante vari ioni metallici (Cu2+, Ni2+, Mg2+, ecc.) dell'idrolisi degli esteri di amminoacidi secondo il meccanismo
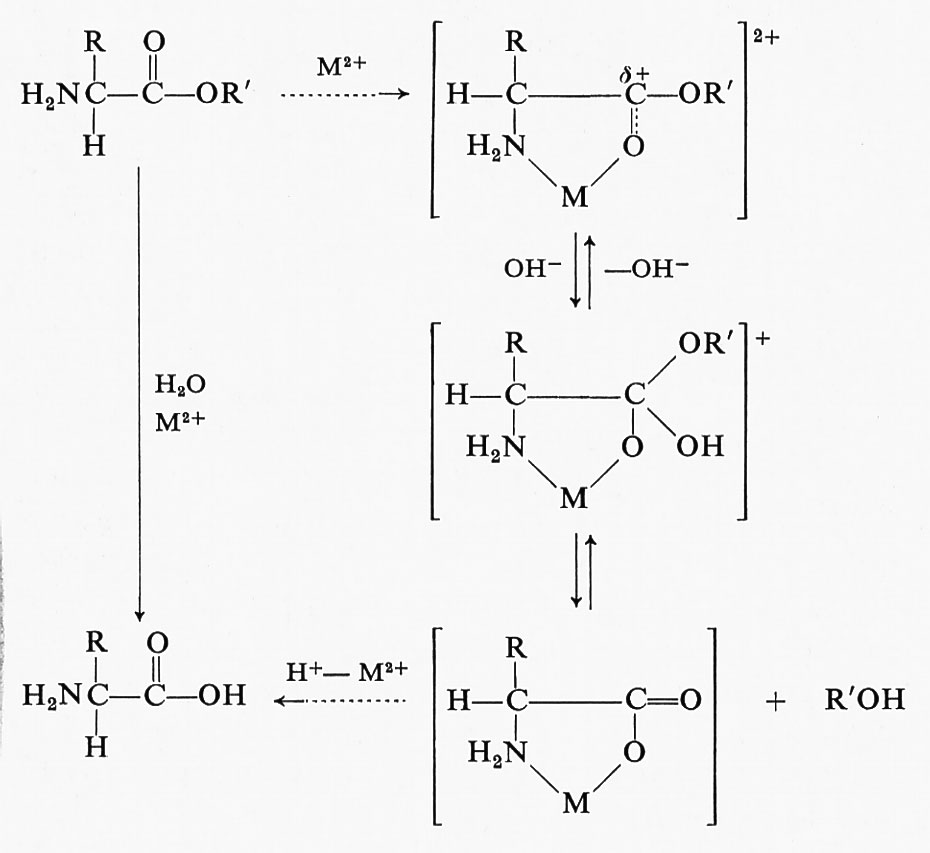
b) catalisi mediante Ni2+, Cu2+, Zn2+, ecc. dell'idratazione della cianofenantrolina. La coordinazione della cianofenantrolina all'Ni2+ aumenta la velocità dell'attacco dell'OH- di un fattore pari circa a 107, secondo il seguente meccanismo proposto:
c) catalisi mediante vari ioni metallici (Cu2+, Zn2+, Co2+, Ni2+, Mn2+, ecc.) della decarbossilazione degli acidi ossalacetici, secondo il meccanismo seguente:
Per reazioni semplici catalizzate da acidi, come quella illustrata dalle equazioni (4-7), si trova frequentemente che le attività catalitiche dei differenti acidi procedono parallelamente alla forza degli acidi, cioè alla loro capacità di ‛protonare' il substrato. Correlazioni quantitative di questo tipo si ritrovano nella legge della catalisi di Brönsted (equazione 42), dove kA è la costante della velocità catalitica della reazione, Ka è la costante di dissociazione acida del catalizzatore, e G e x sono costanti caratteristiche di ogni particolare reazione, ove i valori osservati di x variano tra O e 1:
log kA = log G + x log Ka. (42)
Questa relazione fu dimostrata per la prima volta da Brönsted e Pederson nel 1924 per la decomposizione della nitrammide, catalizzata da un acido, e la si è trovata in seguito valida per molte altre reazioni catalizzate da acidi. Relazioni analoghe tra kB e Kb descrivono la dipendenza di molte reazioni catalizzate da basi dalla forza basica del catalizzatore.
Si è anche constatato che relazioni analoghe tra l'influenza catalizzatrice di differenti ioni metallici e le costanti di equilibrio per la coordinazione dei reagenti agli ioni metallici possono applicarsi a talune reazioni catalizzate da ioni metallici, per esempio all'idrolisi del trifosfato di adenosina (ATP) e all'idroclorurazione dell'acetilene (v. Jones, 1965; v. Siling e Gelbshtein, 1969). Questo comportamento accentua l'importanza, per l'attività catalitica, della coordinazione del substrato agli ioni metallici. Occorre notare allo stesso tempo che, non di rado, i prodotti di queste reazioni nucleofiliche catalizzate da ioni metallici formano complessi più stabili con gli ioni metallici che con i reagenti. In questo caso, la forza addizionale che accompagna la reazione, derivante dall'aumento della stabilizzazione mediante coordinazione, contribuisce indubbiamente ad accrescere la reattività. A conferma di questo vi è l'osservazione che l'ordine delle attività catalitiche di vari ioni metallici bivalenti relativo alla decarbossilazione degli acidi ossalacetici (equazione 41), cioè: Cu2+ > Zn2+ > Ni2+ > Co2+ > Mn2+ > Ca2+, è simile all'ordine delle stabilità dei complessi di questi ioni metallici con l'anione ossalato
che rassomiglia allo ione prodotto
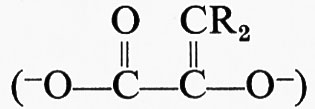
piuttosto che a quello reagente. Pur tuttavia, si suppone che un tale andamento si estenda solo a complessi prodotto-catalizzatore di stabilità intermedia poiché l'eccessiva stabilità vieterebbe la dissociazione del complesso prodotto-catalizzatore e la rigenerazione del catalizzatore stesso. Al limite, la reazione può essere promossa dal ‛catalizzatore', ma cessare di essere realmente catalitica. Questa situazione è esemplificata dallo spostamento di ioni di alogenuri promosso da Ag+, descritto dall'equazione (38), la quale non è realmente catalitica poiché l'inattivazione di Ag+, sotto forma dell'alogenuro altamente stabile, tende a essere irreversibile.
Catalisi di reazioni elettrofiliche mediante perdita di protoni da parte di leganti coordinati
Paradossalmente, in considerazione delle loro cariche positive, gli ioni metallici possono anche catalizzare l'attacco elettrofilico favorendo la perdita di un protone da parte di un legante coordinato, o stabilizzando una forma reattiva (una forma enolica, per esempio) di quest'ultimo (v. Martell, 1968).
Una semplice illustrazione di questo è la catalisi dell'ossidazione di H2 mediante Cu2+ e altri ioni metallici, secondo il meccanismo descritto dall'equazione (23). In questo caso, il ruolo catalitico di Cu2+ può essere interpretato in funzione di un aumento dell'acidità di H2 e della concentrazione efficace della base coniugata reagente, cioè lo ione H-, per stabilizzazione di quest'ultimo mediante coordinazione. Le implicazioni di tutto questo in altre reazioni alle quali partecipa H2, ivi compresa l'idrogenazione catalitica delle olefine, verranno considerate in seguito.
Altri esempi di reazioni di sostituzione elettrofilica catalizzate da ioni metallici attraverso meccanismi comportanti la promozione della perdita di un protone e la stabilizzazione delle forme enoliche dei reagenti, sono la bromurazione dei dichetoni (equazione 43) e quella dei chetoesteri:
L'ordine delle attività catalitiche nel caso della reazione (43) (Cu2+ > Ni2+ > Zn2+ > Mn2+ > Ca2+) corrisponde all'ordine delle stabilità dei complessi acetilacetonati degli stessi ioni metallici.
Catalisi delle reazioni di ossidoriduzione e di radicali liberi mediante trasferimento di elettroni
L'accessibilità di diversi stati di ossidazione per taluni complessi di metalli di transizione (accompagnati in alcuni casi da differenze nel numero di coordinazione) dà origine alla catalisi di una grande varietà di reazioni, specialmente del tipo di ossidoriduzione e di radicali liberi.
Tra gli esempi più semplici vi sono le reazioni nelle quali uno ione metallico o un complesso, che abbia due stati di ossidazione accessibili, catalizza il trasferimento di un elettrone agendo da trasportatore di elettroni. Due di questi esempi sono l'ossidazione di VIII con FeIII catalizzata da CuII (equazioni 44-46) e l'ossidazione di TlI con CeIV catalizzata da AgI (equazioni 47-50):

Alla base di questa catalisi (di cui un altro esempio è rappresentato dalle familiari ossidazioni con persolfato, catalizzate dallo ione argento) è il fatto che il trasferimento di un elettrone dal riducente al catalizzatore, seguito dal trasferimento di un elettrone dal catalizzatore all'ossidante, è più veloce del trasferimento diretto di un elettrone dal riducente all'ossidante. Un requisito ovviamente necessario, ma non sufficiente, per questa catalisi è l'accessibilità di due stati di ossidazione del catalizzatore, nessuno dei quali deve essere troppo stabile rispetto all'altro. Per ragioni che non sono state ancora ben comprese, gli ioni rame e argento sono particolarmente efficaci come catalizzatori di questo tipo.
L'ossidazione e la riduzione monoelettronica di molecole sature si risolvono frequentemente nella generazione di radicali liberi. Conseguenza di ciò è la catalisi di talune reazioni di radicali liberi mediante ioni o complessi di metalli di transizione (come Cu, Co e Mn) che presentino stati di ossidazione variabili. Esempi importanti di queste reazioni sono le reazioni di autossidazione degli idrocarburi (equazione 51), che procedono mediante i meccanismi a radicali liberi, illustrati in parte dalle equazioni (52-59).
Le reazioni responsabili dell'inizio della catalisi, cioè l'ossidazione e la riduzione di ROOH, comportano probabilmente la coordinazione di ROOH agli ioni metallici catalizzatori. L'inattivazione di questi catalizzatori da parte di agenti chelanti, quali l'acido etilendiamminotetraacetico, che sono sovente aggiunti per impedire l'ossidazione, durante la conservazione dei derivati del petrolio e degli oli e dei grassi commestibili, può derivare dall'interruzione di questa coordinazione (v. Lundberg, 1961).
Catalisi dell'ossidazione non radicalica delle molecole mediante coordinazione
Non tutti i casi di catalisi delle reazioni di ossidazione con ioni metallici e complessi implicano meccanismi a radicali liberi, come si è già dimostrato con l'ossidazione di H2 catalizzata da Cu2+, illustrata dalle equazioni (23-25).
L'ossidazione di composti organici può essere anche ottenuta con meccanismi non radicalici, che comportano la catalisi mediante ioni metallici e complessi. Un esempio particolarmente importante è costituito dal processo Wacker, sviluppato in Germania negli anni cinquanta e largamente impiegato nell'industria per l'ossidazione dell'etilene ad acetaldeide. Il processo si realizza in soluzione acquosa, utilizzando due catalizzatori, cioè cloruro di palladio(II) per effettuare l'ossidazione dell'etilene ad acetaldeide, e rame(II) per catalizzare la rigenerazione del palladio(II) mediante ossigeno. Le fasi principali del processo, i cui dettagli non sono stati ancora del tutto chiariti, si ritiene siano:
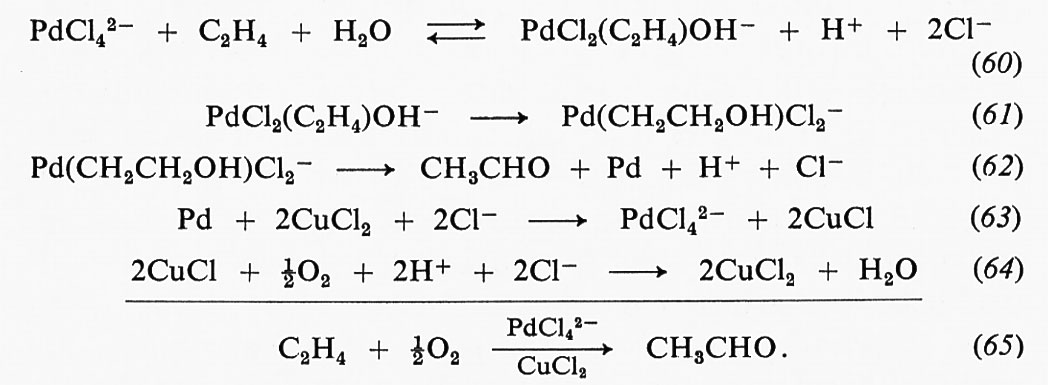
Varianti di questo processo possono essere utilizzate per ossidare altre olefine a chetoni in soluzione acquosa e per ossidare l'etilene a eteri ed esteri vinilici in altri solventi quali, rispettivamente, alcoli e acidi carbossilici (v. Stern, 1967).
Taluni complessi metallici, specialmente di rodio e cobalto (per es. Rh(CO)2C12 e Co(CN)2(PEt8)2(CO)-), catalizzano l'ossidazione del CO attraverso meccanismi del tipo seguente:
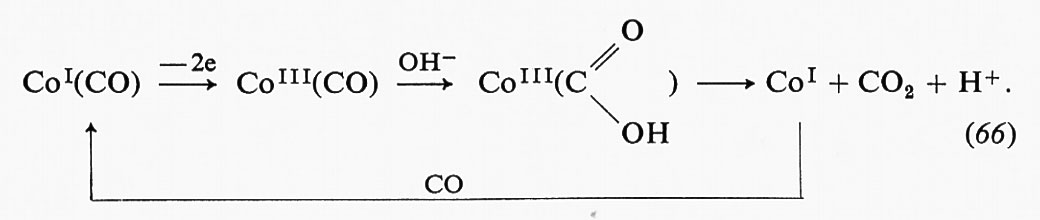
La catalisi delle reazioni di ossidazione può anche essere realizzata con l'attivazione dell'ossigeno molecolare mediante coordinazione. Si conoscono ora molti complessi dell'ossigeno molecolare, formati dalla coordinazione non dissociativa (talvolta reversibile) di O2 a metalli di transizione, quali Co(II), Ir(I), Ni(0) e Pt(0), lo studio dei quali è stimolato dall'interesse che essi suscitano quali possibili modelli per il trasportatore biologico di ossigeno, l'emoglobina. In generale, non sembra che O2 sia attivato in modo significativo per una reazione mediante tale coordinazione. Tuttavia, questa attivazione può verificarsi, e ciò si rispecchia nell'ossidazione delle fosfine catalizzata dal platino(0), secondo il meccanismo descritto dalle equazioni (67-69) (v. Halpern e Pickard, 1970):
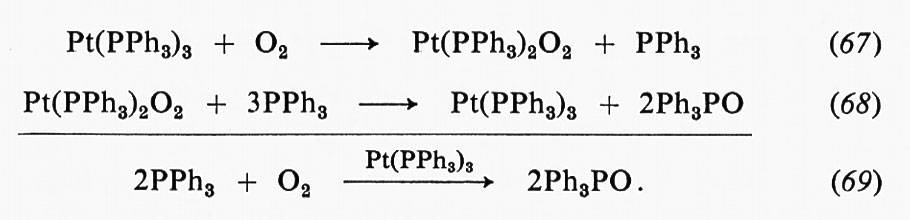
Carbonilazione e decarbonilazione
La carbonilazione e la decarbonilazione di composti organici costituiscono importanti classi di reazioni, la catalisi delle quali può essere realizzata mediante composti dei metalli di transizione.
La carbonilazione degli alogenuri di allile e di benzile ai corrispondenti derivati acilici (che possono a loro volta essere trasformati in esteri per reazione con alcoli) può quindi essere ottenuta senza difficoltà in presenza di quantità catalitiche di nicheltetracarbonile, attraverso meccanismi del tipo di quelli illustrati dall'equazione (70) (v. Chiusoli e Cassar, 1967):
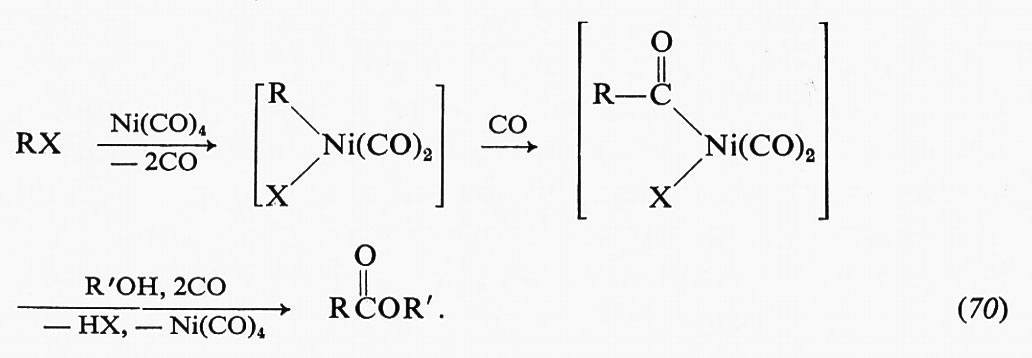
Tra i vari metodi conosciuti per la produzione di acido acetico, in alternativa all'ossidazione dell'etilene descritta dalle equazioni (60-65), vi è un processo di catalisi omogenea e cioè la carbonilazione diretta del metanolo in presenza di complessi di rodio e di promotori ioduranti. Introdotto dalla Monsanto Company nel 1970, qtiesto processo, importante dal punto di vista commerciale, opera in condizioni blande, probabilmente attraverso il seguente tipo di meccanismo (v. Roth e altri, 1971):
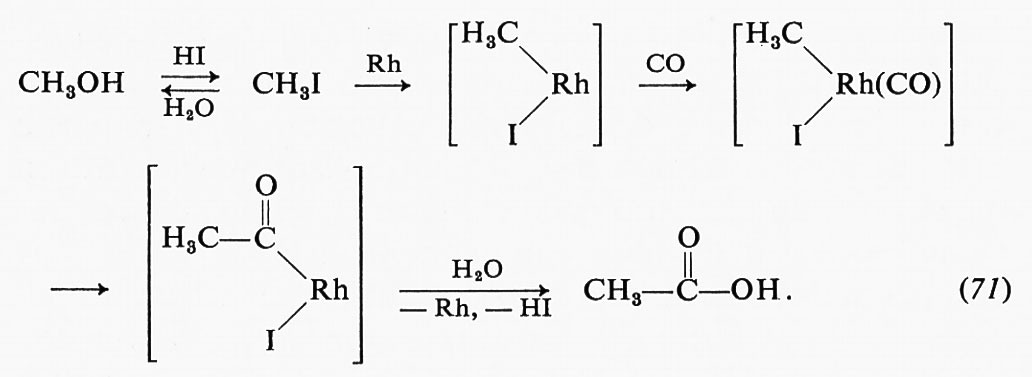
Sono state descritte numerose altre reazioni di carbonilazione, nelle quali complessi metallici d8 e d10 sono particolarmente efficaci come catalizzatori (v. Thompson e Whyman, 1971). Nonostante i meccanismi non siano stati completamente chiariti, si presume che, come nei due casi menzionati in precedenza (equazioni 70 e 71), l'addizione ossidativa (equazione 31) e le reazioni di inserimento migratorio di CO (equazione 34) siano degli stadi essenziali in molti di questi processi.
La decarbonilazione di composti organici, specialmente delle aldeidi e degli alogenuri acilici, può essere anche ottenuta per mezzo di catalizzatori derivati da diversi metalli di transizione, come Rh(PPh3)2 (CO) Cl (=Rh1). Tali reazioni possono essere interpretate secondo meccanismi del tipo seguente, che sono strettamente collegati con il processo inverso a quello di carbonilazione descritto in precedenza (v. Tsuji e Olino, 1969):
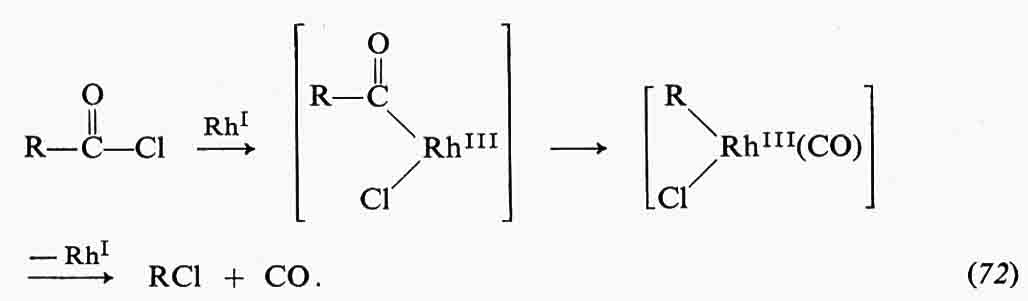
Coordinazione e attivazione catalitica dell'azoto molecolare.
L'attivazione con trasformazione catalitica dell'azoto molecolare in condizioni blande è stata considerata per lungo tempo un importante e stimolante obiettivo scientifico e tecnologico. Lo stimolo, così come le possibilità di realizzazione di questo obiettivo, sono stati rinforzati dalla scoperta che il sistema enzimatico azotoriduttasi effettua, biochimicamente, la riduzione di N2 a NH3 in condizioni blande. Mentre la costituzione e le caratteristiche meccanicistiche di questo sistema enzimatico non sono ancora state del tutto chiarite, è noto che l'enzima contiene ferro e molibdeno e si ritiene generalmente che uno o entrambi questi siti metallici siano implicati nel legame e nella riduzione delle molecole di N2.
Soltanto nel 1965 fu scoperto e caratterizzato da Allen e Senoff il primo complesso stabile contenente la molecola biatomica dell'azoto fungente da legante, precisamente Ru(NH3)5(N2)2+. La struttura e il legame di questo complesso e dei complessi analoghi sono simili a quelli dei ben noti complessi carbonilici di un metallo, contenenti il legante isoelettronico CO. Attualmente si conoscono analoghi complessi terminali di diazoto di molti altri metalli di transizione, quali Mo0, W0, ReI, ReI, FeII, RuII, OsII, CoI, RhI, Ir', Ni0, così come alcuni complessi binucleari contenenti leganti a ponte di diazoto, per esempio [(NH3)5Ru−N≡N−Ru(NH8)5]2+. Questi complessi sono stati preparati con diversi metodi, fra cui la sintesi diretta da azoto molecolare attraverso reazioni del tipo:
Sfortunatamente, non sembra che la coordinazione di N2 ai metalli di transizione, almeno nei complessi finali del tipo descritto in precedenza, abbia come conseguenza una significativa attivazione della molecola di azoto. Sebbene le ricerche indirizzate a raggiungere questo obiettivo proseguano ancora attivamente, i tentativi compiuti per ottenere la riduzione o altre reazioni chimiche dei leganti coordinati N2 in tali complessi si sono mostrati per la maggior parte infruttuosi (v. Chatt e Leigh, 1972).
La riduzione di N2 in condizioni relativamente blande, in presenza di complessi di metalli di transizione, è stata effettivamente ottenuta in sistemi leggermente differenti, aventi con quelli sopra descritti delle relazioni non ancora chiarite. Uno di questi sistemi, descritto per la prima volta da Volpin e Shur nel 1964 (e in seguito studiato, con alcune varianti, da numerosi altri ricercatori), comporta la riduzione di N2 a NH3 o ad ammine da parte di agenti fortemente riducenti quali C2H5MgBr, C6H5Li, Na+C10H8- o alluminio metallico, in presenza di quantità catalitiche di composti di titanio (o di alcuni altri metalli di transizione) come TiCl4 o (C5H5)2TiCl2. Si ritiene che i componenti attivi di questi sistemi siano delle forme ridotte di titanio, forse TiII, ma i meccanismi dettagliati delle reazioni connesse devono essere ancora chiariti. Un altro risultato importante è stato riferito recentemente da Shilov e dai suoi collaboratori, secondo i quali, in presenza di ioni magnesio, i sali di vanadio(II) riducono, in soluzione acquosa, l'N2 a idrazina (come anche ad ammoniaca). Questa riduzione sembra possa essere causata anche da altri agenti riducenti, ad esempio titanio(III) o cromo(II), in presenza di quantità catalitiche di molibdati (v. Chatt e Leigh, 1972; v. Volpin e Shur, 1970).
Idrogenazione e reazioni connesse implicanti idruri metallici
L'inerzia cinetica dell'idrogeno molecolare e la suscettibilità delle sue reazioni alla catalisi sono state per lungo tempo riconosciute come temi d'importanza tanto scientifica quanto tecnologica. Tuttavia, sino a tempi relativamente recenti, tutti gli esempi conosciuti di questa catalisi comportavano catalizzatori eterogenei, specialmente metalli di transizione solidi, come Ni, Pd e Pt, e taluni ossidi metallici, come ZnO e Cr2O3. Fu solo nel 1937 che M. Calvin descrisse il primo esempio definitivo di una reazione di idrogenazione catalizzata omogeneamente, e precisamente la catalisi mediante acetato di rame(I) in soluzione di chinolina nella riduzione di substrati, quali benzochinone e acetato di rame(II). È probabile che il meccanismo sia essenzialmente quello descritto dalle equazioni (20-22).
In seguito, specialmente dal 1950, la possibilità di attivare l'H2 in soluzione è stata dimostrata per gli ioni e i complessi di un gran numero di metalli di transizione, come quelli di CuII, CuI, AgI, HgII, HgI, CoI, CoII, PdII, RhI, RhIII, RuII, RuIII, IrI, Fe0, ecc. In ognuno di questi casi sembra che l'H2 venga scisso dal catalizzatore, con la successiva formazione dell'idruro complesso di un metallo di transizione, che in genere risulta essere sufficientemente reattivo per mantenere un ciclo catalitico. Sono stati riconosciuti tre distinti meccanismi mediante i quali si può realizzare la scissione dell'idrogeno, e precisamente: a) scissione eterolitica, cioè M + H2 → MH- + H+; b) scissione omolitica, cioè 2M + H2 → 2MH; c) ‛inserimento' di un metallo o addizione ossidativa, del tipo
Questi processi sono illustrati dagli esempi in precedenza citati, rispettivamente delle equazioni (23), (29) e (32) (v. Halpern, 1959 e 1969).
In alcuni casi favorevoli è anche possibile ottenere l'idrogenazione omogenea di substrati organici per mezzo di cicli catalitici comportanti la scissione dell'idrogeno mediante uno dei suddetti meccanismi, seguita dal trasferimento al substrato dei leganti degli idruri coordinati che ne risultano. Oggi si conoscono molti di questi processi catalitici; gli esempi seguenti sono stati scelti per illustrare come questa idrogenazione catalitica possa essere ottenuta con dei meccanismi che comportano tutti e tre i tipi di scissione dell'idrogeno sopra riportati (v. Halpern, 1969; v. Kwiatek, 1971; v. Coffey, 1970).
Cloruro di rutenio(II)
Uno dei primi esempi di idrogenazione catalitica omogenea di un composto olefinico, descritto da Halpern, Harrod e James nel 1961, riguarda la catalisi, mediante cloruro di rutenio(II), dell'idrogenazione di doppi legami ‛attivati' (per es., la riduzione dell'acido maleico ad acido succinico) in soluzione acquosa. Questa reazione è stata interpretata secondo il meccanismo seguente, nel quale la fase determinante la velocità è costituita dalla scissione eterolitica di H2 da parte di un complesso di rutenio già contenente il substrato olefinico come legante coordinato:
Pentacianocobaltato
Illustrativa della catalisi dell'idrogenazione delle olefine con meccanismi che comportano la scissione omolitica di H2 è l'idrogenazione di doppi legami coniugati catalizzata da Co(CN)52-. Il meccanismo d'idrogenazione del butadiene, come chiarito da Kwiatek, è illustrato dalle equazioni (76-79). La dipendenza da CN− dell'equilibrio tra gli intermedi (7- e ‛allilici dà ragione del fatto che l'idrogenazione ad alte concentrazioni di CN− produce prevalentemente 1-butene e a basse concentrazioni di CN− trans-2-butene (con un rapporto maggiore, in ciascun caso, dell'80%). Questo tipo di specificità è sovente una caratteristica distintiva dell'idrogenazione omogenea rispetto a quella eterogenea.
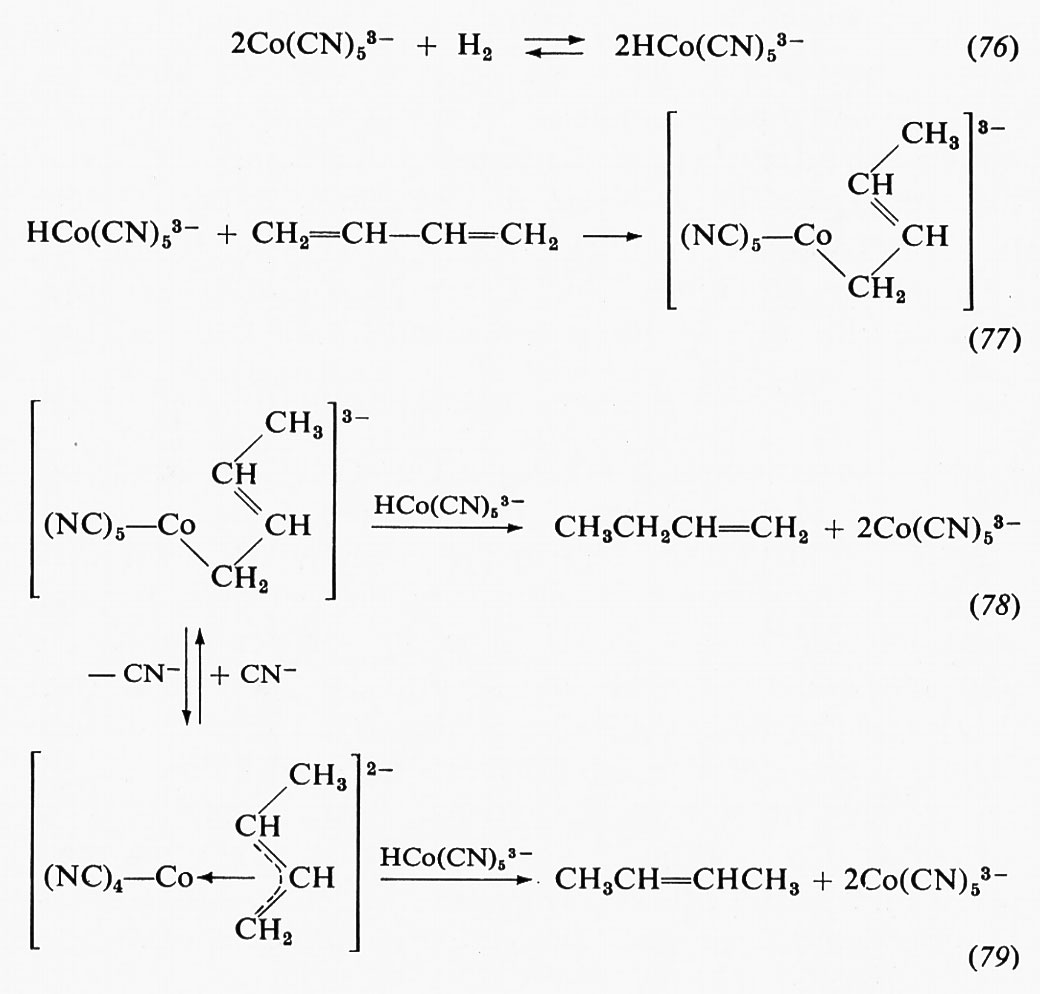
Tris(trifenilfosfina)clororodio(I)
Questo complesso, Rh(PPh3)3Cl, abbreviato in RhL3Cl, scoperto da Wilkinson nel 1965, è uno dei catalizzatori omogenei più efficaci e versatili per l'idrogenazione di olefine e di alchini. Il comportamento di questo sistema catalizzatore è coerente col seguente meccanismo, che probabilmente è anche caratteristico di altri catalizzatori complessi d8, compresi sia quelli di Ir(I) sia quelli di Rh(I).
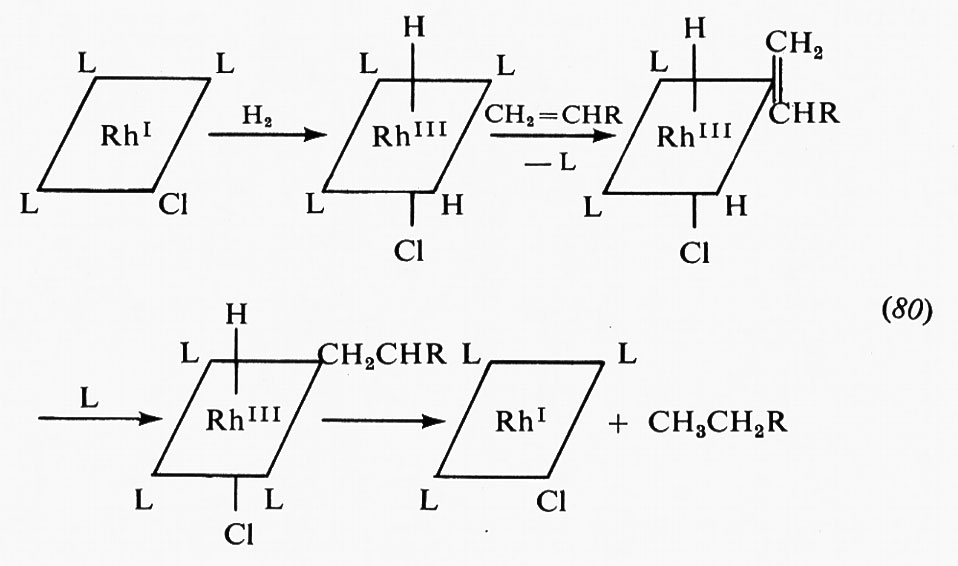
In questo campo, un importante e interessante sviluppo di recente data riguarda la sintesi asimmetrica di composti otticamente attivi mediante l'idrogenazione catalitica omogenea con catalizzatori, specialmente complessi di Rh(I), contenenti come leganti fosfine chirali, cioè *PR1R2R3. Tali sviluppi hanno condotto alla produzione di amminoacidi otticamente attivi, con rese ottiche che raggiungono il 9o%, mediante l'idrogenazione catalitica di acidi amminoacrilici (v. Knowles e altri, 1972). Ciò rappresenta un considerevole progresso nella metodologia sintetica, per la quale sono già state riportate applicazioni pratiche (per es., la sintesi di L-dopa e di L-fenilalanina).
Caratteristiche comuni dei diversi meccanismi dell'idrogenazione catalitica omogenea sopra descritta sono: a) la presenza come intermedi di idruri metallici complessi; b) l'inserimento di olefine nei legami metallo-idrogeno di tali complessi. Queste caratteristiche contribuiscono anche ai meccanismi di svariate altre reazioni di olefine catalizzate omogeneamente, compresa la migrazione del doppio legame e numerose reazioni di addizione, come l'idroformilazione e l'idrosilazione. Gli esempi seguenti sono illustrativi delle possibilità racchiuse in questo vasto campo.
Isomerizzazione delle olefine
La migrazione del doppio legame nelle olefine è catalizzata dai complessi di molti metalli di transizione, tra i quali Co, Rh, Ir, Fe, Ni, Pd, Pt e Ru. Si ritiene che queste reazioni procedano secondo due tipi generali di meccanismi, illustrati rispettivamente dalle equazioni (81) e (82). Il primo di questi meccanismi, che è il più comune, può richiedere un co-catalizzatore, come H2, un acido o un solvente donatore di idrogeno, per trasformare il metallo catalizzatore nella forma attiva di idruro (v. Cramer, 1968):
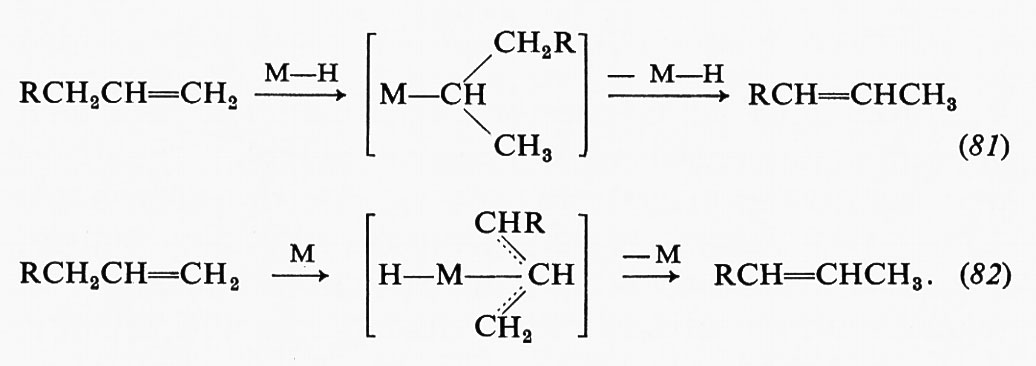
Idroformilazione
L'addizione di H2 e CO alle olefine per formare aldeidi, reazione di interesse scientifico e importanza tecnologica considerevole, è catalizzata da complessi di vari metalli, specialmente Co, Rh e Ru. Il meccanismo generalmente accettato della reazione catalizzata da HCo(CO)4 comprende la successione delle reazioni di inserzione, addizione ossidativa ed eliminazione riduttiva descritte qui sotto (v. Orchin e Rupilius, 1972; v. Paulik, 1972):
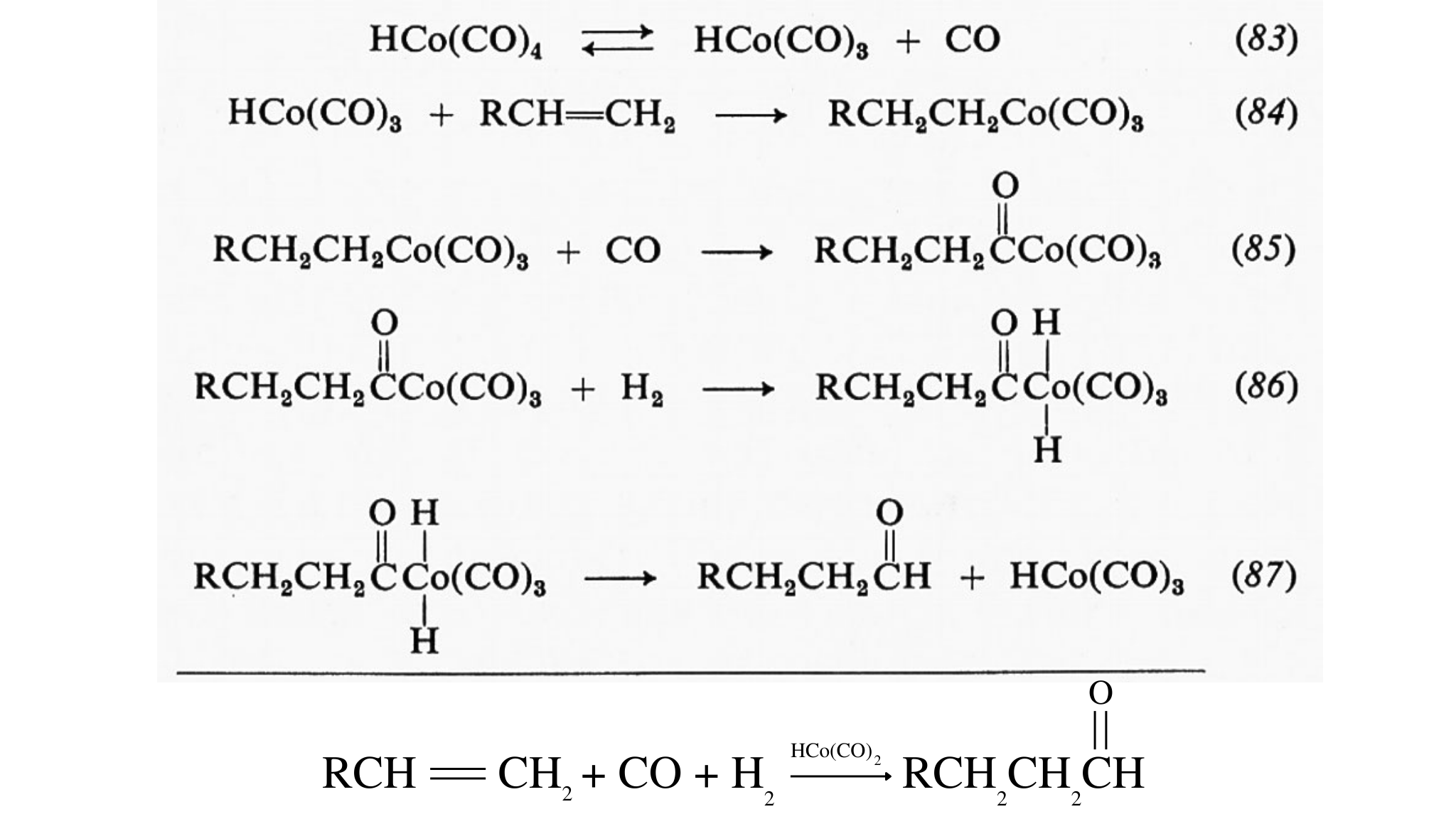
Idrosilazione
La capacità dei metalli di transizione di catalizzare la reazione di addizione di idruri alle olefine è esemplificata dall'idrosilazione, reazione che è catalizzata da complessi del platino, del rodio o del cobalto. Anche in questo caso, come descritto dall'equazione (89), l'addizione ossidativa e l'inserimento dell'olefina sull'idruro metallico sono fasi basilari del meccanismo (v. Chalk, 1970). Altre addizioni di idruri alle olefine catalizzate da metalli di transizione, per esempio le addizioni di HCN catalizzate da Ni(0) e Pd(0), procedono probabilmente secondo meccanismi analoghi:
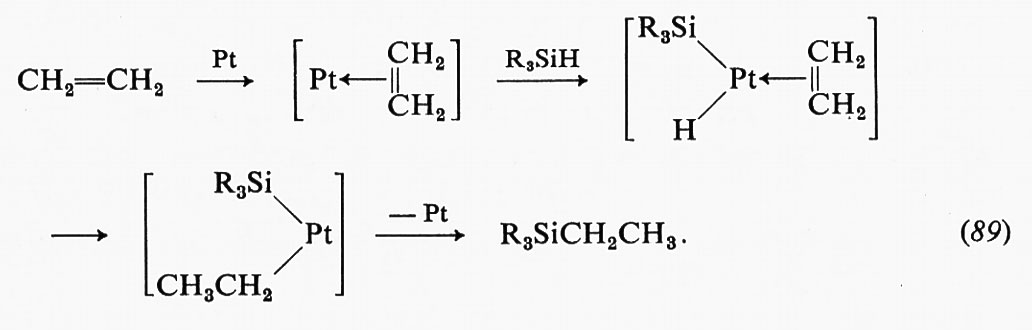
Dimerizzazione, polimerizzazione e ciclooligomerizzazione
La catalisi mediante composti di coordinazione delle reazioni di dimerizzazione, polimerizzazione e ciclooligomerizzazione di olefine, dieni e alchini riguarda un campo ampio e vario della chimica. Il grande interesse pratico e scientifico di tali reazioni si riflette nell'intensa attività e nella continua crescita che hanno caratterizzato, e che continuano a caratterizzare, l'argomento. I pochi esempi relativamente semplici che è possibile citare qui servono a illustrare solo un contesto assai limitato di questo campo vasto e affascinante, ma non riescono a dare l'idea della dimensione complessiva dei suoi fini e delle sue possibilità.
Dimerizzazione delle olefine
La semplice dimerizzazione delle olefine si ottiene con molti catalizzatori, tra cui i complessi di Ti, Pd, Co, Ni e Rh (v. Lefebvre e Chauvin, 1970). Descriveremo qui solo uno di questi sistemi, precisamente il cloruro di rodio che, in soluzioni alcoliche contenenti HCl, catalizza la dimerizzazione dell'etilene, in condizioni moderate, inizialmente a 1-butene, seguita quindi dalla isomerizzazione parziale a 2-buteni. Si ritiene che la reazione proceda secondo il meccanismo seguente, ove si è indicato con L il Cl- un solvente (v. Cramer, 1968):
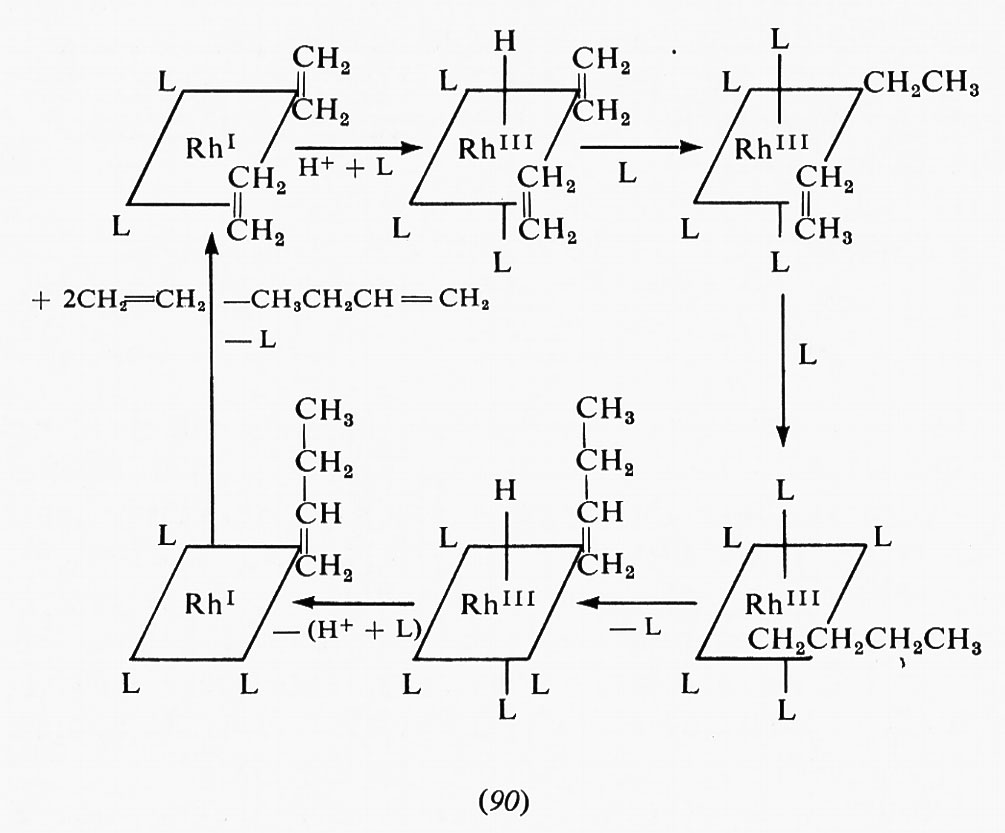
Il meccanismo precedente può anche compiere la codimerizzazione dell'etilene e del butene, con conseguente formazione di oligomeri e di polimeri superiori. Tuttavia, poiché la coordinazione dell'etilene è fortemente favorita rispetto a quella delle olefine superiori, si può ottenere una elevata selettività per la dimerizzazione dell'etilene a 1-butene, relativamente a questa codimerizzazione e alla isomerizzazione del butene che l'accompagna. Varianti dello stesso sistema catalizzatore sono anche efficaci nell'addizione dell'etilene all'1,3-butadiene per la formazione dell'1,4-esadiene (v. Cramer, 1968).
Oligomerizzazione degli 1,3-dieni
Molti catalizzatori contenenti metalli di transizione, specialmente nichel, palladio, rodio, cobalto e ferro, catalizzano le reazioni di oligomerizzazione lineare e ciclica dell'1,3-butadiene e di altri 1,3-dieni, così come la cooligomerizzazione di questi dieni con monoalcheni. È probabile che la maggior parte di queste reazioni proceda attraverso prodotti intermedi π-allilici (v. Keim, 1971).
La formazione di oligomeri lineari del butadiene comporta il trasferimento di un atomo di idrogeno. La varietà di queste reazioni è illustrata dalla formazione del dimero ramificato 3-metileptatriene in presenza di taluni catalizzatori di complessi del cobalto, del dimero lineare n-ottatriene-2,4,6 in presenza di catalizzatori contenenti rodio, di una mescolanza del dimero lineare n-ottatriene-1,3,7 e del trimero n-dodecatetraene-1,3,6,10 in presenza dell'acetato di ‛allilpalladio, ecc. Wilke e i suoi collaboratori hanno mostrato che i complessi del nichel(0) sono catalizzatori particolarmente efficaci e versatili della ciclooligomerizzazione dell'1,3-butadiene, in reazioni caratterizzate da eleva- ti gradi di selettività e stereospecificità, a una varietà di prodotti, quali, ad esempio:
La natura dei prodotti dipende dalle condizioni ed in particolar modo dai leganti (fosfine, ad esempio) coordinati al nichel. Il meccanismo proposto per una di queste reazioni è illustrato qui di seguito (v. Heimbach e altri, 1970):
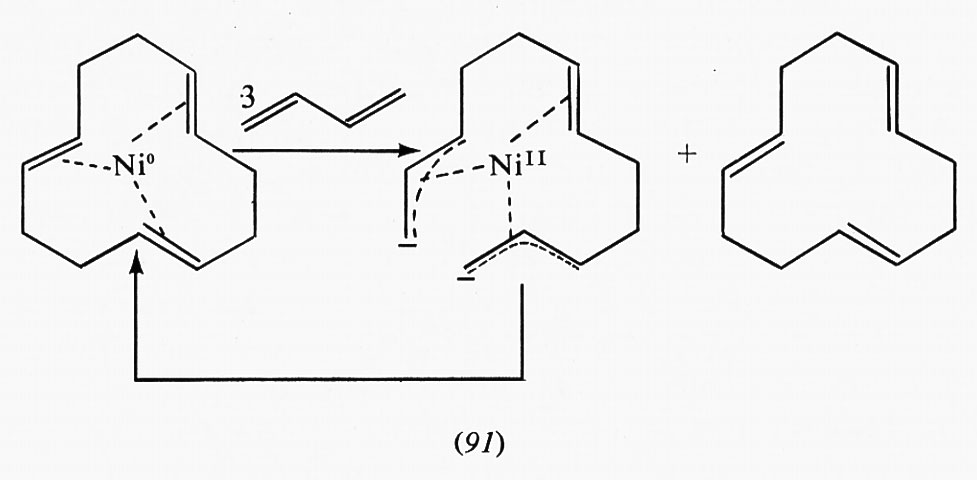
Oligomerizzazione degli alchini
I complessi del nichel sono anche catalizzatori molto efficaci e versatili della oligomerizzazione lineare e ciclica degli alchini. Molti degli eventi che hanno contribuito allo sviluppo spettacolare della scienza moderna della catalisi omogenea e della chimica dei composti organometallici furono ispirati in modo diretto o indiretto dall'importante scoperta, compiuta da W. Reppe nel 1940, che taluni composti del nichel(II), come il cianuro o l'acetilacetonato, catalizzano, con alte rese, la ciclotetramerizzazione dell'acetilene a cicloottatetraene. Fu scoperto in seguito che taluni composti del nichel(0), ad esempio il bis(acilonitrile)-nichel(0), catalizzano anch'essi questa reazione. Nonostante il grande interesse e le varie ricerche che su tali reazioni si sono concentrati, i meccanismi non sono ancora stati chiariti.
Un'altra importante classe di reazioni, catalizzate in modo particolarmente efficiente dai complessi nichelcarbonilici e nichelfosfinici, comprende la ciclotrimerizzazione degli alchini a composti aromatici. Si ritiene che queste reazioni procedano secondo meccanismi del tipo seguente (v. Schrauzer, 1964; v. Hoogzand e Hübel, 1968).
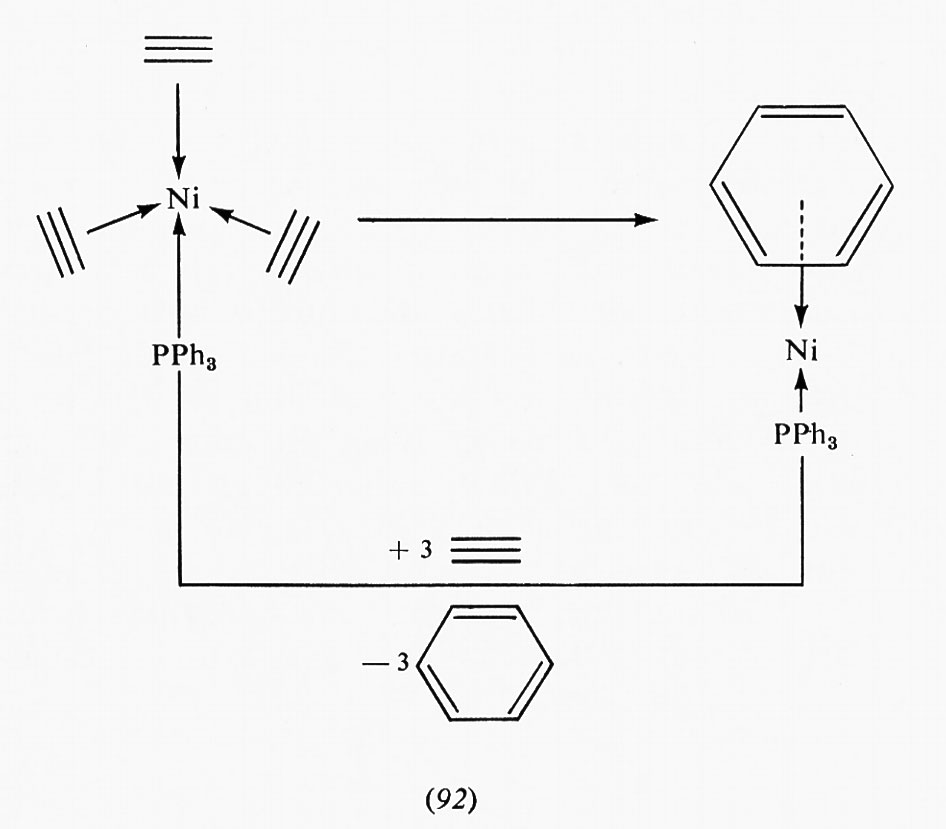
Catalisi di reazioni ‛limitate dalla simmetria' mediante ioni e complessi metallici
Questa sezione tratta della catalisi, mediante composti di un metallo di transizione, di talune reazioni, specialmente di quelle che comportano la formazione o la rottura di legami carbonio-carbonio, i cui andamenti concertati sono termicamente vietati, secondo le regole di Woodward-Hoffmann sulla conservazione della simmetria degli orbitali (v. Woodward e Hoffmann, 1969). Queste reazioni comprendono le 1,2-cicloaddizioni di olefine per la formazione di ciclobutani, le corrispondenti reazioni di cicloreversione e le trasformazioni ‛disrotatorie' del ciclobutene a butadiene.
Si è trovato che talune di queste reazioni, quali ad esempio la isomerizzazione di valenza dei composti del ciclobutano altamente tensionati nei dieni corrispondenti per es.: quadriciclano in norbornadiene, esametilprismano in esametildewarbenzene, cubano in sintricicloottadiene), sono catalizzate da complessi di metalli di transizione, in particolare del rodio(I). A questi effetti catalitici sono state date due interpretazioni alternative, che tuttavia non si escludono necessariamente a vicenda; precisamente: a) l'eliminazione dei vincoli di simmetria negli andamenti concertati, altrimenti vietati, mediante l'interazione del sistema reagente con gli orbitali d del catalizzatore, o, b) la possibilità di andamenti non concertati, altrimenti endotermici in modo proibitivo, mediante la rottura iniziale (o la formazione) di un solo legame carbonio-carbonio attraversò uno stadio di addizione ossidativa. Ragioni a sostegno dei meccanismi del secondo tipo sono state addotte per i riassestamenti del cubano (equazione 93) e del quadriciclano catalizzati dal rodio(I), e sembra probabile che tali meccanismi si possano applicare ad altre reazioni catalizzate dal rodio(I).
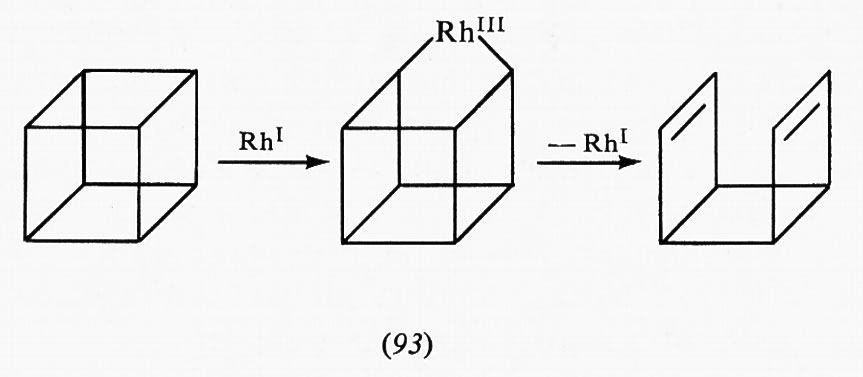
I sali di argento(I) presentano un andamento catalitico del riassestamento degli idrocarburi tensionati leggermente differente, che può anche essere interpretato secondo meccanismi non concertati, che comportano comunque la scissione eterolitica iniziale di un legame carbonio-carbonio e i successivi riassestamenti degli intermedi ioni carbonio. L'equazione (94) illustra quanto sopra relativamente al riassestamento del cubano catalizzato dall'argento(I), che segue un andamento completamente differente (altresì termicamente vietato dalla simmetria, in assenza del catalizzatore) dalla reazione corrispondente catalizzata dal rodio (I) (v. Halpern, 1972).
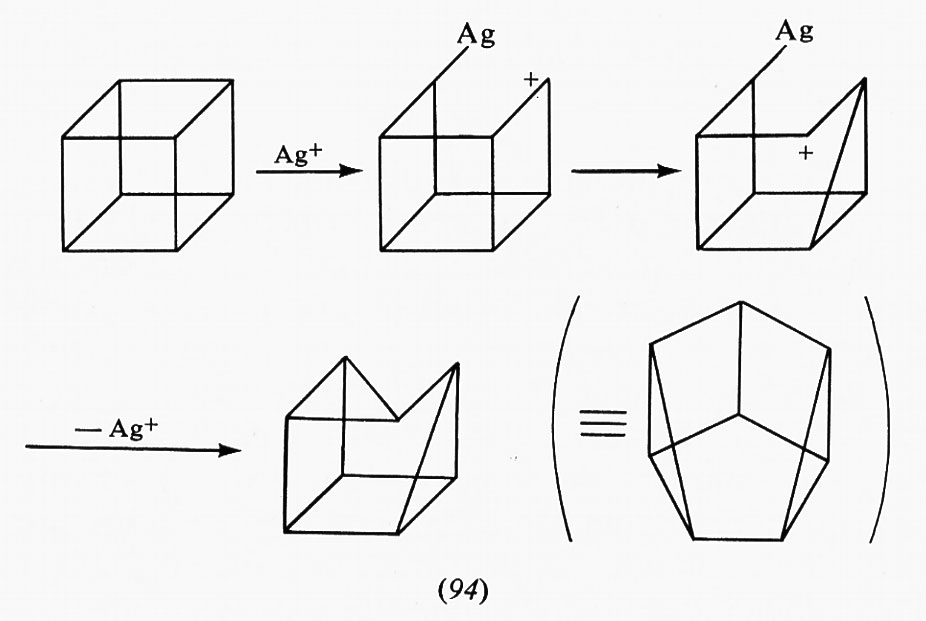
Le differenze nei modi di reagire del RhI e dell'AgI coi legami carbonio-carbonio tensionati sono analoghe ai loro differenti modi di reagire con H2. Così, i complessi di RhI reagiscono tipicamente con H2 mediante addizione ossidativa (equazione 30), mentre Ag+ scinde H2 eteroliticamente tramite una reazione analoga a quella del Cu+ (equazione 20).
Disproporzione delle olefine
Nel contesto dei processi ‛vietati dalla simmetria' dovrebbe essere inclusa una classe di reazioni assai nuove e importanti scoperte da Banks e Bailey nel 1964, precisamente le reazioni di ‛disproporzione delle olefine'. Tali reazioni (per le quali si usano anche le definizioni di ‛dismutazione' e ‛metatesi' delle olefine) comportano la ridistribuzione delle porzioni alchilideniche di due olefine, cioè:
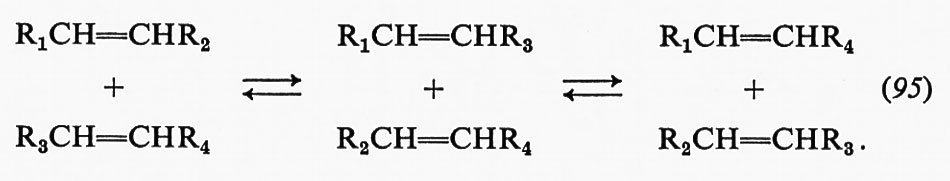
Questa reazione, per la quale non vi è praticamente alcun caso analogo, è indotta da numerosi catalizzatori eterogenei e omogenei, implicanti in modo particolare i componenti del gruppo VI dei metalli di transizione. I catalizzatori eterogenei tipici comprendono i carbonili di molibdeno o di tungsteno od ossidi di questi su supporti di allumina. Tra i sistemi di catalizzatori omogenei vi sono quelli derivanti dalle combinazioni WCl6:C2H5OH:C2H5AlCl2 oppure Mo(NO)2(PPh3)2Cl2 :C2H5AlCl2 (v. Bailey, 1969).
Caratteristica di queste reazioni di disproporzione è quella di fornire delle mescolanze in equilibrio dei costituenti olefinici disponibili. Si è anche mostrato che catalizzatori analoghi sono efficaci nella disproporzione dei dieni e degli alchini. Il processo è stato applicato su scala commerciale per convertire il propilene in etilene e in buteni. Una nuova applicazione riguarda la sintesi di composti carbociclici con più di 12 atomi di carbonio attraverso successive fusioni di olefine cicliche più piccole, del tipo:
Nonostante che i meccanismi di queste reazioni di disproporzione non siano ancora stati chiariti, si ritiene in genere che essi procedano attraverso intermedi coordinati ‛a quattro centri', del tipo:
Reazioni a incastro
La catalisi o la promozione di una reazione può derivare dalla coordinazione simultanea di due o più reagenti a uno ione metallico, in modo che sia facilitato il loro avvicinamento reciproco e il loro orientamento sia reso favorevole per la reazione. Le ciclotrimerizzazioni catalitiche del butadiene e dell'acetilene, illustrate rispettivamente dalle equazioni (91) e (92), quasi di certo presentano caratteristiche proprie di questi ‛effetti a incastro'. Un altro esempio di effetto a incastro è fornito dalla condensazione della o-amminobenzaldeide. In assenza di ioni metallici, i normali prodotti della reazione sono polimeri lineari, mentre in presenza di taluni ioni metallici come Cu2+, Ni2+ e Co2+, tende a effettuarsi la ciclocondensazione, secondo l'equazione (97) (v. Busch, 1964):
I prodotti di queste reazioni ‛a incastro' rimangono di frequente fortemente coordinati al metallo, cosicché le reazioni vengono facilitate, ma non possono essere considerate veramente catalitiche. L'informazione cinetica e meccanicistica relativa alla maggior parte di queste reazioni non è sufficiente per determinare se i fattori cinetici o termodinamici, o entrambi, siano responsabili dell'accrescimento della reattività osservato. Ciò nonostante, l'effetto di incastro è sovente di grande utilità pratica nella sintesi di laboratorio dei leganti macrociclici come le ftalocianine, le porfirine e le corinne. Non è improbabile che ioni metallici abbiano ruoli analoghi nella biosintesi ditali composti.
Miscellanea di fenomeni catalitici e altri a questi connessi
L'ampiezza di trattazione data al soggetto della catalisi omogenea dalla precedente descrizione di un numero selezionato di sistemi catalitici è necessariamente assai poco completa. La trattazione ha messo in evidenza la classificazione dei sistemi catalitici secondo i vari tipi di reazioni riconoscibili e i ruoli distintivi dei catalizzatori. Gli esempi citati sono stati scelti principalmente con la prospettiva di illustrare questi ruoli e sono stati quindi necessariamente limitati ad alcuni dei sistemi più semplici dei quali si siano almeno parzialmente compresi le caratteristiche meccaniche e il ruolo del catalizzatore. Le reazioni descritte sono perciò illustrative del campo della catalisi omogenea ottenibile mediante composti di coordinazione, ma vengono meno, con un margine considerevole, all'assunto di mostrare appieno il loro scopo o le loro possibilità.
L'attuale sviluppo della catalisi omogenea mediante composti di coordinazione è già sufficientemente ampio e avanzato perché siano manifesti taluni principi generali e taluni modelli di reattività. Alcuni di questi sono stati messi in evidenza nei primi capitoli di questo articolo, specialmente nei capp. 2 e 3, che trattano rispettivamente della stabilizzazione dei prodotti intermedi di una reazione per mezzo della coordinazione e dei modelli di reattività connessi con la catalisi omogenea. Tra i temi citati in questi capitoli che ricorrono ripetutamente come caratteristiche comuni di tutte le reazioni catalitiche descritte in seguito, sono: a) l'importanza nei processi catalitici di taluni stadi basilari, specialmente l'addizione ossidativa, l'inserimento migratorio e l'eliminazione riduttiva; b) l'importanza, per l'attività catalitica, di talune configurazioni elettroniche, particolarmente le configurazioni a basso spin d8 e d10.
Processi di inserimento multiplo. - Per semplicità, le reazioni catalitiche in precedenza illustrate comportavano, per la maggior parte, solo uno o due differenti reagenti per ogni singolo caso. Sembra perciò opportuno richiamare l'attenzione sui processi catalitici più complessi, che si possono ottenere mediante l'estensione di alcuni degli schemi meccanicistici che sono già stati descritti. In particolare, si possono ottenere delle sintesi piuttosto complesse attraverso sequenze meccanicistiche, iniziate con uno stadio di addizione ossidativa, seguito da successivi inserimenti di alcune differenti molecole (olefine, dieni, alchini, CO, ecc.) sui risultanti legami metallo-legante, e concluse con una eliminazione riduttiva. I complessi del nichel(0) si sono mostrati catalizzatori particolarmente versatili per tali reazioni, che sono spesso caratterizzate da alti gradi di selettività e di stereospecificità, come nell'esempio seguente (v. Chiusoli, 1970):
Modifica del corso di una reazione mediante neutralizzazione di una posizione reattiva
L'analogo delle note e diffuse applicazioni dei ‛gruppi protettivi' in chimica organica può essere ottenuto neutralizzando le posizioni reattive mediante coordinazione a uno ione metallico. Sebbene non sia di natura catalitica, questo effetto appare qualche volta tale, poiché si esplica in una modifica del corso della reazione in modo che la formazione di taluni prodotti è favorita rispetto a quella di altri. Due esempi di questo tipo sono la modifica del corso dell'alchilazione del cianuro dovuta alla coordinazione con argento(I) (equazioni 99 e 100) e la modifica del corso della bromurazione di un'arsina non satura mediante coordinazione al platino(IV) (equazioni 101 e 102).
Relazione con le catalisi eterogenea e enzimatica
Catalisi eterogenea
La catalisi omogenea e quella eterogenea hanno evidentemente alcuni temi importanti in comune, in special modo nel contesto delle reazioni, come l'idrogenazione e la disproporzione delle olefine, che sono suscettibili di entrambi i tipi di influenza catalitica (v. catalisi eterogenea). A tal proposito, sono degni di nota essenzialmente i punti seguenti.
1. Non inaspettate sono alcune importanti analogie chimiche e meccanicistiche tra taluni sistemi catalitici omogenei e i loro corrispondenti eterogenei. In tal senso, il meccanismo dell'idrogenazione delle olefine in soluzione catalizzata dal cloruro di rutenio, descritta dall'equazione (75), ha molte caratteristiche in comune con quello proposto per l'idrogenazione eterogenea delle olefine sulla superficie dei catalizzatori a base di gel di cromo, cioè
2. A parte il loro interesse intrinseco, grande attenzione è stata posta nello studio dei catalizzatori omogenei quali sistemi da prendere a modello per i loro corrispondenti eterogenei. Il valore di questi catalizzatori omogenei come sistemi modello è accresciuto dalla loro relativa semplicità, in conseguenza della quale le loro caratteristiche chimiche e meccanicistiche sono suscettibili di una spiegazione più dettagliata e definitiva. Inoltre, le significative analogie tra molte caratteristiche delle reazioni catalitiche corrispondenti omogenee ed eterogenee (per es., idrogenazione, carbonilazione, ecc.) tendono a confermare la validità dell'uso dei primi come sistemi modello per i secondi. Affrontato in tal modo il problema, lo studio dei sistemi catalitici omogenei, invero, ha già contribuito a dare una visione e una conoscenza nuove nel campo della catalisi eterogenea.
3. Poiché i comportamenti chimici e strutturali dei sistemi catalitici omogenei sono di solito meglio caratterizzati di quelli eterogenei e più suscettibili di un controllo e di un'analisi sistematici (per es. con la scelta sistematica di leganti con differenti proprietà elettroniche e steriche), è talvolta possibile ottenere con i catalizzatori omogenei dei gradi di selettività e di stereospecificità molto più elevati. Ciò si riflette in alcuni degli esempi già citati, come l'idrogenazione del butadiene catalizzata da Co(CN)53- (equazioni 76-79), l'idrogenazione asimmetrica catalizzata da complessi del rodio contenenti come leganti fosfine chirali, e diverse reazioni catalizzate dal nichel(0), come quelle illustrate dalle equazioni (91) e (98). D'altro canto vi sono processi (specialmente di polimerizzazione) per i quali la stereospecificità risulta dipendere dalle caratteristiche strutturali e topologiche della superficie di un catalizzatore eterogeneo e che non possono essere realizzati in un sistema molecolarmente disperso. Così, è risaputo che complessi solubili che sono chimicamente collegabili a catalizzatori del tipo di Ziegler-Natta - quelli derivati da (πC5H5)2TiCl2, ad esempio - sono piuttosto efficaci nella polimerizzazione dell'etilene, mentre non producono, in generale, polimeri stereoregolari delle α-olefine (v. Arlman, 1966).
4. Benché i catalizzatori omogenei presentino sovente caratteristiche di attività e selettività particolarmente significative, i catalizzatori eterogenei sono spesso preferiti dal punto di vista della convenienza del processo, specialmente in relazione alla separazione tra il prodotto e il catalizzatore. In relazione a ciò, un'estensione logica del campo della catalisi omogenea comporta il tentativo di depositare sulla superficie di supporti solidi dei composti di coordinazione e organometallici, che sono cataliticamente attivi in forma solubile, trasformandoli in tal modo in effettivi catalizzatori eterogenei. Un tentativo a tal fine è quello di legare tali catalizzatori, mediante coordinazione, a resine solide (denominate talvolta ‛leganti polimerici') nelle quali siano stati incorporati adatti siti a capacità legante. Questo modo di procedere ha già fornito risultati promettenti per i catalizzatori della carbonilazione al palladio, per quelli dell'idrogenazione al palladio e al rodio, e dell'idroformilazione al rodio. Soltanto dal 1970 circa si è cominciato a lavorare alacremente lungo queste linee e sebbene questo campo (che abbraccia la zona intermedia tra catalisi omogenea ed eterogenea) sia ancora relativamente nuovo, esso ha già attirato un interesse considerevole e si prevede che la sua portata e la sua importanza siano destinate a crescere (v. Manassen, 1972).
Catailsi enzimatica
I catalizzatori omogenei sono stati a lungo oggetto di interesse quali sistemi modello per gli enzimi. La loro utilità e validità in questo contesto derivano da considerazioni analoghe a quelle già citate, in connessione con i ruoli da essi svolti come modelli per i catalizzatori eterogenei. Le più familiari tra queste analogie sono quelle che intercorrono tra i catalizzatori acido-base e gli enzimi idrolitici, che si ritiene operino secondo meccanismi strettamente analoghi. Si ritiene anche che la catalisi mediante ioni metallici delle reazioni idrolitiche, per esempio l'idrolisi di esteri di amminoacidi (equazione 39) e dell'ATP, siano strettamente collegate col meccanismo di azione di taluni enzimi, alcuni dei quali, significativamente, sono attivati da ioni metallici (per es., la carbossipeptidasi da Zn2+). Si ritiene anche che i ruoli degli ioni metallici, come Cu2+ e Fe3+, nella catalisi delle reazioni di ossidoriduzione siano connessi con la diffusa partecipazione di questi metalli a processi biologici di ossidoriduzione, ad esempio all'azione di diverse ossidasi, quali catalasi, perossidasi, ecc. Un notevole successo è stato ottenuto nella simulazione di molte reazioni catalizzate da enzimi, in special modo di alcuni semplici processi idrolitici e di ossidoriduzione, con catalizzatori-modello sintetici. Sebbene tali modelli abbiano senza dubbio qualche attinenza con gli enzimi corrispondenti e il loro studio sia stato certamente istruttivo, è degno di nota il fatto che le elevate attività e selettività caratteristiche degli enzimi sono di rado ottenute con catalizzatori-modello. Perciò, sebbène lo ione Fe3+ e molti dei suoi complessi catalizzino la decomposizione di H2O2 (in H2O e O2), in nessun caso l'attività catalitica si avvicina a quella dell'enzima catalasi contenente un gruppo eme. Malgrado il considerevole progresso che, specialmente dal 1950 in poi, è stato compiuto nella comprensione del meccanismo d'azione degli enzimi, le cause di questa e di molte altre caratteristiche distintive della catalisi enzimatica non sono state ancora del tutto comprese (v. catalisi enzimatica; v. Bender, 1971; v. Jencks, 1969).
I numerosi problemi insoluti e quelli parzialmente risolti connessi con i temi ora citati continuano ad attrarre l'attenzione sia dei chimici, sia dei biochimici. L'interesse, inoltre, è generalmente centrato sulle possibili relazioni intercorrenti tra alcuni degli sviluppi più recenti nella chimica della coordinazione e dei composti organometallici e i sistemi biochimici. Temi di ricerca particolarmente importanti riguardano i composti ferro-zolfo come modelli delle ferridossine, la coordinazione di O2 ai metalli e i complessi metallo-diossigeno come modelli per l'emoglobina e la coordinazione e l'attivazione catalitica di N2 in relazione al meccanismo di fissazione biochimica dell'azoto.
Con la constatazione fatta nel 1965 che il coenzima B12, un derivato biologicamente attivo della vitamina B12, è un composto organometallico contenente un legame cobalto- carbonio, la chimica dei composti organometallici ha per la prima volta assunto un ruolo importante nel contesto della biochimica. Il ruolo biochimico caratteristico del coenzima B12 consiste nella sua partecipazione alla catalisi di numerose trasposizioni, ciascuna delle quali comporta l'interscambio di un atomo di H e di qualche altro gruppo (OH, NH2, ecc.) tra atomi di carbonio adiacenti. I meccanismi di queste reazioni, così come numerosi complessi-modello del cobalto, che presentano proprietà collegate con quelle della vitamina B12, sono ora oggetto di attenti studi (v. Schrauzer, 1968; v. Abeles, 1971).
Ricerche in atto e problemi non risolti
L'aspetto moderno della catalisi omogenea è caratterizzato in particolar modo dall'interesse che si pone nei sistemi catalitici formati da composti di coordinazione e da composti organometallici dei metalli di transizione. Questo interesse ha dominato il campo della catalisi omogenea sin dal 1950 circa e ha fornito i ‛temi che hanno contribuito agli sviluppi e ai risultati straordinari che hanno caratterizzato tale campo nel corso dei due decenni trascorsi.
Con l'interesse ancora centrato su questi stessi temi generali, il campo della catalisi omogenea continua a essere attivo e fecondo. Attualmente, importanti filoni di ricerca sono volti all'indagine e alla scoperta di nuove reazioni catalitiche, a un più dettagliato chiarimento dei meccanismi di molte reazioni, compresi ancora in modo incompleto (per es., la disproporzione delle olefine) e al conseguimento di una maggior padronanza dei fattori (come la variazione sistematica delle proprietà elettroniche e steriche dei leganti) che controllano l'attività, la selettività e la stereospecificità catalitiche. Questi studi non sono diretti soltanto verso le reazioni catalitiche in quanto tali, ma anche verso la scoperta e la caratterizzazione di nuovi composti di coordinazione e organometallici, che sono interessanti quali catalizzatori potenziali o quali prodotti catalitici intermedi, o il cui studio potrebbe contribuire a una migliore comprensione dei catalizzatori connessi o dei prodotti catalitici intermedi. Il progresso recente in tutti questi settori è stato impressionante e tutto contribuisce a far ritenere che l'attuale procedere della ricerca e il ritmo delle scoperte continuerà per alcuni anni a venire.
Rimangono da risolvere molti problemi importanti, teorici e applicativi. Così, sebbene si siano già compiuti alcuni progressi in questo senso, sono ancora aperti molti problemi e obiettivi stimolanti, di natura tanto scientifica quanto pratica, riguardanti l'attivazione catalitica dell'azoto molecolare. In contrasto con la situazione di altre reazioni catalitiche, come l'idrogenazione e la carbonilazione, lo stato attuale nel campo della fissazione dell'azoto molecolare permette solo una comprensione relativamente limitata dei meccanismi dei diversi sistemi conosciuti di fissazione dell'azoto e una base molto debole per avanzare previsioni circa gli sviluppi futuri. Un altro obiettivo di grande importanza scientifica e pratica, che ancora attende una soluzione, è l'attivazione catalitica omogenea degli idrocarburi saturi. Praticamente, non si è fatto alcun progresso verso questo obiettivo e, sebbene i metodi per attuarlo (quali, per es., l'addizione ossidativa) - che sono stati coronati da successo nell'attivazione catalitica di altre molecole sature, come l'idrogeno molecolare e gli alogenuri alchilici - dovrebbero a priori poter essere estesi anche agli idrocarburi saturi, tali estensioni debbono ancora raggiungere un livello significativo.
Un altro risultato degno di nota è che la crescente portata delle nostre conoscenze nel campo della catalisi omogenea mediante composti di coordinazione è stata accompagnata da un'accresciuta valutazione dei collegamenti tra questo campo e i campi connessi della catalisi eterogenea e di quella enzimatica. Vi è quindi qualche ragione per sperare e presumere che, sebbene la complessità e la difficoltà di questo obiettivo non debbano essere sottovalutate, la comprensione dettagliata che si sta raggiungendo relativamente ai meccanismi catalitici in questi sistemi omogenei piuttosto semplici darà contributi sempre più significativi alla nostra comprensione dei fenomeni connessi nei sistemi eterogenei e in quelli biochimici. Alcuni casi specifici nei quali si può ritenere che quanto detto si sia già avverato riguardano l'idrogenazione catalizzata eterogeneamente, le reazioni di carbonilazione e di idroformilazione delle olefine e, in campo biochimico, la coordinazione dell'ossigeno molecolare, così come taluni aspetti della chimica della vitamina B12. In tale contesto, occorre accennare all'interesse e all'attività crescenti, diretti alla derivazione di nuovi catalizzatori eterogenei da quelli omogenei, ottenuti mediante deposizione di questi sulla superficie di supporti solidi.
Infine, occorrerebbe rivolgere l'attenzione verso le importanti applicazioni tecnologiche della catalisi omogenea. Le reazioni catalitiche omogenee che utilizzano composti di coordinazione dei metalli di transizione hanno già fornito la base per diversi processi chimici commercialmente importanti, come l'idroformilazione delle olefine (processo oxo), l'ossidazione delle olefine catalizzate dal cloruro di palladio (processo Wacker), la disproporzione delle olefine e la carbonilazione del metanolo ad acido acetico. È da ritenere senz'altro che il numero e la portata di tali applicazioni continueranno a crescere sensibilmente e che la catarsi omogenea avrà un ruolo fondamentale nello sviluppo della tecnologia chimica nel corso dei restanti decenni del XX secolo.
Bibliografia
Abeles, R., Studies on mechanism of action of B12 coenzymes, in ‟Advances in chemistry series", 1971, C, pp. 346-364.
Arlman, E. J., Ziegler-Natta catalysis. IV. The stereospecificity in the polymerization of olefins and conjugated dienes in relation to the crystal structure of TiCl3, in ‟Journal of catalysis", 1966, V, pp. 178-189.
Ashmore, P. G., Catalysis and inhibition of chemical reactions, London 1963.
Bailey, G. C., Olefin disproportionation, in ‟Catalysis reviews", 1969, III, pp. 37-60.
Bell, R. P., Acid-base catalysis, London 1941.
Bell, R. P., The proton in chemistry, Ithaca, N. Y., 1959.
Bender, M. L., Mechanisms of homogeneous catalysis from protons to proteins, New York 1971.
Bird, C. W., Transition metal intermediates in organic synthesis, New York 1967.
Busch, D. H., The significance of complexes of macrocyclic ligands and other syntheses by ligand reactions, in ‟Record of chemical progress", 1964, XXV, pp. 107-126.
Chalk, A. J., Olefin hydrosilation by group VIII metal complexes, in ‟Transactions of the New York Academy of Sciences", 1970, XXXII, pp. 481-488.
Chatt, J., Leigh, G. J., Nitrogen fixation, in ‟Chemical Society reviews", 1972, I, pp. 121-144.
Chiusoli, G. P., Stereoselectivity in organic syntheses involving nickelcoordinated intermediates, in Aspects of homogeneous catalysis (a cura di R. Ugo), vol. I, Milano 1970, pp. 77-105.
Chiusoli, G. P., Cassar, L., Nickel catalyzed reactions of allyl halides and related compounds, in ‟Angewandte Chemie" (International edition), 1967, VI, pp. 124-133.
Coffey, R. S., Recent advances in homogeneous hydrogenation of carbon-carbon multiple bonds, in Aspects of homogeneous catalysis (a cura di R. Ugo), vol. I, Milano 1970, pp. 13-75.
Cramer, R., Transition metal catalysis exemplified by some rhodium-promoted reactions of olefins, in ‟Accounts of chemical research", 1968, I., pp. 186-191.
Dainton, F. S., Chain reactions, London 19662.
Halpern, J., The catalytic activation of hydrogen in homogeneous, heterogeneous and biological systems, in Advances in catalysis, vol. XI (a cura di D. D. Eley e altri), New York 1959, pp. 301-370.
Halpern, J., Catalysis by coordination compounds, in ‟Annual reviews of physical chemistry", 1965, XVI, pp. 103-124.
Halpern, J., Homogeneous catalysis by coordination compounds, in ‟Advances in chemistry series", 1968, LXX, pp. 1-24.
Halpern, J., Coordination compounds in homogeneous catalysis, in ‟Pure and applied chemistry", 1969, XX, pp. 59-75.
Halpern, J., Oxidative addition reactions of transition metal complexes, in ‟Accounts of chemical research", 1970, III, pp. 386-392.
Halpern, J., Catalysis of the rearrangements of strained hydrocarbons by transition metal compounds, in Proceedings of the fourteenth international conference of coordination chemistry, Toronto 1972, pp. 698-703.
Halpern, J., Pickard, A. L., Mechanism of the tris(triphenylphosphine) platinum(0)-catalyzed oxidation of triphenylphosphine, in ‟Inorganic chemistry", 1970, IX, pp. 2798-2800.
Heck, R. F., Insertion reactions of metal complexes, in ‟Advances in chemistry series", 1965, XLIX, pp. 181-219.
Heimbach, P., Jolly, P. W., Wilke, G., π-Allylnickel intermediates in organic synthesis, in ‟Advances in organometallic chemistry", 1970, VIII, pp. 29-96.
Hoogzand, C., Hübel, W., Cyclic polymerization of acetylenes by metal carbonyl compounds, in Organic syntheses via metal carbonyls (a cura di I. Wender e P. Pino), vol. I, New York 1968, pp. 343-371.
Jencks, W. P., Catalysis in chemistry and enzymology, New York 1969.
Jones, M. M., Kinetic Patterns of ligand reactivity, in ‟Advances in chemistry series", 1965, IL, pp. 153-180.
Jones, M. M., Ligand reactivity and catalysis, New York 1968.
Keim, W., Allyl system in catalysis, in Transition metals in homogeneous catalysis (a cura di G. N. Schrauzer), New York 1971, pp. 59-91.
Knowels, W. S., Sabacky, M. J., Vineyard, B. D., Catalytic asymmetric hydrogenation, in ‟Chemical communications", 1972, pp. 10-11.
Kwiatek, J., Hydrogenation and dehydrogenation, in Transition metals in homogeneous catalysis (a cura di G. N. Schrauzer), New York 1971, pp. 13-57.
Lappert, M. F., Prakai, B., Insertion reactions of compounds of metals and metalloids involving unsaturated substrates, in ‟Advances in organometallic chemistry", 1967, V, pp. 225-319.
Lefebvre, G., Chauvin, Y., Dimerization and co-dimerization of olefin compounds by coordination catalysis, in Aspects of homogeneous catalysis (a cura di R. Ugo), vol. I, Milano 1970, pp. 107-203.
Lundberg, W. O., Autoxidation and antioxidants, voll. I e II, New York 1961.
Manassen, J., The catalytic activity of organic and metallo-organic compounds in heterogeneous systems, in ‟Fortschritte der chemischen Forschung", 1972, XXV, pp. 1-38.
Martell, A. E., Catalytic effects of metal chelate complexes, in ‟Pure and applied chemistry", 1968, XVII, pp. 129-178.
Orchin, M., Rupilius, W., On the mechanism of the oxo reaction, in ‟Catalysis reviews, 1972, VI, pp. 85-131.
Paulik, F. E., Recent developments in hydroformylation catalysis, in ‟Catalysis reviews, 1972, VI, pp. 49-84.
Roth, J. F., Craddock, J. H., Hershman, A., Paulik, F. E., Low pressure process for acetic acid via carbonylation of methanol, in ‟Chemical technology", 1971, pp. 600-605.
Schrauzer, G. N., Some advances in the organometallic chemistry of nickel, in ‟Advances in organometallic chemistry", 1964, II, pp. 1-48.
Schrauzer, G. N., Organocobalt chemistry of vitamin B12 model compounds (cobaloximes), in ‟Accounts of chemical research", 1968, I, pp. 97-103.
Siling, M. I., Gel'bshtein, A. I., Catalysis and coordination interaction, in ‟Russian chemical reviews", 1969, XXXVIII, pp. 249-260.
Stern, E. W., Reactions of unsaturated ligands in palladium (II) complexes, in ‟Catalysis reviews", 1967, I, pp. 73-172.
Thompson, D. T., Whyman, R., Carbonylation, in Transition metals in homogeneous catalysis (a cura di G. N. Schrauzer), New York 1971, pp. 147-222.
Tsuji, J., Ohno, K., Decarbonylation reactions using transition metal compounds, in ‟Synthesis", 1969, pp. 157-169.
Volpin, M. E., Shur, V. B., Chemical fixation of molecular nitrogen, in Organometallic reactions (a cura di E. I. Becker e M. Tsutsui), vol. I, New York 1970, pp. 55-117.
Woodward, R. B., Hoffmann, R., The conservation of orbital symmetry, in ‟Angewandte Chemie" (International edition), 1969, VIII, pp. 781-853.