
Macromolecole, struttura delle
Macromolecole, struttura delle
SOMMARIO: 1. Introduzione. 2. Generalità: a) costituzione, configurazione e regolarità; b) unità costitutive di macromolecole; c) coordinate cartesiane e coordinate interne; d) modelli; e) principio di equivalenza e strutture elicoidali; f) energia conformazionale. 3. Metodi di determinazione strutturale: a) diffrazione dei raggi X; b) risonanza magnetica nucleare. 4. Risultati: a) macromolecole di sintesi; b) acidi nucleici; c) omopolipeptidi e proteine fibrose; d) proteine globulari. 5. Considerazioni conclusive. □ Bibliografia.
1. Introduzione
Il termine ‛macromolecola' è entrato nel vocabolario scientifico nel secondo decennio di questo secolo, quando le proprietà di alcune sostanze naturali, quali la cellulosa, furono correttamente interpretate in termini di una struttura chimica formata da un numero elevato di unità identiche, legate covalentemente l'una all'altra in maniera sequenziale, come anelli di una lunghissima catena. Nel corso di settanta anni di intensa attività scientifica, i principî chimico-fisici che regolano la struttura delle grosse molecole sono stati messi a fuoco attraverso lo studio sistematico di un numero elevato di macromolecole sia naturali e di interesse biologico, sia costruite in laboratorio attraverso nuovi metodi sintetici. Dopo le prime corrette intuizioni sulla struttura cristallina della cellulosa, già agli inizi degli anni cinquanta furono proposti i modelli strutturali degli acidi nucleici e di alcune proteine fibrose (v. acidi nucleici, vol. I; v. proteine, vol. V); contemporaneamente furono messe a punto le procedure sperimentali per la sintesi in laboratorio di macromolecole stereoregolari e per la determinazione della loro struttura spaziale (v. polimeri, vol. V; v. materiali: materiali polimerici, vol. VIII). Poco dopo, dalle mappe di densità elettronica costruite a partire dai dati di diffrazione dei raggi X da cristallo singolo emergevano le prime sorprendenti immagini strutturali di una proteina globulare, la mioglobina. Sorprendenti perché, mentre le strutture proposte fino ad allora per le macromolecole lineari erano coerenti con il principio secondo cui alla ripetitività della struttura chimica corrispondeva anche una ripetitività della struttura spaziale, il modello della mioglobina - una proteina capace di formare, a differenza delle proteine fibrose, bellissimi cristalli singoli - era paradossalmente privo di qualsiasi apparente regolarità strutturale, relegando tutto a un tratto nel campo della fantasia tutti i complessi modelli strutturali a gabbie geometriche precedentemente proposti e basati, appunto, sulla supposta regolarità geometrica delle unità costituenti le proteine globulari. Gli anni successivi hanno registrato un fortissimo sviluppo dei metodi chimico-fisici per lo studio strutturale; le metodologie cristallografiche, agevolate dallo sviluppo tecnologico e in particolare dal vertiginoso incremento delle potenzialità di calcolo e di gestione dell'enorme quantità dei dati sperimentali, hanno assunto importanza primaria nella delucidazione strutturale dei sistemi macromolecolari. Negli ultimi anni, la risonanza magnetica nucleare ha acquistato un ruolo autonomo, contribuendo in maniera sostanziale alle conoscenze strutturali, ed è presumibile che essa svolgerà funzioni sempre più essenziali nel prossimo futuro.
2. Generalità
a) Costituzione, configurazione e regolarità.
Un polimero è formato dal concatenamento, mediante legami covalenti, di unità generalmente piccole (monomeri); le unità hanno eguale costituzione se sono formate dagli stessi atomi e presentano gli stessi legami tra atomi, indipendentemente dalla loro posizione spaziale. Un polimero regolare è formato da unità con uguale costituzione (omopolimeri), che si susseguono in catena mantenendo la stessa orientazione (testa-coda). I copolimeri sono macromolecole formate da due o più unità costituzionalmente diverse, che si susseguono secondo un ordine definito (ad es., copolimeri alternati o a blocchi) o in maniera casuale.
Per una data costituzione, la configurazione di una molecola definisce la posizione spaziale relativa dei legami, senza tener conto delle diversità dovute unicamente a rotazione intorno a legami singoli (v. stereochimica, vol. VII). Configurazioni diverse sono possibili quando in catena sono presenti centri di stereoisomeria, quali doppi legami (con possibile configurazione cis o trans) o atomi di carbonio tetraedrici legati a sostituenti diversi. Un polimero regolare è anche stereoregolare se la configurazione delle unità successive in catena varia secondo una legge definita. La cellulosa, la guttaperca e la gomma naturale (v. cap. 2, § b) sono esempi di polimeri naturali stereoregolari. Tra i polimeri di sintesi, il nailon e il polietilene sono esempi di polimeri le cui unità costituzionali possono avere una sola configurazione e per i quali non ha senso parlare di stereoregolarità.
Per un centro di stereoisomeria formato da un carbonio tetraedrico C, i due legami in catena adiacenti al carbonio possono essere distinti, da un punto di vista configurazionale, con un segno (+/-) in accordo con la seguente convenzione. Si consideri un polimero avente in catena la sequenza di atomi A, C e B, e sia C un centro di stereoisomeria con sostituenti R′ ed R″, di cui il primo più pesante del secondo, posti rispettivamente al di sotto e al di sopra del piano individuato dai tre atomi A, C, B, come riportato nella parte a dello schema seguente:
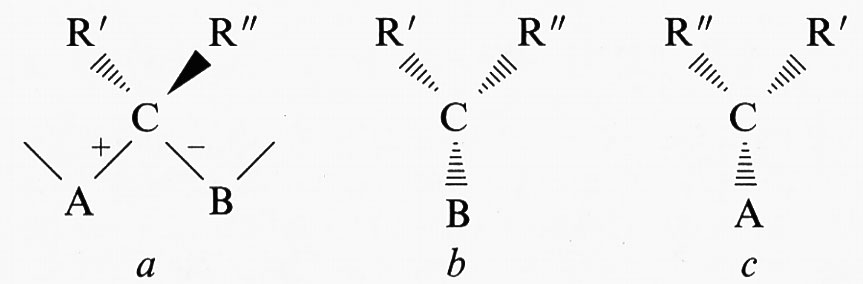
Nelle parti b e c dello stesso schema sono riportate le vedute prospettiche quali risultano guardando, rispettivamente, da A lungo il legame A−C e da B lungo il legame B−C. Il legame A−C è contrassegnato con il segno + se gli atomi B, R′ ed R″ in b si succedono secondo una rotazione oraria, mentre è contrassegnato con il segno - se gli stessi atomi si succedono secondo una rotazione antioraria. Analogamente, il segno del legame B−C si può ottenere prendendo in considerazione la successione degli atomi A, R′, e R″ rappresentati in c. Per definizione, se il legame A−C ha segno +, il legame B−C ha segno - e viceversa. Due centri di stereoisomeria hanno la stessa configurazione se sono uguali i segni dei due legami adiacenti in catena, sono invece enantiomorfi se i segni sono opposti. I polimeri vinilici con unità costitutiva −CH2−CHR−, dove R è un generico sostituente diverso da H, posseggono un centro di stereoisomeria in catena, perché un carbonio è legato a due sostituenti diversi H e R. Per un concatenamento regolare, sono possibili due sequenze stereoregolari: la sequenza isotattica (v. fig. 1A), in cui la coppia di segni +/- si sussegue in maniera regolare, e la sequenza sindiotattica (v. fig. 1B), che presenta inversione dei segni configurazionali nel passare da un centro di stereoisomeria al successivo. Le denominazioni derivano dal fatto che, nella forma planare estesa della catena, i sostituenti R rispetto al piano individuato dagli atomi di carbonio della catena si trovano tutti dalla stessa parte nella sequenza isotattica e alternativamente da lati opposti nella sequenza sindiotattica. Successioni non regolari delle coppie di segni +/- e -/+ danno luogo a polimeri non stereoregolari o atattici.
Nonostante la presenza di centri di stereoisomeria tutti aventi la stessa configurazione, una catena stereoregolare isotattica di un polimero vinilico (in assenza di ulteriori centri di stereoisomeria in R) non è otticamente attiva; per questo i centri sono anche chiamati ‛pseudochirali'. Benché i segmenti di catena sui due lati dell'atomo di carbonio asimmetrico, indicati con una spezzata nello schema seguente,

siano in generale di diversa lunghezza, gli effetti sull'attività ottica dovuti ad atomi lontani sono praticamente nulli e il polimero è otticamente inattivo; tuttavia sono possibili due configurazioni distinguibili che corrispondono a una diversa tatticità. In un polipeptide formato dal concatenamento di amminoacidi L (v. cap. 2, § b), i gruppi CO e NH legati al carbonio asimmetrico sono diversi e il polimero isotattico possiede attività ottica.
b) Unità costitutive di macromolecole.
La maggior parte delle macromolecole sintetiche o naturali può essere ottenuta mediante reazioni di addizione o di condensazione. Nel primo caso il riordinamento dei legami chimici nella molecola del monomero lascia libere due valenze che permettono il concatenamento con altri monomeri senza eliminazione di atomi. Nella reazione di condensazione il monomero, che possiede due gruppi funzionali, può reagire con un altro monomero con eliminazione di una molecola piccola, spesso di acqua. Qui di seguito vengono descritti i blocchi costitutivi di alcune macromolecole, sia sintetiche che naturali; la disamina ha solo scopo esemplificativo, per permettere al lettore di seguire più agevolmente l'esposizione degli aspetti strutturali.
Polimeri di sintesi. - Numerosi tipi di polimeri sintetici - polipropilene, polivinilcloruro, 1,2-polibutadiene, ecc. - possono essere ottenuti per reazione di addizione a partire da monomeri vinilici CH2=CHR al variare del sostituente R. L'apertura del doppio legame determina la formazione dell'unità costitutiva −CH2−CHR−, che può essere schematizzata come −A−B−. Per A ≠ B (R ≠ H), il monomero ha una polarità costituzionale e la reazione di inserzione del monomero nella catena in crescita può avvenire in due modi diversi, −A−B−A−B− e A−B−B−A−, denominati testa-coda e testa-testa, rispettivamente. Se il tipo di inserzione è casuale, si ha disordine spaziale; se l'inserzione avviene prevalentemente in un modo, si ha la formazione di un polimero regolare. Per il polietilene, in cui il sostituente R è un atomo di idrogeno, A e B sono uguali e le due situazioni sono indistinguibili.
Un'altra fonte di irregolarità dipende dal fatto che alcuni monomeri possono dare luogo a unità costitutive diverse; ciò si verifica, ad esempio, nella reazione di addizione a partire da dieni coniugati. Da butadiene CH2=CH−CH=CH2 si può avere 1,2-polibutadiene o 1,4-polibutadiene, aventi rispettivamente le unità costitutive

L'inserzione testa-testa o testa-coda è fonte di ulteriore irregolarità nel primo caso, mentre è irrilevante nel secondo. Nell'addizione 1,4 il polimero contiene un doppio legame in catena, centro di stereoisomeria, che può essere cis o trans (1,4-cis- o 1,4-trans-polibutadiene). Se le unità sono tutte cis o tutte trans il polimero si dice cis-tattico o trans-tattico, rispettivamente. Più complessa è la situazione nel caso dell'isoprene (2-metil-butadiene) CH2=C(CH3)−CH=CH2; da esso si originano tre unità costitutive diverse, di cui due danno luogo a un concatenamento 1,2:
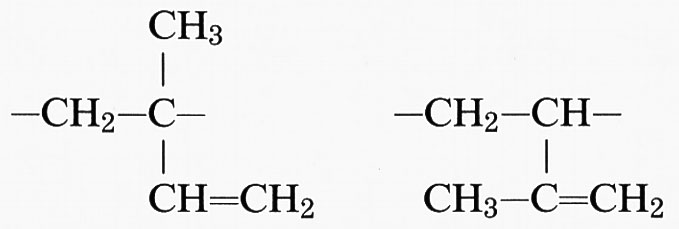
Nell'addizione 1,4 si forma un doppio legame nella catena principale del polimero, che quindi può essere cis- o trans-tattico. Entrambe le forme si trovano in natura e sono note, rispettivamente, con il nome di gomma naturale e di guttaperca:
Nei polimeri di condensazione ottenuti da monomeri bifunzionali, irregolarità legate a inserzioni testa-coda o testa-testa possono esistere solo se i due gruppi funzionali sono uguali e il monomero ha una polarità costituzionale. Un esempio è rappresentato dai polieteri, che possono essere ottenuti per eliminazione di H2O dai dioli. Utilizzando un diolo del tipo HO−CH(R)−CH2−OH, una inserzione testa-testa in una catena in crescita secondo lo schema testa-coda crea una irregolarità strutturale, come già visto per i polimeri vinilici. Quando i gruppi funzionali sono diversi, il modo di inserzione del monomero è determinato dall'orientazione dell'ultimo monomero in catena: è questo il caso dei poliesteri e delle poliammidi, ottenuti rispettivamente per condensazione di monomeri del tipo HO−A−COOH e H2N−A−COOH (A indica un generico gruppo):
−O−A−COOH+HO−A−COOH →
→ −O−A−CO−O−A−COOH+H2O
−NH−A−COOH+H2N−A−COOH →
→ −NH−A−CO−NH−A−COOH+H2O
Poliammidi molto diffuse possono essere ottenute anche per copolimerizzazione alternata di una diammina con un acido bicarbossilico; il nailon 66 è un copolimero alternato con unità ripetitiva
[−NH−(CH2)6−NH−CO−(CH2)4−CO−]n
Acidi nucleici. - Le unità costitutive degli acidi nucleici sono nucleotidi, il cui scheletro è formato da uno zucchero con struttura ciclica furanica a cinque termini e un gruppo fosfato (v. fig. 2); per l'acido desossiribonucleico (DNA) lo zucchero è il β-2′-desossiribosio, mentre per l'acido ribonucleico (RNA) è il β-ribosio. In entrambi i casi il concatenamento dello zucchero con i gruppi fosfati adiacenti avviene attraverso gli ossigeni O5′ e O3′, e si assume convenzionalmente che la direzione di catena sia quella che va dall'atomo O5′ all'atomo O3′; lo zucchero, inoltre, è legato mediante un legame β-glicosidico a un composto eterociclico azotato (base), di tipo purinico o pirimidinico, che costituisce la catena laterale. Nel DNA le possibili basi puriniche sono adenina e guanina e le basi pirimidiniche sono timina e citosina; nell'RNA la timina è sostituita dall'uracile. Gli acidi nucleici sono copolimeri di quattro nucleotidi (diversi solo per la catena laterale), che si susseguono secondo una sequenza definita.
A causa della repulsione tra i sostituenti degli atomi di carbonio, l'anello a cinque termini dello zucchero non è planare, ma può assumere varie forme nello spazio: le più comuni sono quelle che presentano un atomo fuori dal piano determinato dagli altri quattro atomi dell'anello (forme ‛a busta') o due atomi adiacenti spostati su lati opposti rispetto al piano determinato dagli altri tre atomi (forme ‛a mezza sedia' o twist). Nel primo caso le denominazioni Y-endo e Y-eso indicano che l'atomo Y è fuori dal piano e spostato, rispettivamente, nello stesso verso dell'atomo C5′ o nel verso opposto. I dati sperimentali indicano che le forme più stabili sono le due forme ‛a busta' C3′-endo e C2′-endo (v. fig. 3).
Polipeptidi e proteine. - Le unità costitutive delle proteine sono gli α-amminoacidi H2N−CαH(R)−COOH, che posseggono un gruppo amminico e un gruppo carbossilico legati allo stesso atomo di carbonio tetraedrico, chiamato Cα, e sono distinguibili tra di loro per il tipo di sostituente R legato al carbonio Cα. Per condensazione tra il gruppo carbossilico e il gruppo amminico di due α-amminoacidi si forma un legame ammidico o peptidico: la catena risultante prende il nome di catena polipeptidica. Il carbonio Cα è legato a quattro sostituenti diversi e pertanto è un centro di stereoisomeria; le due forme stereoisomere possibili sono chiamate forma D (Destro) e forma L (Levo). Le strutture proteiche sono formate quasi tutte esclusivamente da 20 α-amminoacidi nella forma L. Il gruppo −NH−CHR−CO− prende il nome di residuo di amminoacido, e viene indicato con un simbolo a una o a tre lettere come G o Gly per la glicina (l'unico α-amminoacido non otticamente attivo, essendo il sostituente R un atomo di idrogeno), A o Ala per l'alanina, ecc. Occasionalmente, in piccoli peptidi si riscontra la presenza di D-α-amminoacidi o L-α-amminoacidi non canonici. Con esclusione della prolina, in cui il sostituente R è un anello pirrolidinico che chiude sull'atomo di azoto adiacente al Cα, le proteine sono rappresentabili dallo schema seguente (per gli angoli di torsione ϕ, ψ e ω, vedi cap. 2, § c, e cap. 4, § c):
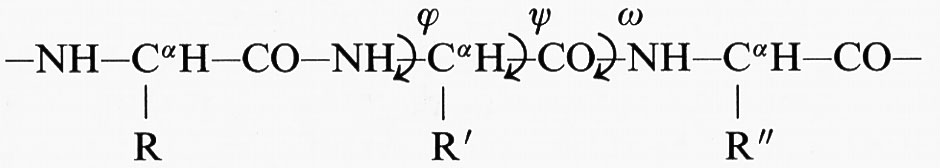
I gruppi NH, CαH e CO definiscono il corso della catena principale, mentre i sostituenti R, R′, R″ rappresentano le catene laterali dello scheletro polipeptidico. Il gruppo −CO−NH− è detto ‛gruppo peptidico'; il legame peptidico tra l'atomo di carbonio e quello di azoto (entrambi in uno stato di ibridazione trigonale planare) presenta parziale carattere di doppio legame, che impone una configurazione cis o trans del gruppo peptidico. Nella stragrande maggioranza dei casi esso si trova in configurazione trans.
Omopolipeptidi (polipeptidi formati da un solo tipo di amminoacido) possono essere facilmente ottenuti per sintesi. Tutte le proteine sono copolimeri la cui sequenza amminoacidica è solo parzialmente ripetitiva (come spesso capita nelle proteine fibrose) o senza nessun ordine apparente ma esattamente programmata (proteine globulari). In entrambi i casi la ripetitività chimica è comunque esatta per la catena principale.
c) Coordinate cartesiane e coordinate interne.
Con i metodi cristallografici vengono determinate le tre coordinate di ogni atomo rispetto a una terna cartesiana fissa; tuttavia, rispetto a una terna di riferimento arbitraria, sono sufficienti 3n - 6 parametri (coordinate interne) per definire completamente la struttura spaziale di una molecola non lineare composta da n atomi (v. stereochimica, vol. VII). Le coordinate interne sono costituite da una opportuna selezione di distanze di legame, angoli di valenza e angoli di rotazione interna della molecola (v. fig. 4). Per questi ultimi vale la seguente definizione: dati quattro atomi A, B, C, D sequenzialmente legati, l'angolo di rotazione interna, o angolo di torsione, intorno al legame B−C è l'angolo di cui bisogna ruotare il legame C−D in senso orario per sovrapporlo al legame A−B, quando si guardi nella direzione del legame C−B da C verso B. Angoli di torsione vicini a 0°, ± 60°, ± 120° e 180° sono rispettivamente indicati con i termini cis (o ‛sinperiplanare'), gauche (o ‛sinclinale'), ‛anticlinale' e trans (o ‛antiperiplanare'), oppure con i simboli C, G±, A±, T.
L'uso delle coordinate interne permette di dividere i parametri strutturali in tre categorie che hanno gradi di variabilità considerevolmente diversi da molecola a molecola. È noto infatti che le distanze di legame sono approssimativamente indipendenti dall'intorno dei due atomi legati, se non per particolari situazioni elettroniche che caratterizzano il tipo di legame (legami singoli, doppi, ecc.); per questo motivo, le lunghezze di legame possono essere considerate abbastanza indipendenti dalla particolare molecola in cui la coppia di atomi si trova. Ciò è vero anche per gli angoli di valenza, pur se questi presentano un grado di variabilità che, per quanto limitata, è maggiore. Queste caratteristiche delle distanze di legame e degli angoli di valenza permettono di utilizzare i valori misurati in molecole piccole per lo studio strutturale di macromolecole, per le quali il numero dei dati sperimentali è relativamente piccolo rispetto al numero di parametri che definiscono il modello strutturale.
Per valori fissi di distanze e angoli di valenza, la struttura di una molecola risulta completamente definita dagli angoli di rotazione interna, la cui sistematica variazione permette di ottenere tutte le possibili strutture: la struttura generata assegnando specifici valori agli angoli di torsione è chiamata anche ‛conformazione' (v. molecole: Analisi conformazionale delle grandi molecole, vol. IV).
d) Modelli.
Coordinate interne e coordinate cartesiane sono facilmente interconvertibili. Le prime sono particolarmente utili per molecole piccole o per descrivere strutture locali di macromolecole; le coordinate cartesiane sono più adatte per una rappresentazione globale della struttura. La struttura di una molecola può essere rappresentata in maniera schematica da un modello tridimensionale costruito con sfere di dimensioni opportune (che definiscono posizione e tipo degli atomi nella molecola), connesse da bacchette, che definiscono la sequenza dei legami chimici; fotografie del modello danno quindi una rappresentazione della molecola. Alternativamente, il modello può essere disegnato per proiezioni lungo opportune direzioni, a partire dalle coordinate cartesiane (v. figg. 1 e 9). Un'impressione più realistica dello spazio effettivamente occupato dagli atomi può essere ottenuta usando per ogni sfera un raggio pari al raggio di van der Waals dell'atomo che essa rappresenta (v. figg. 7, 13 e 19). Viceversa, quando motivi di chiarezza lo richiedono, le sfere possono essere eliminate del tutto e le posizioni degli atomi risultano allora definite dalle intersezioni delle linee che rappresentano i legami (v. fig. 17). Un notevole miglioramento si raggiunge con l'uso di disegni formati da due proiezioni dello stesso modello, lungo direzioni reciprocamente inclinate di circa 6°, poste fianco a fianco a distanza di circa 6 cm: con l'ausilio di una lente che facilita la sovrapposizione delle due proiezioni (l'occhio esercitato può farne facilmente a meno), si ottiene una nitida visione tridimensionale del modello (v. figg. 11, 12 e 17). Anche in questo modo, tuttavia, la rappresentazione può non essere adeguata a illustrare e analizzare modelli complessi come le proteine globulari, e può essere vantaggioso introdurre ulteriori semplificazioni, che mettano in evidenza le caratteristiche più importanti della macromolecola. Ad esempio, il corso della catena può essere sommariamente schematizzato riportando per ogni unità costitutiva uno solo degli atomi che formano l'unità stessa e collegando in sequenza con delle linee gli atomi tra di loro (nelle proteine globulari di norma l'atomo riportato è il carbonio Cα); in altri casi un semplice nastro, che corre approssimativamente lungo gli atomi dello scheletro, può efficacemente rappresentare la catena e le conformazioni locali (v. figg. 15, 18 e 20-24). Un'ulteriore schematizzazione è data dalla sostituzione dei tratti a conformazione elicoidale con cilindri di pari lunghezza coassiali all'asse dell'elica (v. fig. 21), e dei tratti a conformazione β con nastri aventi una freccia terminale che indica la direzione di catena (v. figg. 18 e 22-24). Questi disegni sono particolarmente utili per il confronto tra molecole diverse e per una classificazione delle loro caratteristiche strutturali. Tutte queste rappresentazioni sono ora facilmente ottenibili mediante calcolatori elettronici con terminali grafici: le figg. 15, 20, 22C e 24 sono state ottenute con il programma MOLSCRIPT (v. Kraulis, 1991). Lo sviluppo delle tecniche di grafica molecolare computerizzata ha rappresentato un importante presupposto per i rilevanti successi registrati nel campo della struttura delle macromolecole in questi ultimi anni.
e) Principio di equivalenza e strutture elicoidali.
Una considerevole mole di informazioni strutturali su macromolecole stereoregolari deriva da dati di diffrazione dei raggi X su fibre; questi dati indicano che nella maggioranza dei casi la struttura delle macromolecole stereoregolari può essere descritta da una successione di unità strutturalmente equivalenti, legate cioè da un'operazione di simmetria, che può essere esatta (vale a dire compresa nelle operazioni di simmetria del cristallo) o approssimata. Molto spesso l'unità strutturalmente indipendente coincide con l'unità costitutiva; questo implica che l'equivalenza chimica delle unità costitutive si traduce in un'equivalenza della conformazione e i parametri strutturali descrittivi di una macromolecola si riducono a quelli che definiscono la conformazione dell'unità indipendente.
È interessante esplorare un po' più in dettaglio le conseguenze del principio di equivalenza. Per un polimero isotattico, ogni generica unità i in catena è legata all'unità successiva i+1 attraverso un'operazione che, senza perdere di generalità, può essere decomposta in una rotazione di un angolo ϕ intorno a un ben determinato asse nello spazio, seguita da una traslazione h lungo lo stesso asse; sotto l'ipotesi di equivalenza, la stessa operazione trasforma l'unità i +1 nell'unità i +2, e così via. L'asse comune a queste trasformazioni è detto anche asse della macromolecola. Una trasformazione con ϕ = 0 è una traslazione pura; una trasformazione con h = 0 e ϕ ≠ 0 è incompatibile con una catena polimerica, perché la ripetuta applicazione di essa genera una struttura che si ricopre periodicamente. Per ϕ e h diversi da zero, la catena assume una forma elicoidale, il cui asse coincide con l'asse della macromolecola ed è un elemento di simmetria di rototraslazione (elicogira) della catena stessa (v. fig. 5). La quantità n = 2π/ϕ rappresenta il numero di monomeri per spira dell'elica; se n è intero, i monomeri i e i +n sono collegati da una traslazione lungo l'asse dell'elica. Se n non è intero, questo si verifica solo dopo N giri e un numero di monomeri M = nN = N 2π/ϕ. La simmetria della struttura elicoidale, quindi, risulta caratterizzata da M monomeri in N giri (spesso, infatti, è indicata con il simbolo M/N) e da un periodo di ripetizione lungo l'asse elicoidale c = h M. Il parametro c prende il nome di ‛periodo di identità' della struttura, mentre la grandezza p = c/N si chiama ‛passo dell'elica'. Gli elementi di simmetria roto-traslazionali sono frequenti nei cristalli; essi sono però compatibili con gli elementi di simmetria traslazionale del cristallo solo se n assume i valori 2, 3, 4 o 6; per una macromolecola isolata questa limitazione non esiste.
Per un polimero sindiotattico le unità monomeriche si alternano con configurazioni opposte; in questo caso l'operazione che trasforma un'unità nella successiva è una riflessione in un piano che contiene l'asse della macromolecola, seguita da una traslazione lungo l'asse pari a metà del periodo di identità (slittopiano). Se le due direzioni di catena sono equivalenti, come nel caso dei polimeri vinilici, sono anche possibili strutture con simmetria elicoidale di tipo M/N e un asse di simmetria binario normale all'asse della macromolecola (in questo caso il numero di monomeri nel periodo di identità è 2M).
Sulla base del principio di equivalenza e nell'approssimazione della geometria fissa, le variabili conformazionali indipendenti per un polimero vinilico stereoregolare sono due angoli di torsione (non si considerano qui gli angoli di torsione necessari per definire eventualmente la conformazione della catena laterale). Per una catena isotattica la conformazione è caratterizzata da una sequenza ripetitiva di angoli di torsione θ1, θ2, θ1, θ2 ... . Per una catena sindiotattica si possono avere conformazioni caratterizzate dalla sequenza θ1, θ2, - θ1, - θ2 (con la restrizione θ1 = π) o dalla sequenza θ1, θ1, θ2, θ2. Per ogni insieme di angoli di torsione indipendenti è possibile calcolare tutti i parametri geometrici che definiscono la struttura della macromolecola (direzione dell'asse, simmetria, periodo di identità).
f) Energia conformazionale.
La massima stabilità di un sistema è legata al valore minimo del potenziale termodinamico ‛energia libera'. Per un sistema allo stato solido, in cui di norma è presente una sola conformazione, il contributo conformazionale all'entropia è praticamente nullo e quello dei moti vibrazionali interni della molecola dipende poco dalle interazioni con altre molecole nel cristallo; trascurabile è inoltre la differenza tra entalpia ed energia interna. Sulla base di queste assunzioni, il calcolo dell'energia interna, per tutte le possibili conformazioni di una molecola, permette di definire le conformazioni più stabili. È stato inoltre verificato sperimentalmente che la struttura di molte macromolecole lineari allo stato solido corrisponde da vicino a quella con valori più bassi della sola energia intramolecolare; è pertanto possibile, in prima approssimazione, trascurare le interazioni intermolecolari, nell'ipotesi che esse abbiano una importanza secondaria rispetto ai contatti intramolecolari. Naturalmente, in presenza di conformazioni approssimativamente isoenergetiche, il ruolo di queste interazioni può diventare determinante per la stabilizzazione di una forma rispetto alle altre.
In principio il calcolo dell'energia richiedeva metodi quantomeccanici; ancora oggi ciò è possibile, ma solo nel caso di sistemi con relativamente pochi atomi. Un grosso lavoro è stato fatto per la ricerca di funzioni analitiche capaci di riprodurre accuratamente l'energia potenziale per molecole complesse, utilizzando parametri derivati da studi teorici di molecole piccole o dal confronto diretto con i dati sperimentali disponibili. Nei calcoli di energia, di norma, si assume che questa possa essere ottenuta come somma di vari contributi che tengono conto dell'energia di deformazione delle lunghezze di legame e degli angoli di valenza (non considerati nell'approssimazione della geometria fissa), delle interazioni di non legame, elettrostatiche e di legami idrogeno.
Quando il principio di equivalenza conformazionale può essere applicato, l'energia conformazionale, nell'approssimazione della geometria fissa, è funzione solo degli angoli di torsione dell'unità ripetitiva; in questo caso è possibile costruire mappe di energia che danno un quadro compatto delle conformazioni elicoidali energeticamente stabili della macromolecola. Con l'aumentata potenza dei calcolatori elettronici a disposizione, l'uso della geometria variabile è diventato molto più comune, e questo permette di tener conto di una certa variabilità degli angoli di valenza, specialmente in gruppi stericamente affollati. Naturalmente l'equivalenza conformazionale si verifica di norma solo in presenza dei vincoli imposti da molecole vicine in uno stato cristallino ordinato e non nello stato amorfo o in soluzione. Le proteine globulari sono un caso a parte: la stereoregolarità della catena principale è qui accoppiata a una specifica sequenza di catene laterali diverse, spesso formate da gruppi carichi o polari che influenzano gli angoli di torsione della catena e, attraverso le interazioni con l'ambiente esterno e con gli altri atomi della macromolecola, determinano il ripiegamento della proteina in una struttura globulare compatta e ben definita anche in soluzione. In questo caso il numero di variabili torsionali diventa enorme e, anche sulla base di ragionevoli semplificazioni, un campionamento sistematico dello spazio conformazionale per la ricerca delle conformazioni più stabili richiederebbe tempi di calcolo proibitivi; nel calcolo dovrebbero essere anche incluse le interazioni con il solvente, il che pone ulteriori formidabili problemi di calcolo. Partendo da una conformazione iniziale, è possibile, in principio, ottenere la conformazione di minima energia utilizzando uno dei numerosi metodi matematici per la ricerca dei minimi di una funzione a più variabili. Tuttavia, l'energia potenziale presenta numerosi minimi relativi e qualsiasi processo di minimizzazione converge al minimo relativo più vicino; l'uso della geometria variabile, oltre ad aumentare i tempi di calcolo, peggiora anche il problema dei minimi relativi. I processi di minimizzazione sono spesso utilizzati per definire la stabilità relativa di un certo numero selezionato di conformazioni. Va ricordato che l'approccio teorico è importante non solo come metodo di predizione delle conformazioni più stabili di una macromolecola, ma anche per una più coerente interpretazione dei dati sperimentali: anche le tecniche di indagine strutturale più potenti, quando applicate a sistemi macromolecolari, fanno largo uso dei criteri energetici per trasformare le informazioni strutturali in modelli atomici accurati.
3. Metodi di determinazione strutturale
a) Diffrazione dei raggi X.
L'uso di microscopi a vari livelli di ingrandimento permette di osservare dettagli sempre maggiori fino a un limite di risoluzione massimo, legato alla lunghezza d'onda della radiazione. Quando si è interessati alla struttura di una molecola, ovvero alla determinazione delle posizioni relative degli atomi che la costituiscono, è necessario ricorrere alla radiazione X con lunghezza d'onda confrontabile con le distanze interatomiche (~ 1 Å). Anche se non è possibile focalizzare con lenti i fasci di raggi X, una immagine della molecola può essere ottenuta analizzando le intensità diffratte da cristalli della sostanza in esame. Un cristallo singolo è una porzione di solido, formato da una periodica ripetizione di un insieme di atomi lungo tre direzioni non complanari dello spazio; i tre vettori di traslazione, a, b e c, definiscono un parallelepipedo che prende il nome di ‛cella elementare'. Per un cristallo molecolare il gruppo di atomi nella cella elementare, che rappresenta l'unità ripetitiva fondamentale, può essere costituito da una o più molecole. Quando i raggi X incidono su un cristallo, l'onda diffratta dagli atomi in una cella elementare si combina in maniera coerente con quella diffratta dagli atomi in altre celle elementari per dare luogo a una intensità totale non nulla solo per alcune direzioni, determinate dai parametri della cella elementare. Per ognuna di queste direzioni (‛riflesso di Bragg'), l'intensità diffratta dal cristallo è del tutto equivalente a quella diffratta dal contenuto di una sola cella elementare amplificata milioni di volte. Ogni riflesso è caratterizzato da una ampiezza, proporzionale alla radice quadrata dell'intensità, misurabile con lastre fotografiche o contatori elettronici di radiazione, e da un angolo di fase che non è possibile misurare (v. anche cristallografia, vol. X).
A partire dalle intensità diffratte è possibile calcolare, mediante metodi matematici e con l'uso di calcolatori elettronici, la funzione ‛densità elettronica' delle molecole nella cella elementare. Ogni massimo della funzione definisce la posizione di un atomo e il suo valore numerico è proporzionale al numero di elettroni dell'atomo. Atomi con elevato numero atomico danno un grosso contributo all'intensità diffratta e sono di conseguenza ben localizzabili nella mappa di densità elettronica; viceversa, gli atomi di idrogeno danno uno scarso contributo alla diffrazione e sono, quindi, più difficilmente localizzabili. Va inoltre precisato che, mentre le caratteristiche più grossolane della densità elettronica sono definite dai riflessi con una piccola angolazione rispetto al fascio incidente, i dettagli di essa sono sempre meglio delineati man mano che aumenta l'angolazione dei riflessi inclusi nel calcolo. La massima risoluzione della densità è data dalla semilunghezza d'onda, λ/2, della radiazione X; per la radiazione emessa da un anticatodo di rame (una delle radiazioni più frequentemente adoperate negli esperimenti di diffrazione) λ/2 ha un valore di 0,77 Å, più che sufficiente per separare i massimi più vicini della densità elettronica, quelli cioè corrispondenti ad atomi direttamente legati. Per le macromolecole, tuttavia, la risoluzione effettiva è ben al di sotto di quella massima, sia per la presenza di disordine, sia a causa di una elevata mobilità degli atomi anche nello stato solido, che provoca una rapida diminuzione delle intensità diffratte all'aumentare dell'angolo di deviazione rispetto al fascio incidente. In realtà, per questi sistemi, di norma si riescono a ottenere cristalli singoli solo per quelle classi di macromolecole aventi un ben definito peso molecolare e la cui costituzione chimica favorisce il raggiungimento di una struttura ripiegata compatta; in pratica, cristalli singoli adatti per una indagine cristallografica sono ottenibili quasi esclusivamente per proteine globulari. La maggior parte delle macromolecole lineari, anche stereoregolari, possiede sempre una certa variabilità di peso molecolare e numerosi difetti costituzionali e/o di stereoregolarità, che difficilmente permettono la crescita di cristalli singoli. Solitamente esse danno luogo a fibre, caratterizzate da piccole regioni cristalline (‛cristalliti') orientate con uno dei lati della cella elementare (coincidente con l'asse della macromolecola) in modo parallelo all'asse di fibra, mentre gli altri due assi sono orientati in modo casuale; queste regioni sono intervallate da zone più o meno amorfe.
Un diagramma di fibra presenta riflessi diffusi a forma di semiarchi - la cui estensione dipende dal grado di orientamento dei cristalliti lungo l'asse di fibra - e zone di intensità diffusa causata dalle regioni amorfe; la risoluzione è generalmente bassa per la presenza di disordine di vario tipo anche nelle regioni cristalline. Anche se le informazioni presenti in un diagramma di fibra sono di gran lunga inferiori a quelle ottenibili da un cristallo singolo, esse sono spesso sufficienti a definire la struttura della macromolecola. Nel caso di strutture elicoidali, il diagramma di diffrazione mostra una caratteristica distribuzione delle intensità, lungo strati normali all'asse di fibra, direttamente collegata ai parametri che definiscono la struttura elicoidale della macromolecola; dalla spaziatura tra gli strati e dalla distribuzione delle intensità sugli strati è infatti possibile calcolare il periodo di identità c, il numero di monomeri M e il numero di spire N nel periodo di identità. Un esame minuzioso dell'intero spettro permette inoltre di definire con una certa sicurezza i dettagli dell'impacchettamento delle macromolecole nel cristallo.
Cristalli singoli di proteine globulari possono essere ottenuti da una soluzione della proteina per lenta evaporazione del solvente, o per lenta diffusione di un non solvente o, anche, per lenta variazione della temperatura. Scegliendo opportunamente le condizioni sperimentali (tipo di solvente, pH della soluzione, temperatura), sono stati ottenuti cristalli di centinaia di proteine diverse, di complessi di proteine con molecole piccole e di complessi tra proteine, e anche cristalli di alcuni virus; in molti casi la crescita di cristalli di dimensioni di qualche frazione di millimetro, adatti per esperimenti di diffrazione, può richiedere anche diversi mesi e rappresenta lo stadio più incerto dei metodi cristallografici.
Per il calcolo della densità elettronica è necessario conoscere non solo il valore dell'intensità di ogni riflesso, ma anche il valore del relativo angolo di fase che, come è stato già detto, non può essere misurato sperimentalmente. Il problema è stato affrontato in vario modo nel passato e in parte risolto, a partire dagli anni sessanta, mediante metodi matematici che permettono di ottenere una stima degli angoli di fase direttamente dalle intensità dei riflessi (metodi diretti); questi metodi sono però applicabili con successo solo a cristalli formati da molecole piccole. Per cristalli di proteine, il problema fu risolto quando M. Perutz nel 1956 dimostrò che era possibile determinare le fasi confrontando le intensità diffratte dal cristallo della proteina nativa con le intensità diffratte da cristalli isomorfi, che differiscono da quelli della proteina nativa per la presenza di uno o più atomi con elevato numero atomico. Un cristallo di proteina può essere considerato un sistema ‛bifasico', in cui solo una parte del volume (40 ÷ 60%) è occupato in maniera compatta dalle molecole di proteina, mentre per la restante parte è occupato dalla soluzione di cristallizzazione. Gli ampi canali tra molecole permettono la diffusione all'interno del cristallo di opportuni reagenti chimici, che possono reagire con specifici gruppi presenti sulla superficie della molecola senza alterarne l'impacchettamento. Questa proprietà dei cristalli di proteine, oltre a fornire una via per la preparazione di derivati isomorfi, ha anche altre importanti conseguenze: a causa della bassa energia reticolare dei cristalli (scarsi contatti intermolecolari) e dell'estesa superficie di contatto delle molecole con la soluzione, la struttura della proteina nel cristallo risulta poco influenzata dalle forze di impacchettamento e quindi simile a quella che la proteina assume in soluzione. Tuttavia, per gli stessi motivi, i cristalli di proteine sono estremamente fragili e relativamente più disordinati dei cristalli formati da molecole piccole; ne consegue che la struttura molecolare ha una risoluzione più bassa. Negli ultimi quindici anni la cristallografia di proteine è diventata un metodo maturo grazie a importanti innovazioni tecniche e metodologiche, quali: 1) maggiore comprensione dei processi di cristallizzazione; 2) sviluppo di rivelatori ad area per la registrazione dei dati di diffrazione, notevolmente più sensibili e accurati delle lastre fotografiche, che permettono la misura simultanea di numerosi riflessi; 3) uso di sorgenti molto intense di raggi X (luce di sincrotrone); 4) metodi per registrare i dati di diffrazione a temperature molto basse (criocristallografia), che aumentano notevolmente la stabilità dei cristalli nel fascio dei raggi X (un effetto particolarmente importante quando si usano sorgenti molto intense) e possono in alcuni casi portare a una risoluzione più alta; 5) sviluppo di complessi programmi di calcolo che, accoppiati con sistemi di grafica al calcolatore, permettono una facile gestione della enorme quantità di dati sperimentali, una più veloce interpretazione della densità elettronica in termini di un modello molecolare e un'efficiente ottimizzazione del modello stesso in rapporto ai dati sperimentali e stereochimici (affinamento della struttura). In particolare, la luce di sincrotrone ha cambiato radicalmente le prospettive stesse del metodo: la possibilità di variare la lunghezza d'onda della radiazione è importante per la registrazione di effetti anomali di diffrazione, che sono preziosa fonte di informazioni sugli angoli di fase; la potenza della radiazione permette inoltre l'uso di cristalli anche di dimensioni molto piccole (inferiori a 100 µm) e/o la riduzione dei tempi di esposizione del cristallo (particolarmente importante per proteine molto sensibili alla radiazione); le intensità diffratte possono essere misurate in tempi brevissimi, permettendo così di seguire in tempo reale le variazioni che possono intervenire in una proteina in seguito a reazioni con effettori specifici.
b) Risonanza magnetica nucleare.
Negli ultimi venti anni è andata sempre crescendo l'importanza delle informazioni strutturali ottenibili mediante i metodi di risonanza magnetica nucleare (RMN). Molti nuclei, e tra questi il protone 1H, hanno un momento di dipolo magnetico associato a un momento angolare intrinseco di spin. L'applicazione di un campo magnetico esterno genera una serie di livelli energetici, che corrispondono alle possibili orientazioni del momento angolare di spin. La spettroscopia di risonanza magnetica misura le transizioni tra questi livelli indotte da una radiazione elettromagnetica con frequenza che corrisponde alla differenza di energia tra i livelli. Le frequenze delle transizioni dipendono dal campo magnetico applicato e, per valori del campo dell'ordine di 104 ÷ 105 gauss (104 gauss = 1 tesla), le frequenze cadono nell'intervallo delle radiofrequenze dello spettro elettromagnetico; per esempio, per il protone 1H la frequenza di transizione occorre a circa 42,6 MHz per un campo di 104 gauss. Tuttavia, non tutti i protoni presenti in una molecola mostrano risonanza alla stessa frequenza; infatti, per un effetto di schermo causato dalla distribuzione di carica elettronica, l'effetto magnetico su uno specifico nucleo di una molecola, a parità di campo magnetico esterno, dipende dall'intorno chimico del protone; la frequenza caratteristica di ogni protone, rispetto a uno zero di riferimento, è nota come ‛spostamento chimico'. Quando lo spettro RMN è osservato ad alta risoluzione, la riga di assorbimento, dovuta alla risonanza di ogni protone, spesso risulta separata in più righe per effetto della interazione con protoni vicini; la separazione tra le righe dipende da una costante chiamata ‛costante di accoppiamento'. Per molecole in soluzione l'effetto è dovuto a un accoppiamento indiretto degli spin nucleari, trasmesso attraverso gli elettroni di legame, e diminuisce rapidamente all'aumentare del numero di legami che separano i due nuclei; i valori delle costanti di accoppiamento tra protoni separati da tre legami possono essere correlati all'angolo di torsione intorno al legame centrale e sono quindi una preziosa fonte di informazioni strutturali. Un secondo aspetto importante per lo studio della struttura tridimensionale è legato a esperimenti che mettono in evidenza effetti dovuti a interazioni dipolo-dipolo: questi effetti sono fortemente dipendenti dalla distanza tra i protoni coinvolti (3 ÷ 6 Å) e forniscono informazioni sulle distanze tra atomi non vicini nella sequenza chimica, e quindi sul ripiegamento della macromolecola.
Naturalmente il primo stadio di una risoluzione strutturale consiste in una corretta assegnazione delle singole risonanze a specifici protoni della macromolecola. La risoluzione di questo problema, nel campo delle macromolecole, è stata resa possibile da una serie di sviluppi tecnici e metodologici, quali: 1) la costruzione di spettrometri con campi magnetici elevati, che permettono di raggiungere frequenze dell'ordine di 400 ÷ 800 MHz; 2) l'uso della trasformata di Fourier (FT), che permette di ottenere non solo lo spettro in funzione delle frequenze, a partire dalla misura (in funzione del tempo) dell'assorbimento di un impulso di brevissima durata di radiazione a radiofrequenza, ma anche, a parità di tempo, un incremento notevolissimo della sensibilità e accuratezza della misura; 3) l'estensione di questa tecnica con l'aggiunta di un secondo o anche di un terzo impulso, applicati in successione secondo varie metodologie (spettri bi- e tridimensionali).
Una volta risolto il problema della corretta assegnazione, è possibile raccogliere tutte le informazioni riguardanti le distanze tra protoni, che, insieme alle informazioni sugli angoli di torsione, possono essere utilizzate per ottenere una conformazione complessiva della macromolecola; la conformazione così ricavata può essere successivamente affinata utilizzando anche criteri energetici. Poiché i dati RMN sono ottenuti in soluzione, essi contengono informazioni riguardanti anche fluttuazioni nel tempo della conformazione dominante, che è possibile simulare mediante metodi di dinamica molecolare: i risultati di queste simulazioni sono espressi in termini di una serie di conformazioni, piuttosto che di una struttura unica che è di norma il risultato della determinazione strutturale mediante diffrazione dei raggi X. Le tecniche RMN possono inoltre essere applicate a macromolecole per le quali non è possibile ottenere cristalli singoli e in condizioni che sono più vicine a quelle fisiologiche.
4. Risultati
a) Macromolecole di sintesi.
Molti polimeri lineari mostrano proprietà tipiche di sostanze completamente amorfe, come si può evincere, per esempio, dalle caratteristiche del diagramma di diffrazione e dall'assenza di un punto di fusione netto. A bassa temperatura i polimeri amorfi sono duri e fragili: le catene sono rigidamente bloccate e gli unici movimenti atomici sono quelli determinati dai moti vibrazionali. All'aumentare della temperatura lo stato amorfo presenta una transizione del secondo ordine, detta transizione vetrosa; al disopra della temperatura di transizione iniziano moti coordinati delle catene polimeriche che portano a un graduale rammollimento del materiale. Per questi sistemi, la quasi totalità delle informazioni strutturali (in termini di modi di concatenamento delle unità costitutive, difetti, gruppi terminali, irregolarità di tipo stereochimico, composizione dei copolimeri) è stata ottenuta mediante indagini spettroscopiche. In particolare, la spettroscopia vibrazionale e quella di risonanza magnetica hanno fornito informazioni dettagliate concernenti microirregolarità di catena che sono di notevole importanza per la comprensione dei meccanismi di crescita della macromolecola. Tuttavia, una descrizione della struttura dei polimeri amorfi mediante modelli che permettano di collegare la struttura microscopica alle proprietà macroscopiche è ancora notevolmente incompleta e rappresenta un tema di ricerca tra i più attuali e stimolanti.
Contrariamente ai polimeri amorfi, i polimeri cristallini presentano immagini di diffrazione tipiche di sostanze ben ordinate e punti di fusione entro intervalli molto ristretti di temperatura. Tuttavia, anche i polimeri cristallini contengono quantità variabili di materiale amorfo e presentano, a temperatura più bassa di quella di fusione, una transizione vetrosa che è imputabile alla parte amorfa. Quando i polimeri sono cristallizzati da fuso, si formano cristalliti microscopici che sono parte integrante della massa polimerica. In queste condizioni il polimero mostra un diagramma di diffrazione simile a quello di una polvere cristallina; in seguito a stiramento, i cristalliti tendono a orientarsi lungo la direzione di stiro e danno luogo a un diagramma di diffrazione di fibra. Per lenta precipitazione da soluzioni diluite spesso è possibile ottenere cristalli singoli lamellari, di dimensioni piccole ma sufficienti per poterli osservare mediante microscopia elettronica: le lamelle hanno dimensioni di qualche µm con uno spessore dell'ordine di un centinaio di Å (v. fig. 6A). Mediante diffrazione di elettroni è stato possibile accertare che gli assi di catena sono normali alla lamella; questo risultato sorprendente, in considerazione della lunghezza media delle catene (~ 104 Å) e dello spessore delle lamelle, implica che le catene devono ripiegarsi varie volte nel cristallo. Anche se il ripiegamento può avvenire in maniera regolare, secondo un motivo che coinvolge poche unità monomeriche e collega tratti adiacenti nel cristallo (v. fig. 6B), è probabile che in generale si possano verificare concatenamenti più complessi, in cui sono coinvolti segmenti molto più lunghi che formano regioni di materiale amorfo: per esempio, copolimeri a blocchi di poliossietilene [−CH2−CH2−O−]n e polistirene atattico [−CH2−CH(C6H5)−]m, anche con elevate percentuali di quest'ultimo, formano cristalli singoli lamellari in cui le sequenze non stereoregolari di polistirene sono espulse dalla matrice cristallina e trovano posto nel ripiegamento irregolare alla superficie del cristallo. Nella cristallizzazione da fuso si formano all'interno della matrice cristallina strutture approssimativamente sferiche, che vengono chiamate ‛sferuliti'. Studi di diffrazione elettronica hanno mostrato che gli sferuliti incominciano a formarsi a partire da lamelle cristalline aventi l'asse normale alla lamella orientato ortogonalmente alla direzione radiale di crescita degli sferuliti; catene che attraversano più lamelle formano forti legami tra strati successivi. Il ripiegamento della catena e il rientro nella lamella avvengono in maniera molto più irregolare che nella cristallizzazione da soluzione diluita. Il ripiegamento delle macromolecole durante la cristallizzazione dipende dalla cinetica del processo e, in condizioni che si avvicinano all'equilibrio termodinamico, la lunghezza dei tratti non ripiegati aumenta: cristalli di polietilene ottenuti, dopo una lunga permanenza a elevata temperatura, per lento raffreddamento e sotto elevate pressioni presentano spessori comparabili o maggiori della lunghezza media delle catene. In rari casi la polimerizzazione allo stato solido, a partire da un cristallo singolo del monomero, può dare luogo a polimeri ordinati con catene impacchettate fianco a fianco senza ripiegamenti, secondo un ordine dettato dall'impacchettamento cristallino del monomero: cristalli ben ordinati di poliossimetilene [−CH2−O−]n possono essere ottenuti per polimerizzazione da cristalli singoli di triossano, una molecola ciclica con formula chimica [−CH2−O−]3.
La scoperta della sintesi stereospecifica da parte di G. Natta e collaboratori agli inizi degli anni cinquanta ha aperto un vasto campo per l'applicazione dei metodi di indagine strutturale sia sperimentali che teorici, con l'ambizione ultima di poter disegnare polimeri con proprietà fisiche predeterminate. Il controllo della stereospecificità nell'addizione successiva di unità monomeriche alla catena in crescita, partendo dalle più svariate unità costitutive, ha fornito una ricca serie di campioni stereoregolari adatti per un dettagliato studio strutturale e ha evidenziato il ruolo della struttura spaziale come importante anello di collegamento tra la costituzione chimica dei materiali e le loro proprietà macroscopiche. Il successo di questi studi è certamente dovuto alla forte semplificazione introdotta dalla stereoregolarità della catena polimerica, che permette da un lato la formazione di uno stato solido ordinato, e quindi l'applicazione di metodi molto potenti di indagine quali la diffrazione dei raggi X, e dall'altro riduce drasticamente i tempi di calcolo necessari per effettuare un'analisi conformazionale della macromolecola. L'applicazione di questi metodi, fortemente complementari tra di loro, ha permesso di derivare una serie di principî strutturali utili anche per sistemi in cui il principio di equivalenza non può essere applicato (polimeri stereoregolari in soluzione, polimeri amorfi; v. Corradini e altri, 1980).
Il polietilene, uno tra i polimeri più importanti, possiede una struttura cristallina, estesamente studiata, trans-planare con simmetria 2/1 (v. fig. 7). La molecola di polietilene è stata anche una delle prime macromolecole a essere studiate mediante i metodi dell'analisi conformazionale, con risultati in perfetto accordo con i dati cristallografici: la catena può essere schematizzata come (A)n e il principio di equivalenza impone un solo angolo di torsione θ indipendente. Per il politetrafluoroetilene (CF2)n l'energia conformazionale presenta due minimi simmetrici per valori di θ vicini a 165° e 195°, corrispondenti a eliche enantiomorfe destre e sinistre con simmetria 13/6 e ~ 2,2 unità monomeriche per spira (v. fig. 7). I dati cristallografici hanno mostrato che allo stato solido questo polimero presenta una transizione a 19 °C, che comporta un leggero svitamento dell'elica, la quale adotta una simmetria 15/7; la forma trans-planare, che ha energia leggermente maggiore, è stata trovata in campioni del polimero sottoposti a pressioni elevate.
Di particolare interesse, per l'uso combinato delle tecniche di diffrazione e dell'analisi conformazionale, sono i risultati sui polimeri vinilici stereoregolari (v. Corradini e altri, 1980). Per essi l'energia conformazionale è funzione di due angoli di torsione in catena; la mappa calcolata per il polipropilene isotattico è mostrata nella fig. 8. I minimi di energia a θ1 = - 180°, θ2 = 60° e θ1 = - 60°, θ2 = 180° sono simmetrici e corrispondono a eliche aventi chiralità opposte con 3 monomeri per giro (simmetria 3/1), che possono essere schematizzate con le sequenze conformazionali (T G+)3 (elica sinistra) e (G-T)3 (elica destra). Nella fig. 9A è riportato il modello dell'elica destra del polipropilene isotattico, che è in ottimo accordo con i dati di diffrazione da fibre orientate. L'energia conformazionale di molti polimeri vinilici isotattici presenta minimi vicini a quelli trovati per il polipropilene. Piccole differenze nella posizione dei minimi possono portare a strutture elicoidali con periodi di identità leggermente diversi e con un numero di monomeri per passo compreso tra 3 e 4. Quando il sostituente R (v. cap. 2, § b) è abbastanza ingombrante, può essere necessario tener conto esplicitamente del termine di deformazione degli angoli di valenza nel calcolo dell'energia conformazionale; per il poli-α-butene (R = −CH2CH3), l'inclusione di questo termine nei calcoli di energia causa la separazione del minimo più profondo in più minimi, che spiegano l'esistenza di strutture elicoidali molto simili con simmetria 3/1, 11/3 (3,65 monomeri per spira) e 4/1, trovate in tre distinte forme cristalline del polimero. Per il polipropilene con sequenza sindiotattica, l'energia conformazionale presenta tre minimi approssimativamente isoenergetici: due di essi, simmetrici e con sequenze conformazionali (TTG-G-)2 e (G+G+TT)2, corrispondono a eliche enantiomorfe con simmetria 2/1 e asse di rotazione binario ortogonale all'asse della macromolecola (v. fig. 9B); il terzo minimo corrisponde a una struttura trans-planare. Questo risultato spiega il polimorfismo allo stato solido del polimero, che presenta due modificazioni cristalline corrispondenti alle due conformazioni predette teoricamente; più frequentemente, nei polimeri il polimorfismo è causato da una diversa modalità di impacchettamento delle molecole piuttosto che da una sostanziale diversità della simmetria molecolare.
In presenza di gruppi polari, che possono dare luogo a formazione di legami idrogeno intra- o intercatena, le strutture energeticamente più favorite sono quelle che permettono la formazione del massimo numero di legami idrogeno: questi polimeri presentano punti di fusione molto più alti. Un esempio di struttura con legami idrogeno intercatena è mostrato nella fig. 10, dove è raffigurata la struttura di una poliammide (nailon 66) avente una catena quasi completamente estesa.
Anche le regioni ordinate dei polimeri cristallini spesso presentano un certo grado di disordine che può influenzare profondamente le proprietà meccaniche. Il disordine può essere dovuto a spostamento più o meno casuale delle catene lungo la direzione del loro allungamento, a rotazione rispetto all'asse di catena, alla presenza nella stessa catena di tratti elicoidali destri e sinistri e, in generale, a difetti di stereoregolarità. Il politetrafluoroetilene, di cui si è già parlato, mostra per esempio una seconda transizione a circa 30 °C, legata presumibilmente a un disordine posizionale delle catene. Lo studio del disordine delle zone cristalline può fornire utili indicazioni per i modelli dei polimeri amorfi.
b) Acidi nucleici.
Lo scheletro degli acidi nucleici è formato dalla ripetizione di sei atomi (−P−O5′−C5′−C4′−C3′−O3′−)n, due dei quali, gli atomi C4′ e C3′, fanno parte anche della struttura ciclica del pentosio (v. fig. 2). Assumendo l'equivalenza conformazionale dei nucleotidi, la conformazione della catena è definita da cinque angoli di torsione (contrassegnati da lettere greche in fig. 2), ai quali va aggiunto l'angolo χ che definisce l'orientazione della base rispetto allo zucchero. L'angolo di torsione δ intorno al legame C4′−C3′ può variare entro intervalli ristretti e i valori che può assumere sono strettamente legati alla conformazione dello zucchero (~ 80° per la conformazione C3′-endo del ribosio e ~ 140° per quella C2′-endo). Le interazioni attrattive e repulsive, tra atomi non direttamente legati, limitano notevolmente i valori assunti dagli altri angoli di torsione; inoltre, i dati strutturali su oligonucleotidi indicano che essi sono correlati. L'angolo χ intorno al legame glucosidico può assumere valori vicini a 0° (conformazione sin) o a 180° (conformazione anti): le basi puriniche possono adottare entrambe le conformazioni, mentre le basi pirimidiniche di norma prediligono la conformazione anti. Un altro fattore che diminuisce drasticamente il numero di stati conformazionali energeticamente accessibili agli acidi nucleici è l'accoppiamento antiparallelo di due catene con sequenze complementari, cioè di sequenze di nucleotidi tali per cui a una base purinica G o A su una catena corrisponde rispettivamente una base pirimidinica C o T (U) sull'altra, e viceversa; la complementarità delle basi permette la formazione di legami idrogeno specifici tra A e T (U) e tra C e G. L'unicità e l'importanza di questo accoppiamento, rispetto ad altri possibili, risiedono nei parametri geometrici che collegano le coppie di basi all'atomo C1′ delle rispettive catene: le coppie A/T, T/A, C/G e G/C sono da questo punto di vista sufficientemente isomorfe da poter essere interscambiate senza perturbare significativamente la struttura delle catene; pertanto, nella condizione di complementarità delle due catene, la mancanza di regolarità nella successione delle basi non invalida sostanzialmente il principio di equivalenza conformazionale.
La comprensione di tutti questi fattori, che limitano drasticamente le possibili conformazioni dell'unità nucleotidica, congiuntamente alle caratteristiche del diagramma di diffrazione da fibra, portò nel 1953 J. D. Watson e F. H. C. Crick a proporre la struttura a doppia elica per il DNA. Oltre alla formazione dei legami idrogeno tra le coppie di basi, uno dei fattori più importanti di stabilizzazione per questa struttura è la forte sovrapposizione tra le coppie di basi successive lungo l'asse dell'elica: questo effetto dipende dalla geometria della sovrapposizione ed è maggiore, per geometrie simili, per la coppia C/G rispetto alla coppia A/T. Per questo motivo, i DNA con un contenuto più alto di basi C e G sono più stabili e fondono a temperature più alte. Il volume di ingombro della doppia elica è assimilabile a quello di un cilindro, lungo la cui superficie laterale è possibile individuare due solchi elicoidali di dimensioni diverse: in entrambi i solchi ognuna delle quattro possibili coppie di basi è esposta al solvente e può essere riconosciuta in maniera univoca per la diversità degli atomi esposti. Molecole di dimensioni opportune possono interagire specificamente con le basi senza alterare sostanzialmente la struttura a doppia elica.
Fino all'inizio degli anni ottanta, le informazioni strutturali sugli acidi nucleici sono state ottenute essenzialmente dai dati di diffrazione su fibre orientate di DNA, RNA e polinucleotidi sintetici, oltre che dai dati di diffrazione su cristallo singolo di mono- e dinucleotidi. Con lo sviluppo di metodi nuovi per la sintesi di frammenti di DNA, aventi lunghezza e sequenza controllata, è stato possibile produrre oligonucleotidi autocomplementari in quantità e purezza sufficienti per la preparazione di cristalli singoli; gli studi cristallografici ad alta risoluzione di questi nucleotidi, oltre a fornire informazioni dettagliate sulle strutture già note del DNA, hanno anche permesso la scoperta di un nuovo tipo di struttura sinistrorsa, chiamata struttura Z (v. Wang e Rich, 1985). In condizioni di bassa forza ionica è stabile la forma B (v. Dickerson e altri, 1985), caratterizzata da una simmetria 10/1 e da una conformazione C2′-endo del desossiribosio; le basi giacciono in piani praticamente normali all'asse dell'elica e lo spostamento assiale h, tra coppie di basi successive, è di 3,38 Å (v. fig. 11). Nella forma A, stabile per bassi valori di umidità relativa, il desossiribosio adotta la conformazione C3′-endo e ciò porta a un leggero svitamento della struttura, che assume la simmetria 11/1, e a una modifica della orientazione delle basi, il cui asse risulta inclinato di circa 20° rispetto all'asse dell'elica; la struttura diventa più larga e più corta (h = 2,56 Å) e mostra anche notevoli variazioni nella larghezza e nella profondità dei solchi sulla superficie laterale della doppia elica. Queste variazioni sono anche responsabili del diverso grado di idratazione delle due strutture: nella forma B il solco maggiore è rivestito da un monostrato di acqua di idratazione, mentre il solco minore, più profondo e stretto, contiene una catena di molecole di acqua ben ordinate, legate l'una all'altra mediante legami idrogeno. È stata avanzata l'ipotesi che questa struttura di molecole di acqua svolga un ruolo importante per la stabilizzazione della forma B in condizioni di elevata umidità relativa (~ 95%) e che la sua rottura sia un primo passo necessario per la transizione alla forma A (v. Dickerson e altri, 1985).
La scoperta della forma Z sinistrorsa è strettamente legata alla risoluzione della struttura cristallina di oligonucleotidi con sequenze alternanti delle basi C e G. Anche se strutture a doppia elica sinistrorse furono prese in considerazione nel periodo in cui fu proposta la struttura B, il buon accordo di questa con i dati sperimentali portò al convincimento generale che essa fosse la forma dominante nei sistemi biologici. Invero, la forma Z può essere ottenuta da soluzioni con elevata forza ionica e solo con sequenze alternanti di nucleotidi purinici e pirimidinici, tra le quali la sequenza CG è la più favorevole. L'unità strutturale indipendente della forma Z è una coppia di nucleotidi: il nucleotide purinico adotta una conformazione C3′-endo per il desossiribosio e sin per la base, quello pirimidinico preferisce una conformazione C2′-endo per lo zucchero e anti per la base. La doppia elica sinistrorsa ha una simmetria 6/1 (con 12 nucleotidi per spira) e un passo h per nucleotide di 3,70 Å (v. fig. 12); essa è molto più sottile e più alta della forma B (v. fig. 13). L'alternarsi dei parametri conformazionali lungo la catena conferisce un caratteristico andamento a zig-zag, da cui deriva il nome di forma Z. Gran parte del DNA nelle cellule è nella forma B; vi sono tuttavia indicazioni che la forma Z esista in vivo e contribuisca alla regolazione dell'espressione genica attraverso l'interazione con proteine specifiche.
L'acido ribonucleico (RNA) differisce dal DNA per la presenza del ribosio al posto del 2′-desossiribosio e della base uracile (U) al posto della timina (T). Le strutture a doppia elica dell'RNA sono simili alla forma A del DNA; la forma B, mai osservata nell'RNA, è destabilizzata da interazioni troppo strette tra il gruppo ossidrilico in posizione 2′ con la base e i gruppi fosfati vicini. Addotti ibridi RNA-DNA assumono la conformazione A comune a entrambe le molecole. Di norma le molecole di RNA sono molto più corte delle molecole di DNA e non si aggregano. Le molecole di RNA transfer (tRNA) sono formate da ~ 80 nucleotidi e assumono una conformazione ripiegata tenuta insieme dallo stesso tipo di interazioni non covalenti che stabilizzano la struttura a doppia elica del DNA. Nel tRNA le coppie di basi complementari, legate da legami idrogeno, fanno parte di nucleotidi che appartengono alla stessa catena; quattro segmenti corti della molecola hanno una struttura a doppia elica, lasciando due triplette di basi non accoppiate che svolgono un ruolo essenziale nel riconoscimento di una molecola di RNA messaggero e di una molecola di amminoacido. Per il modo specifico di ripiegamento della catena polinucleotidica, la molecola di tRNA ricorda il comportamento delle proteine globulari.
c) Omopolipeptidi e proteine fibrose.
La conformazione di una catena polipeptidica è definita da tre angoli di torsione, di solito indicati con ϕ (intorno al legame N−Cα), ψ (intorno al legame Cα−C) e ω (intorno al legame peptidico C−N tra due residui successivi). Quest'ultimo, per le caratteristiche elettroniche del gruppo peptidico, può assumere di norma valori prossimi a 0° o a 180°; gruppi peptidici cis (ω ≈ 0°) sono stati trovati sperimentalmente in peptidi ciclici o quando il residuo successivo è una prolina; in tutti gli altri casi i valori sperimentali di ω sono distribuiti intorno a 180°, con spostamenti medi da tale valore inferiori a 10°. Quando può essere applicato il principio di equivalenza (catene omopolipeptidiche) e l'angolo ω può essere fissato a 180°, l'energia conformazionale è funzione solo di ϕ e ψ. Per una sequenza stereoregolare, la catena assume una conformazione elicoidale; a differenza dei polimeri vinilici stereoregolari, le conformazioni elicoidali destre e sinistre hanno energia diversa per la presenza (a eccezione della glicina) dell'atomo di carbonio asimmetrico Cα. Nella fig. 14 è riportata l'energia conformazionale della poli-L-alanina, i cui minimi riproducono le conformazioni sperimentali più frequentemente riscontrate per le proteine fibrose; queste proteine sono caratterizzate da una sequenza a lunghi tratti ripetitivi e hanno caratteristiche strutturali abbastanza simili a quelle dei polimeri stereoregolari (v. Ramachandran e Sasisekharan, 1968; v. Liquori, 1969).
Il minimo di energia a ϕ ≈ - 60°, ψ ≈ - 50° corrisponde alla struttura a elica proposta agli inizi degli anni cinquanta da L. Pauling per spiegare i dati di diffrazione di alcune proteine fibrose, quali la miosina, la cheratina dei capelli e il fibrinogeno: queste proteine producono un diagramma di diffrazione con caratteristiche simili, detto di tipo α. Tratti più o meno lunghi di struttura α-elicoidale molto di frequente si riscontrano come parte dell'organizzazione strutturale di proteine globulari. L'α-elica ha simmetria 18/5 (3,6 residui per giro) con un periodo di identità c = 27 Å e un passo h = 1,5 Å per residuo; essa risulta fortemente stabilizzata, oltre che da interazioni di van der Waals, anche da legami idrogeno tra atomi della catena principale e in particolare tra il gruppo carbonilico CO di un generico residuo i e il gruppo ammidico NH del residuo i +4 (v. fig. 17). Il minimo di energia a esso simmetrico (ϕ ≈ 60°, ψ ≈ 50°) corrisponde all'α-elica sinistra che, per un amminoacido L, è meno stabile dell'elica destra di un ammontare che dipende dalle caratteristiche chimiche e dall'ingombro della catena laterale; l'α-elica sinistra è stata osservata solo raramente in alcuni omopolipeptidi di sintesi (per residui aventi la configurazione opposta D, la stabilità relativa delle due eliche si inverte). Anche se la conformazione ad α-elica è alla base della struttura di diverse proteine fibrose, i dettagli strutturali possono essere differenti nei vari casi. Piccole variazioni periodiche degli angoli di torsione determinano una regolare distorsione dell'asse dell'elica, che a sua volta descrive un'elica (superelica); evidenze sperimentali per questa regolare distorsione sono state riscontrate per la struttura della cheratina. Per la miosina è stata proposta una struttura formata dall'intreccio di due supereliche intorno allo stesso asse, che serve probabilmente a conferire una buona resistenza meccanica.
Eliche multiple sono state trovate solo occasionalmente in polimeri sintetici, mentre la loro presenza è frequente in macromolecole biologiche. Un esempio tipico è rappresentato dal collageno, una delle più importanti proteine strutturali dei Vertebrati. La catena del collageno ha una sequenza fortemente ripetitiva del tipo (Gly-X-Y)n, in cui X è essenzialmente un residuo di prolina (Pro) e Y può essere un qualsiasi amminoacido, anche se molto spesso è un residuo di prolina o di idrossiprolina (Hyp). Per questa proteina si hanno informazioni strutturali molto accurate: il modello ottenuto sulla base dell'interpretazione di diagrammi di diffrazione su fibre ha trovato conferma nei risultati di una estesa analisi conformazionale per polimeri regolari aventi la sequenza (Gly-Pro-Pro)n e (Gly-Pro-Hyp)n, sfruttando il principio di equivalenza. Successivamente, è stato anche possibile sintetizzare peptidi modello, relativamente corti, le cui strutture, determinate mediante diffrazione di raggi X su cristallo singolo, hanno pienamente confermato il modello precedentemente proposto e hanno fornito informazioni dettagliate sulla distribuzione delle molecole di acqua legate. La struttura del collageno è formata da tre eliche sinistrorse con simmetria 10/3, che si intrecciano intorno allo stesso asse (v. fig. 15); la tripla elica risultante è destrorsa, con la glicina che punta verso l'interno (la mancanza di spazio per catene laterali più ingombranti richiede necessariamente un residuo di glicina in questa posizione), mentre le catene laterali degli altri due residui della tripletta ripetitiva puntano verso l'esterno. Non vi sono legami idrogeno nell'elica singola, ma la tripla elica è stabilizzata da legami idrogeno tra il gruppo NH della glicina e il gruppo CO della prolina di una catena adiacente. La conformazione dei residui cade nella zona di bassa energia posta a ϕ ≈ - 60°, ψ ≈ 150°, che è rappresentativa anche della conformazione trovata per il polimero della prolina (poliprolina II trans).
L'ampio minimo di energia nella regione ϕ ≈ - 160° e ψ ≈ 160° corrisponde a catene estese di tipo β con simmetria 2/1 e passo per residuo di ≈ 3,6 Å , molto vicino al valore che si otterrebbe per una catena completamente estesa (ϕ = ψ = 180°). La conformazione β è caratteristica di una classe di proteine che danno uno spettro di diffrazione detto appunto β, quali la fibroina della seta, la cheratina delle piume di uccello, ecc. I gruppi CO e NH di ogni residuo si proiettano alternativamente da lati opposti rispetto alla direzione di allungamento della catena e formano legami idrogeno trasversali con catene vicine, in modo simile a quello mostrato nella fig. 10 per il nailon 66: si ha così uno strato di catene, legate da una fitta rete di legami idrogeno, in cui gli assi di catena hanno la stessa orientazione (strati β paralleli) o, alternativamente, orientazioni opposte (strati β antiparalleli). La struttura tridimensionale è formata da una successione di strati, ruotati di 180° intorno all'asse di catena. Le catene laterali di residui successivi nella catena polipeptidica emergono da facce opposte dello strato e svolgono un ruolo rilevante per l'impacchettamento degli strati. La fibroina della seta ha la sequenza (Gly-X)n sostanzialmente ripetitiva, dove X è serina o alanina; per l'alternanza sistematica della glicina, le catene laterali del residuo X emergono tutte da una stessa faccia dello strato β, mentre le catene laterali di glicina, con ingombro molto più piccolo, emergono dalla faccia opposta. Ciò permette un buon impacchettamento tra le facce simili di due strati contigui; la presenza di residui con catene laterali più ingombranti provoca invece la formazione di regioni amorfe.
d) Proteine globulari.
Struttura secondaria. - Per le sequenze eteropeptidiche di proteine globulari, l'analisi conformazionale non può essere condotta con la condizione del postulato di equivalenza, ma richiederebbe un'esplorazione sistematica della coppia di angoli ϕ e ψ di ogni residuo in catena, con tempi di calcolo proibitivi. Inoltre, andrebbe preso in considerazione anche il contributo energetico del solvente, che svolge un ruolo importante nella stabilizzazione della struttura globulare. Per analizzare le strutture energeticamente permesse di una catena polipeptidica è utile prendere in considerazione l'unità strutturale Cα−CO−NH−CαHR−CO−NH−Cα, la cui energia conformazionale - ponendo fisso a 180° l'angolo di torsione intorno ai due legami peptidici - dipende solo da una coppia di angoli ϕ e ψ e può dare utili indicazioni sulle preferenze conformazionali dettate dalle interazioni locali. La mappa per l'unità della L-alanina è abbastanza simile a quella presentata nella fig. 14, calcolata per una catena infinita; i minimi energetici risultano più larghi, specialmente nelle regioni α-elicoidali, per l'assenza degli effetti cooperativi legati alla ripetizione della stessa struttura in un'elica infinita. Questi risultati indicano che le interazioni locali (interne all'unità strutturale sopra considerata) sono di per sé sufficienti a limitare considerevolmente le conformazioni accessibili ai singoli residui di una catena polipeptidica (v. Liquori, 1969).
La complessità dell'approccio teorico nel caso delle proteine globulari è stata felicemente compensata dalla quantità sempre crescente di dati sperimentali ottenuti essenzialmente grazie ai metodi di diffrazione su cristallo singolo. Le strutture di proteine note e depositate presso la Brookhaven Protein Data Bank (PDP), che erano solo 30 nel 1980 e circa 250 nel 1990, sono ormai arrivate a più di 6.800, con un ritmo di crescita divenuto esponenziale negli ultimi anni. La disponibilità di un numero così elevato di dati strutturali ha permesso una serie di analisi statistiche, che tra l'altro hanno fornito criteri per diverse classificazioni delle strutture proteiche (v. Richardson e Richardson, 1989). La distribuzione delle conformazioni sperimentali dei singoli residui (v. fig. 16) mostra un'elevata concentrazione nelle zone che corrispondono a bassi valori dell'energia conformazionale locale. Dai dati sperimentali emerge, inoltre, che certe successioni regolari di conformazioni, più o meno estese, sono altamente favorite e danno luogo a elementi strutturali caratteristici, che vengono indicati come elementi di struttura secondaria di una proteina (con la dizione ‛struttura primaria' si intende la sequenza amminoacidica). Questi motivi strutturali sono correlati a specifiche sequenze amminoacidiche; sulla base di queste e di altre correlazioni è stato possibile sviluppare diversi metodi che permettono di predire, con sempre maggior successo, elementi di struttura secondaria di una proteina a partire dalla sua struttura primaria.
Uno dei motivi più noti e caratteristici di struttura secondaria è la struttura ad α-elica: i tratti in conformazione α-elicoidale possono estendersi da pochi residui fino a comprenderne 30-35 e sono spesso localizzati in regioni relativamente ricche di metionina, acido glutammico, leucina, alanina e povere di prolina, glicina, tirosina e serina. In particolare, la prolina può essere trovata all'inizio di una α-elica ma raramente all'interno, sia perché l'azoto immidico in catena non può formare il legame idrogeno tipico della struttura α-elicoidale, sia perché la catena laterale dà luogo a interazioni steriche sfavorevoli; quando è interna all'elica, la prolina provoca un ripiegamento dell'asse dell'elica (v. fig. 17). Il gruppo peptidico ha un momento dipolare causato dalla polarità dei gruppi NH e CO; tutti i legami idrogeno i +4 → i hanno la stessa orientazione rispetto all'asse dell'elica e i momenti dipolari dei singoli gruppi si sommano per dare un momento dipolare netto positivo nella direzione N-terminale dell'elica. Questo momento di dipolo può spiegare la maggiore probabilità di trovare i residui carichi negativamente nella regione N-terminale e quelli carichi positivamente nella regione C-terminale; inoltre, è nella zona N-terminale che tendono a legarsi preferenzialmente molecole cariche negativamente.
Un secondo esempio comune di struttura secondaria è rappresentato dalla struttura β formata da catene antiparallele, parallele o miste, di cui la prima è di gran lunga la più frequente; di norma la lunghezza delle catene β è inferiore a quella delle α-eliche, anche se le catene associate possono essere numerose. Mioglobina ed emoglobina, le prime proteine studiate cristallograficamente, sono prive di strutture β; con l'aumentare del numero di strutture proteiche note, è apparso chiaro che le strutture β rappresentano una parte sostanziale dell'organizzazione secondaria di una proteina, della quale occupano di norma la parte interna. I dati strutturali indicano che strati β planari sono assai rari; catene β affiancate tendono ad avvitarsi nello spazio, sempre nello stesso senso, per effetto di interazioni di non legame tra le catene laterali (v. fig. 18).
I tratti in α-elica e in struttura β sono connessi da segmenti di catena di varia lunghezza aventi struttura non regolare, nei quali i gruppi polari NH e CO sono spesso esposti all'esterno, dove interagiscono con il solvente. Queste regioni sono costituite prevalentemente da gruppi polari e carichi, e sono quelle in cui più spesso si accumulano variazioni di sequenza in proteine omologhe. Molti di questi tratti sono formati da un piccolo numero di residui (2-5), classificabili in una quantità limitata di conformazioni, e permettono bruschi cambi di direzione della catena: tipico il motivo ‛a forcina di capelli', spesso riscontrato come connessione tra due catene β antiparallele.
Dominî e struttura terziaria. - Il modo in cui gli elementi di struttura secondaria si combinano per dare luogo a uno specifico ripiegamento della catena in una struttura compatta definisce la struttura terziaria della proteina. Il ripiegamento è tale che l'interno della proteina risulta costituito quasi esclusivamente da residui idrofobici e la superficie esterna da residui idrofilici (polari o carichi). In linea con la diversità di funzioni che le varie proteine svolgono, la struttura tridimensionale è varia e complessa; tuttavia, essa risulta meno variabile della struttura primaria: sono note proteine che presentano strutture terziarie simili, pur con sequenze molto diverse. Un certo tipo di ripiegamento coinvolge in alcuni casi tutta la catena polipeptidica; in altri casi, e con una frequenza che aumenta con la lunghezza di catena, questa si ripiega formando più unità strutturali compatte, chiamate dominî, che si ritrovano con caratteristiche sostanzialmente simili anche in proteine molto diverse tra loro. Esistono proteine con dominî ben separati nello spazio e legati solo da uno o pochi segmenti di catena flessibili, altre nelle quali le interazioni tra i dominî sono numerose e danno luogo a una estesa superficie di contatto. La suddivisione in dominî, specie per catene polipeptidiche molto lunghe, può facilitare il processo di ripiegamento nella struttura globulare nativa; inoltre, movimenti relativi dei dominî possono contribuire a rendere più efficiente e selettiva la funzione della proteina. La fig. 19 riporta la struttura dell'enzima esochinasi, in presenza e in assenza del substrato glucosio: il movimento dei due dominî, che si richiudono intorno alla molecola di glucosio, permette un efficiente sequestro del substrato, con formazione di un sito idrofilico altamente selettivo; l'esclusione dell'acqua dal sito attivo risulta determinante per favorire la specifica reazione catalizzata dall'enzima. Altre proteine nelle quali la flessibilità nell'orientazione dei dominî sembra avere un ruolo biologico importante sono quelle che formano il rivestimento esterno dei Virus e gli anticorpi.
La struttura terziaria di una catena polipeptidica, o dei dominî che la compongono, può essere classificata considerando il tipo e il numero di elementi di struttura secondaria, l'architettura o la posizione spaziale relativa di questi elementi, la topologia o il modo in cui essi sono connessi tra di loro. Sulla base di queste caratteristiche, è possibile distinguere tre classi principali di strutture: α antiparallele, β antiparallele e α/β parallele; la classificazione non rispecchia solo la presenza dominante di un determinato tipo di struttura secondaria, ma anche il grado di importanza che essa ricopre nella stabilità complessiva del dominio. Secondo una recente stima, il numero di famiglie di proteine presenti in natura ammonta a poco più di un migliaio, di cui non più di un quarto è stato studiato dal punto di vista strutturale. Non è pertanto sorprendente la scoperta di nuovi ripiegamenti, anche se spesso la novità della struttura consiste in una diversa topologia più che in una diversa architettura.
La classe di strutture α antiparallele è caratterizzata da un insieme di α-eliche strettamente impacchettate con formazione di una regione idrofobica interna, mentre le regioni di connessione e i gruppi polari di eliche esterne interagiscono con il solvente. Molto comune è la sottoclasse di proteine formate da un fascio di quattro eliche approssimativamente isoorientate (v. fig. 20A) e quella delle globine (mioglobina, emoglobina, ficocianine, ecc.) in cui gli assi delle eliche a contatto formano un angolo di circa 50°. Nelle globine otto eliche sono disposte in modo da formare una tasca, dove trova posto il gruppo prostetico eme che lega reversibilmente una molecola di ossigeno (v. fig. 20B). Va sottolineato che nei vari organismi le globine mostrano una diversità di sequenza superiore anche al 75%, pur conservando praticamente inalterata la struttura terziaria; mutazioni anche rilevanti sono compensate da piccoli movimenti relativi dei tratti elicoidali. Alcune proteine di membrana sono organizzate in fasci di eliche antiparallele con asse perpendicolare alla membrana e con lunghezza pari allo spessore della membrana (v. fig. 21; v. Deisenhofer e altri, 1989).
La classe di strutture β antiparallele è caratterizzata da strati di catene β antiparallele (generalmente da 4 a 10); come è stato detto, ogni strato tende ad avvitarsi nello spazio e spesso dà luogo alla formazione di una struttura detta ‛a barile', con le catene laterali idrofobiche che puntano all'interno del barile, il quale costituisce il nucleo della struttura e funge spesso da contenitore per un ligando; si possono individuare più sottoclassi, a seconda della topologia di connessione delle catene che formano il barile. Possono essere annoverate nella classe di strutture β antiparallele molte proteine, diverse delle quali svolgono funzioni di trasporto di piccole molecole (v. biochimica, vol. X). Nella fig. 22A è riportata la struttura della proteina che lega il retinolo: ogni tratto β della catena costituisce una doga del barile, che è collegata alla precedente da una stretta connessione. Recentemente, questa semplice struttura è stata riscontrata in alcune proteine di membrana, come per esempio le porine, trovate nella membrana esterna di batteri gram-negativi, mitocondri e cloroplasti; la membrana protegge la cellula da agenti dannosi quali proteasi, antibiotici, tossine, ecc., e le porine, incluse nella membrana, permettono il passaggio di piccole molecole idrofiliche che sono nutrienti della cellula o prodotti di rifiuto. La porina rappresentata nella fig. 22B è formata da 16 tratti β, tutti antiparalleli, avvitati, come di norma, in senso destro per formare una struttura chiusa a barile, con formazione di un canale interno attraverso cui possono passare piccole molecole senza alcun impedimento sterico. Esistono, e sono anche più numerose delle prime, altre strutture a barile più complesse in cui una doga (o più di una) del barile è connessa alla quarta doga nella sequenza, piuttosto che a quella immediatamente vicina (v. fig. 22C): questa topologia ricorda il motivo decorativo che si trova su antichi vasi greci e, per questo, è denominata ‛barile a chiave greca'. La larga diffusione di questo motivo è probabilmente legata a un favorevole percorso di ripiegamento, che inizia con la formazione di due catene β affiancate le quali, ripiegandosi, formano il motivo a chiave greca. Nella classe delle strutture β antiparallele sono comprese alcune strutture, anch'esse molto diffuse, in cui lo strato β è solo parzialmente avvitato nello spazio: mentre un lato dello strato è coperto da eliche o altri tipi di connessioni, l'altro lato è in contatto con il solvente o interagisce con altri dominî o subunità.
La classe α/β è formata da proteine che includono sequenze, approssimativamente alternate, di tratti α e tratti β che possono generare una struttura chiusa a barile o una struttura aperta. Nel primo caso è molto frequente la formazione di un barile costituito da otto catene β parallele con i tratti di connessione a elica situati all'esterno del barile; nel secondo si ha uno strato di catene β parallele con le eliche disposte da entrambi i lati dello strato. Molti enzimi glicolitici hanno una struttura α/β: un esempio classico è fornito dalla triosofosfatoisomerasi (TIM), in cui le otto catene β formano la struttura a barile interna, circondata da otto α-eliche (v. fig. 23). La lunghezza e la conformazione delle regioni di connessione possono variare notevolmente e conferiscono specificità di funzione alle varie proteine con questo tipo di struttura terziaria. Molto diffuso è anche il ripiegamento a struttura aperta: a questa sottoclasse appartiene il dominio che lega nucleotidi, trovato in numerose deidrogenasi e nella flavodossina, e formato da sei segmenti paralleli β interconnessi da quattro α-eliche.
Struttura quaternaria. - Molte proteine sono oligomeriche, sono cioè formate da poche (anche solo due) subunità la cui relazione spaziale definisce la loro struttura quaternaria. L'associazione di più subunità in una struttura unica è una caratteristica diffusa dell'organizzazione cellulare per i vantaggi che offre: fornisce un più favorevole rapporto superficie/volume, maggiore economicità nella informazione genetica ed efficienza nell'eliminazione di errori, e inoltre facilita i processi connessi con l'evoluzione molecolare. Nei numerosi esempi di proteine formate da più subunità identiche, queste tendono a disporsi nello spazio secondo precise relazioni di simmetria risultanti da specifiche interazioni tra le subunità. Deviazioni dalla esatta simmetria possono spesso verificarsi a livello della struttura terziaria, per interazioni repulsive che hanno luogo vicino agli elementi di simmetria; queste possono essere rilassate mediante una variazione conformazionale di uno dei segmenti delle due subunità che vengono a trovarsi a stretto contatto nella struttura quaternaria. In oligomeri formati da subunità identiche è rara l'aggregazione non simmetrica: un esempio è fornito dalla struttura dimerica dell'esochinasi, le cui subunità sono poste in relazione da una rotazione di 156° e da una traslazione di 13,8 Å lungo l'asse di rotazione, anziché da un asse di rotazione di 180° con componente traslazionale nulla. Proteine formate da subunità non uguali non hanno i requisiti per un'aggregazione simmetrica; tuttavia se le subunità sono simili, come nel caso delle emoglobine di Mammiferi formate da quattro subunità uguali a coppie (α2β2), esse sono spesso legate da operazioni di simmetria, anche se solo approssimate.
In diverse proteine oligomeriche l'affinità di una subunità verso un ligando è influenzata dalla presenza delle altre subunità (effetti cooperativi); nell'emoglobina la cooperatività è positiva, in quanto il legarsi di una molecola di ossigeno a un gruppo eme aumenta l'affinità degli altri gruppi eme per l'ossigeno. Gli effetti cooperativi possono essere mediati da piccole molecole che interagiscono con siti della proteina diversi da quello che lega il substrato; tali molecole sono chiamate ‛effettori allosterici' e la proteina è definita ‛allosterica'. La regolazione dell'attività catalitica o di trasporto attraverso gli effetti cooperativi tra subunità è tra i vantaggi più significativi determinati dall'associazione in strutture oligomeriche. Secondo il modello proposto da J. Monod, J. Wyman e J.-P. Changeux (v., 1965), gli effetti cooperativi sono legati all'esistenza di due diverse strutture quaternarie in mutuo equilibrio: una struttura T (o tesa) avente una modesta affinità verso il substrato, in cui le subunità sono vincolate da forti interazioni che si oppongono alle variazioni (generalmente piccole) della struttura terziaria, necessarie per accomodare il substrato; una struttura R, più attiva, in cui questi vincoli sono rilassati. Un effettore allosterico è capace di spostare l'equilibrio tra le due forme, favorendo la forma attiva R (attivatore) o la forma tesa T (inibitore). Nella transizione da T a R la simmetria è conservata e tutte le subunità in un dato stato sono equivalenti (transizione concertata). Il modello sequenziale di D. E. Koshland (v., Koshland e altri, 1966) non impone questa restrizione, e ogni subunità può variare la sua struttura terziaria nel legarsi con il substrato e quindi influenzare l'attività delle subunità vicine. Anche se il modello concertato fu sviluppato quando era nota solo la struttura a bassa risoluzione dell'emoglobina, allo stato attuale sono note in dettaglio le strutture di diversi enzimi che presentano strutture quaternarie diverse e riconducibili agli stati T e R predetti dal modello allosterico; alcuni esempi sono forniti dagli enzimi glicogenofosforilasi, asparticotranscarbammilasi e fosfofruttochinasi. Per ognuno di questi enzimi sono noti le strutture ad alta risoluzione delle forme T e R e, in parte, i meccanismi stereochimici che permettono la transizione tra le due forme.
Sotto il profilo evoluzionistico l'acquisizione di una struttura quaternaria a partire da una proteina monomerica è un processo lungo. Esistono esempi in cui è sufficiente una singola sostituzione per la stabilizzazione di aggregati: l'emoglobina S - forma mutata dell'emoglobina umana responsabile dell'anemia a cellule falciformi, con una singola sostituzione in posizione 6 della catena β - forma lunghi aggregati elicoidali la cui struttura è stata estesamente studiata (v. Perutz, 1992). Tuttavia, in generale sono necessarie più sostituzioni per lo sviluppo nel monomero di una regione superficiale che possa favorire la formazione di una struttura aggregata attraverso una serie di contatti energeticamente favorevoli con la stessa o una diversa regione di un altro monomero. Recentemente è stato scoperto che alcune proteine dimerizzano attraverso un processo mai osservato in precedenza, che comporta lo scambio reciproco della posizione spaziale di una parte della catena tra le due subunità; in altre parole, un segmento di catena T avente una caratteristica struttura secondaria, quale un tratto α-elicoidale N- o C-terminale o anche un intero dominio della struttura terziaria del monomero, si stacca dalla rimanente parte B del monomero e va a occupare una posizione equivalente rispetto a B del secondo monomero. Si ha così la formazione di una estesa superficie di contatto T/B, identica a quella formata nella struttura monomerica, che stabilizza la forma aggregata in quanto i dominî T e B appartengono a monomeri diversi; questo scambio dei dominî tra le due subunità può essere reso possibile da una piccola modifica in un tratto relativamente corto della catena polipeptidica, che funziona da cerniera per lo scambio. La prima proteina nella quale è stato osservato un tale meccanismo è una ribonucleasi dimerica, che possiede un'elevata omologia di sequenza con la ribonucleasi pancreatica, enzima formato da una sola subunità (v. Mazzarella e altri, 1993). La struttura tridimensionale dell'enzima dimerico, pur essendo molto simile a quella della ribonucleasi pancreatica bovina, presenta il tratto N-terminale in α-elica scambiato tra le due subunità (v. fig. 24). Dopo questo primo risultato, sono state scoperte altre proteine che si aggregano mediante questo meccanismo; è probabile che lo scambio di dominî rappresenti, nel percorso evolutivo, una scorciatoia che permette di acquisire di una maggiore complessità strutturale e nuove funzioni biologiche.
5. Considerazioni conclusive
La notevole mole di dati strutturali su macromolecole sintetiche, ottenuti con svariati metodi chimico-fisici, ha svolto un ruolo determinante nell'indirizzare la ricerca verso la produzione di numerosi nuovi prodotti di largo interesse applicativo. Le conoscenze strutturali sono state, per esempio, particolarmente utili nello sviluppo di polimeri capaci di dare luogo alla formazione di uno stato ‛liquido cristallino', in cui la disposizione ordinata delle macromolecole si estende lungo una o due direzioni dello spazio anche per distanze relativamente grandi. Altri esempi riguardano i polimeri conduttori e quelli che presentano proprietà ottiche non-lineari, un campo in fase di rapido sviluppo finalizzato alla produzione di prodotti ad alta tecnologia, e infine materiali con elevata resistenza meccanica e termica ed elevata stabilità verso agenti corrosivi. Le nuove metodologie di sintesi permettono ora la produzione di macromolecole costituite da una gamma sempre più varia di unità monomeriche; tuttavia, resta ancora lontana la possibilità di sintetizzare polimeri con una sequenza programmata di unità costitutive diverse. Anche per quest'ultimo aspetto, i risultati più straordinari sono stati ottenuti in anni recenti nel campo delle macromolecole biologiche ed estremamente interessanti appaiono le prospettive future, a giudicare dal numero di ricercatori e di mezzi che la comunità scientifica internazionale sta sempre più massicciamente dedicando allo sviluppo di ricerche in questo campo. L'uso di sorgenti di raggi X molto potenti, quali la luce di sincrotrone, ha permesso di affrontare lo studio di sistemi molto complessi; il modello mostrato nella fig. 21, ad esempio, rappresenta il centro di reazione che fa parte del fotosistema di un batterio fotosintetizzante - un complesso formato da una molecola di citocromo c, tre subunità e diversi gruppi prostetici - la cui risoluzione strutturale ha permesso una prima importante ricostruzione dell'organizzazione dei sistemi coinvolti nella fotosintesi. Diversi gruppi di ricercatori sono impegnati nello studio strutturale delle subunità grande e piccola di ribosomi batterici mediante metodi cristallografici e di ricostruzione di immagine da microscopia elettronica. I ribosomi sono la sede della sintesi proteica e il ribosoma di un batterio è formato da circa 2,5 × 105 atomi (corrispondente a un peso molecolare di circa 2,3 × 106 dalton); di essi circa i due terzi fanno parte di 3 catene di RNA ribosomiale, mentre i restanti sono distribuiti in circa 57 proteine differenti.
La delucidazione strutturale di proteine coinvolte nella regolazione dell'espressione genica e dei complessi tra queste e oligomeri di DNA ha mostrato l'esistenza di motivi strutturali comuni - quali il motivo elica-gomito-elica (helix-loop-helix) e quello che lega ioni zinco (zinc-finger), che riconoscono specifiche sequenze nucleotidiche - e ha portato a una migliore comprensione del modo in cui queste proteine si legano al DNA, provocando deformazioni locali della struttura a doppia elica (v. anche chimica bioinorganica, vol. X). Le curvature della forma canonica B, provocate da specifiche sequenze nucleotidiche, alterano la larghezza relativa dei solchi della doppia elica e sembrano essere importanti nel primo stadio dei processi di riconoscimento tra DNA e proteine specifiche. Alcune regolarità nelle distorsioni dalla forma canonica B sono osservabili nelle strutture cristalline di oligonucleotidi a doppia elica e possono essere predette teoricamente con i metodi dell'analisi conformazionale (v. De Santis e altri, 1988). Ancora modeste sono le conoscenze sulle interazioni tra proteine e RNA: i complessi tra tRNA e le corrispondenti amminoacilsintetasi svolgono un ruolo primario nel processo di traduzione del codice genetico nella sequenza amminoacidica di una proteina e saranno un importante tema di ricerca nel prossimo futuro. Lo sviluppo dei metodi di risoluzione strutturale si è felicemente coniugato con quello dei metodi che, attraverso le tecniche del DNA ricombinante (v. biologia molecolare, vol. VIII), hanno reso attuabili modifiche mirate della struttura primaria delle proteine: la determinazione strutturale e la mutagenesi sito-specifica fanno ora parte, sempre più di frequente, dei programmi integrati di ricerca di numerosissimi laboratori universitari e industriali, e hanno rivoluzionato gli studi sulle relazioni tra struttura e funzione, permettendo una verifica diretta delle ipotesi riguardanti il ruolo funzionale di residui specifici. Questa attività di ricerca ha già portato a risultati di vasta portata nel campo industriale, agricolo e in quello medico (v. biotecnologie, voll. VIII e X) e lascia prevedere successi ancora più importanti nei prossimi anni.
BIBLIOGRAFIA
Corradini, P., Guerra, G., Pirozzi, B., Present status of the configurational and conformational analysis of stereoregular polymers, in Preparation and properties of stereoregular polymers (a cura di R. W. Lenz e F. Ciardelli), Dordrecht-Boston 1980, pp. 317-352 e pp. 387-405.
Deisenhofer, J., Huber, R., Michel, H., The structure of the photochemical reaction center of Rhodopseudomonas viridis and its implications for function, in Prediction of protein structure and the principles of protein conformation (a cura di G. D. Fasman), New York 1989, pp. 99-116.
De Santis, P., Giglio, E., Liquori, A. M., Ripamonti, A., Van der Waals interactions and the stability of helical polypeptide chains, in ‟Nature", 1965, CCVI, pp. 456-458.
De Santis, P., Palleschi, A., Savino, M., Scipioni, A., A theoretical model of DNA curvature, in ‟Biophysical chemistry", 1988, XXXII, pp. 305-317.
Dickerson, R. E., Kopka, M. L., Pjura, P., Base sequence, helix geometry, hydration, and helix stability in B-DNA, in Biological macromolecules and assemblies, vol. II, Nucleic acids and interactive proteins (a cura di F. A. Jurnak e A. McPherson), New York 1985, pp. 37-126.
Koshland, D. E., Nemethy, G., Filmer, D., Comparison of experimental binding data and theoretical models in protein containing subunits, in ‟Biochemistry", 1966, V, pp. 365-385.
Kraulis, P. J., MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures, in ‟Journal of applied crystallography", 1991, XXIV, pp. 946-950.
Liquori, A. M., Stereochemical code of aminoacid residues in polypeptides and proteins, in Symmetry and function of biological systems at the macromolecular level. Proceedings of the XI Nobel symposium (a cura di A. Engström e B. Strandberg), Stockholm-New York 1969.
Mazzarella, L., Capasso, S., Demasi, D., Di Lorenzo, G., Mattia, C. A., Zagari, A., Bovine seminal ribonuclease: structure at 1.9 Å resolution, in ‟Acta crystallographica" (Section D), 1993, XLIX, pp. 389-402.
Monod, J., Wyman, J., Changeux, J.-P., On the nature of allosteric transitions: a plausible model, in ‟Journal of molecular biology", 1965, XII, pp. 88-118.
Perutz, M., Protein structure: new approaches to disease and therapy, New York 1992.
Ramachandran, G. N., Sasisekharan, V., Conformation of polypeptides and proteins, in Advances in protein chemistry (a cura di M. L. Anson e J. T. Edsall), New York 1968, pp. 283-437.
Richardson, J. S., Richardson, D. C., Principles and patterns of protein conformation, in Prediction of protein structure and the principles of protein conformation (a cura di G. D. Fasman), New York 1989, pp. 1-98.
Wang, A. H.-J., Rich, A., The structure of the Z form of DNA, in Biological macromolecules and assemblies, vol. II, Nucleic acids and interactive proteins (a cura di F. A. Jurnak e A. McPherson), New York 1985, pp. 127-170.