
Biotecnologie
Biotecnologie
sommario: 1. Introduzione. 2. Reazione di polimerizzazione a catena: a) principî generali; b) applicazioni. 3. Analisi dei genomi: a) principî e problematiche generali; b) sequenziamento del genoma di lievito; c) mappatura del genoma di topo; d) mappatura del genoma umano. 4. Terapia genica: a) aspetti tecnologici; b) applicazioni in medicina umana. 5. Biotecnologie e risorse genetiche: a) aspetti della biodiversità; b) applicazioni biotecnologiche nel settore della biodiversità. □ Bibliografia.
1. Introduzione
Nei primi anni novanta le biotecnologie hanno ricevuto un notevole impulso, sia dalle nuove conoscenze di base in campo biologico, sia dallo sviluppo e dall'ottimizzazione di tecniche che in parte rappresentano la naturale evoluzione di tecnologie in precedenza descritte (v. biotecnologie, vol. VIII) e in parte costituiscono un capitolo del tutto nuovo nella storia di questo settore.
Processi o prodotti derivati dall'impiego delle biotecnologie sono ormai utilizzati con grande successo in aree che vanno dalla nutrizione alla conservazione della variabilità biologica, dalla depurazione di ambienti contaminati ai modelli animali che simulano malattie umane fino alla terapia genica di malattie quali il cancro o l'AIDS. Contemporaneamente, alcune discipline che prima rivestivano un ruolo marginale sono divenute strategiche per la ricerca biotecnologica: la bioinformatica, ad esempio, è ormai uno strumento necessario per il progresso di queste ricerche e la mole di dati da immagazzinare, elaborare, confrontare sta diventando così imponente da spingere organizzazioni transnazionali come l'Unione Europea a cercare di armonizzare le procedure e i prodotti informatici risultanti da questo settore.
Il progresso, a volte tumultuoso, in alcuni settori si ripercuote quasi immediatamente in altri, rendendo particolarmente arduo il compito di mantenersi aggiornati. A prescindere dall'evoluzione dei prodotti farmaceutici già immessi in commercio (v. tab. I), è in corso di valutazione clinica una serie notevole di altri ritrovati terapeutici il cui sviluppo è stato reso possibile dall'impiego delle biotecnologie (v. tab. 2). Si è ritenuto quindi utile dedicare questo articolo di aggiornamento alla trattazione di alcuni grandi settori di sicuro interesse strategico, in quanto il loro evolversi è necessariamente legato al miglioramento di conoscenze e tecnologie che tra loro interagiscono perseguendo un obiettivo comune. I capitoli del presente articolo sono pertanto dedicati il primo esclusivamente alla reazione di polimerizzazione a catena del DNA, una tecnica che sta segnando gli anni novanta e che del tutto verosimilmente continuerà a influenzare la ricerca e le applicazioni della biologia ancora per lungo tempo; gli altri tre capitoli sono dedicati, rispettivamente, alla mappatura e al sequenziamento di genomi di Eucarioti, alla terapia genica e infine alle biotecnologie per lo studio e la difesa delle risorse genetiche.
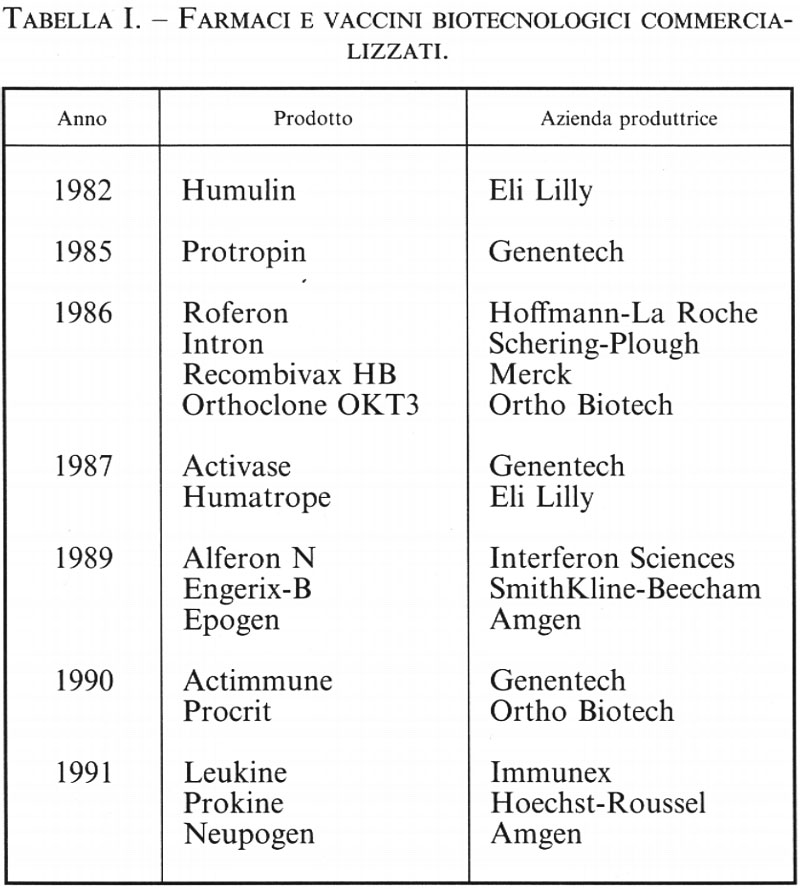

Due considerazioni fondamentali valgono a giustificare questa scelta che, a prima vista, può sembrare riduttiva; anzitutto, in questi campi sono impegnati migliaia di ricercatori e centinaia di aziende biotecnologiche di varie dimensioni in tutto il mondo; in secondo luogo alcuni di questi progetti presuppongono accordi internazionali, dato che impiegano ingenti risorse finanziarie erogate dai paesi più industrializzati. I settori che verranno presi in considerazione sono in pratica classificabili come big science projects, un'espressione inglese usata per definire l'ordine di grandezza di iniziative che impiegano discipline d'avanguardia e possono pertanto divenire delle vere e proprie pietre miliari nella storia dell'umanità e quindi influenzarne il futuro. Progetti come il sequenziamento del genoma umano sono analoghi per importanza a quelli relativi alla fusione nucleare o all'identificazione delle particelle subnucleari, obiettivi che presuppongono la conduzione e il coordinamento di subprogetti in discipline molto diverse tra loro.
2. Reazione di polimerizzazione a catena
a) Principî generali
L'area tecnologica che ha avuto il maggior impulso e che ha esercitato un'influenza determinante tanto sulla ricerca di base quanto su quella applicata rimane senza dubbio la tecnologia degli acidi nucleici, in virtù del fatto che essa ha potuto beneficiare dello sviluppo della tecnica chiamata reazione di polimerizzazione a catena (PCR, Polymerase Chain Reaction).
Nella storia del progresso scientifico è spesso accaduto che le aspettative alimentate dalle scoperte non trovassero, per vari motivi, una realizzazione adeguata e molti avanzamenti tecnologici sono così rimasti fine a se stessi. La PCR, al contrario, costituisce senza ombra di dubbio uno degli esempi più eclatanti di come una metodologia possa assumere un ruolo fondamentale nella ricerca biologica. La specificità della reazione, che offre la possibilità di ottenere copie teoricamente infinite di un particolare gene - e quindi dei ‛difetti' a esso associati -, ha trovato immediati riscontri in ogni settore delle biotecnologie: dalla diagnostica di laboratorio alla microbiologia, all'oncologia, alle malattie ereditarie, alla medicina legale, alla paleontologia, ecc.
La PCR consiste nell'amplificazione enzimatica in vitro di una definita sequenza di DNA. Questa amplificazione viene ottenuta mediante cicli ripetuti di polimerizzazione di desossiribonucleotidi in cui il prodotto di reazione diventa il reagente dell'amplificazione successiva, rendendo così possibile che il ciclo si ripeta un numero n di volte. Per poter utilizzare questo metodo è necessario conoscere la sequenza del DNA della regione interessata all'amplificazione, o almeno di due tratti limitati di DNA ai suoi estremi 3′ e 5′, allo scopo di sintetizzare due brevi sequenze di DNA complementari che servono da innesco o iniziatore (primer) della reazione di amplificazione.
Prima di descrivere in dettaglio la PCR, è importante ricordare che i legami idrogeno nella molecola del DNA possono essere distrutti dal calore o da valori elevati di pH. Tuttavia, in seguito a raffreddamento o ad abbassamento del pH i due filamenti tendono a riassociarsi o ibridare l'uno con l'altro, tornando ad assumere la conformazione a doppia elica.
Fisiologicamente, la separazione dei due filamenti della doppia elica del DNA avviene durante la sua replicazione. I desossiribonucleotidi liberi si appaiano in modo complementare nel momento in cui le due catene di DNA vengono separate, fungendo da stampo per la sintesi di filamenti complementari.
La reazione di sintesi del DNA è catalizzata da un enzima, una DNA-polimerasi, scoperto nel 1955 da A. Kornberg (v., 1974). Le DNA-polimerasi possono allungare una corta sequenza di nucleotidi, il primer, legando in successione i desossiribonucleotidi trifosfati all'estremità 3′ del primer in crescita. Il primer si trova ovviamente già ibridato alla sequenza complementare del filamento che fa da stampo. Sfruttando questo principio si ha la sintesi unidirezionale di DNA, una metodologia ampiamente usata (v. Sanger e altri, 1987) per risolvere la sequenza del DNA dei geni finora identificati.
Alcuni anni fa, precisamente nel 1986, Kary Mullis - allora biochimico presso un'azienda biotecnologica californiana, la Cetus Corporation - ebbe l'idea di usare un paio di primers complementari a due sequenze nucleotidiche su ciascun filamento di DNA e delimitanti una regione genomica di interesse (v. Mullis e Faloona, 1987). Una reazione di questo genere porta alla sintesi, in direzioni opposte, di due molecole di DNA a doppia elica che, denaturate, possono poi diventare lo stampo per altri filamenti in una successione esponenziale. Questo è il semplice principio che sta alla base della reazione di polimerizzazione a catena o PCR.
Un ciclo di amplificazione consiste schematicamente di tre fasi ed è preceduto da una fase in cui il DNA del campione in esame viene estratto e digerito con diversi enzimi di restrizione secondo metodiche già descritte (v. biotecnologie, vol. VIII). La prima fase prevede la denaturazione ad alta temperatura della doppia elica di DNA, allo scopo di separare i due filamenti. La seconda fase consiste nel legame dei primers, in precedenza aggiunti in eccesso alla miscela di reazione, alle sequenze di DNA a essi complementari. Il terzo stadio è rappresentato dalla sintesi del DNA a opera dell'enzima DNA-polimerasi.
In virtù della loro alta concentrazione, i primers si legano rapidamente alle sequenze loro complementari, anticipando, quando la temperatura viene abbassata, la loro riassociazione. A seguito di un successivo riscaldamento della miscela di reazione, anche le catene di DNA neosintetizzate si denaturano aprendosi: viene così reso disponibile l'attacco dei primers per la sintesi di altre catene complementari. Il ciclo quindi riprende nuovamente. In una PCR in cui la resa di amplificazione risulti ottimizzata si può raggiungere un'amplificazione di un fattore 106.
Le prime reazioni di polimerizzazione a catena sono state sviluppate utilizzando il frammento di Klenow della DNA-polimerasi estratta da Escherichia coli. Tuttavia, durante la fase di denaturazione ad alta temperatura la polimerasi veniva inattivata ed era perciò necessario aggiungere nuovo enzima all'inizio del successivo ciclo di amplificazione, il che rendeva il metodo macchinoso e soggetto a errori. Attualmente viene invece impiegata una DNA-polimerasi termostabile, che resiste a temperature di 95 °C, purificata dal batterio Thermus aquaticus, un microrganismo che vive normalmente in sorgenti calde a temperature tra 70 e 75 °C (v. Erlich, 1989). Con l'introduzione di questo nuovo tipo di DNA-polimerasi è stato possibile evitare di aggiungere continuamente enzima alla miscela di reazione e, cosa più importante, automatizzare completamente le varie fasi della PCR. Essa infatti viene ora allestita mediante appositi strumenti programmabili, in grado di variare opportunamente le temperature di reazione all'interno della microprovetta contenente tutti i reagenti necessari al mantenimento della reazione.
Il prodotto finale della reazione di polimerizzazione a catena è una certa quantità di DNA specificamente amplificato, che deve però essere evidenziato e distinto dal DNA non amplificato. A tale scopo si usano varie tecniche: la più comune consiste in una separazione dei frammenti mediante elettroforesi in gel di agarosio, a cui fa seguito una colorazione del DNA con etidio bromuro che evidenzia la banda del prodotto della PCR, in genere molto ben visibile. Questo metodo di visualizzazione può essere integrato o sostituito dall'uso di ibridazioni secondarie con sonde specifiche marcate radioattivamente.
Recentemente sono stati sviluppati i sistemi ELISA (Enzyme-Linked Immunosorbent Assay) che, mediante l'uso di anticorpi monoclonali specifici nei confronti del DNA a doppia elica, sono in grado di determinare i prodotti di amplificazione (v. Mantero e altri, 1991). In questo caso, il DNA prodotto di amplificazione viene ‛catturato' da una sonda a DNA, complementare a sequenze del DNA amplificato, adese a pozzetti di micropiastre. Successivamente questo legame viene rivelato da anticorpi anti-DNA a doppia elica marcati con perossidasi. Questi sistemi sono di uso molto più semplice e hanno il vantaggio di riuscire ad analizzare centinaia di campioni in poche ore.
A causa dell'estrema sensibilità della PCR, è necessario fare particolare attenzione a evitare le possibili contaminazioni delle diverse miscele di reazione, o della delicata strumentazione, da parte dei prodotti di amplificazione di reazioni precedenti. Vi è inoltre il rischio di contaminazioni crociate fra i campioni da analizzare (v. Kwok e Higuchi, 1989). Per questo motivo viene suggerito di usare ambienti separati per la preparazione dei campioni, per l'approntamento dei reagenti necessari e per l'allestimento e l'analisi dei prodotti della PCR. Ciascun ambiente dovrebbe avere una dotazione propria di apparecchiature da utilizzare esclusivamente per le operazioni da eseguire in quella particolare zona.
b) Applicazioni
L'utilizzazione delle sonde ad acidi nucleici nel laboratorio clinico rappresentava fino a poco tempo fa un obiettivo ancora piuttosto lontano, in quanto tali tecniche, nate per il laboratorio di ricerca, erano macchinose, lunghe da allestire e richiedevano personale specificamente addestrato. La PCR ha invece facilitato l'impiego delle tecniche di diagnostica molecolare anche nel laboratorio generale. Attualmente sono molto numerose le applicazioni di questa tecnica in vari settori della diagnostica umana (v. Eisenstein, 1990).
Uno dei campi di applicazione più importanti, sia dal punto di vista clinico-epidemiologico che da quello della ricerca, è la microbiologia, come risulta dalla tab. III in cui sono elencati una serie di microrganismi identificabili per mezzo della PCR. Questa tecnica può essere usata per l'identificazione, in un campione di materiale infetto, di un qualsiasi patogeno di cui sia conosciuta anche solo una limitata porzione della sequenza genomica.
Sono virtualmente analizzabili mediante PCR tutte le malattie genetiche ereditarie di cui sia stato identificato il locus genetico implicato (v. Narayanan, 1992), quali ad esempio le varie forme di talassemia, di emofilia, la distrofia muscolare di Duchenne e altre (v. tab. 4). La possibilità di rilevare il DNA anche con quantità limitate di materiale cellulare ha conferito un ruolo di particolare rilievo a questa tecnologia nella diagnostica prenatale: in questo caso sono sottoposte a PCR cellule fetali ottenute da amniocentesi o campioni di villi coriali.


La risoluzione della sequenza di regioni di geni amplificati con PCR ha poi consentito di individuare malattie associate a variazioni di singoli nucleotidi nei geni che codificano per gli antigeni di istocompatibilità.
Un settore che si è recentemente avvalso della PCR è quello della medicina legale, specialmente nei casi in cui i reperti contengano solo pochissime cellule. Tale tecnica ha infatti consentito di tipizzare geneticamente cellule da singoli capelli o dal sedimento urinario e da singoli spermatozoi (v. Erlich e altri, 1991).
La PCR ha permesso di conseguire rilevanti risultati anche nel campo dell'oncologia (v. tab. 4). L'identificazione di diversi elementi genetici, come oncogeni e geni oncosoppressori, fattori di crescita e loro recettori, ne ha chiarito il ruolo in relazione alla genesi e all'evoluzione delle malattie neoplastiche, rendendo possibile un approccio molecolare all'oncologia (v. Albertini e altri, 1993). Le principali applicazioni in questo ambito, che rendono unica la PCR come strumento diagnostico, includono sia la possibilità di identificare minimi cambiamenti strutturali, mutazioni puntiformi o riassetti, sia la capacità di individuare la quota minima di malattia residua, cioè le cellule tumorali resistenti alla chemioterapia.
3. Analisi dei genomi
a) Principî e problematiche generali
Lo straordinario sviluppo delle tecniche di biologia molecolare ha aperto ampie prospettive nella direzione di uno dei grandi sogni della moderna biologia, vale a dire l'analisi dei genomi degli esseri viventi.
Verso la metà degli anni ottanta, la comunità scientifica aveva iniziato a pianificare una serie di progetti riguardanti il sequenziamento sistematico di genomi, con lo scopo di allestire uno strumento in grado di portare la ricerca biologica in ambiti finora inesplorati. Tra i progetti più ambiziosi figurava la mappatura e il sequenziamento del genoma di Homo sapiens: tale progetto, denominato Progetto Genoma (v. genoma), ha in effetti fornito la prima occasione per una dettagliata discussione sulle implicazioni strategiche di una iniziativa del genere (v. U.S. Department of Health and Human Services, 1990), ponendo altresì una serie di problemi di tipo tecnologico che, data la mole dell'impresa, è paragonabile a quella sollevata dal programma spaziale culminato negli anni settanta con le missioni umane sulla Luna.
Fu subito chiaro che i progetti di mappatura e sequenziamento dei genomi, considerando i costi, la complessità, la necessità di pluridisciplinarietà e le ricadute di amplissimo respiro, non potevano essere affrontati da una sola istituzione, per quanto di grandi dimensioni e di alto livello organizzativo; era invece necessaria un'ampia collaborazione che prevedesse lo sviluppo di reti di laboratori interconnessi e coordinati da centri in grado di raccogliere tutte le informazioni che i singoli laboratori avrebbero messo a disposizione.
Qualunque sia il genoma che si intenda sequenziare, vale una regola generale: deve essere messo a punto il programma di sequenziamento. A questo scopo sono necessari due strumenti: uno per rendere possibile l'orientamento spaziale e l'altro che permetta di assegnare a ogni tratto sequenziato la giusta collocazione all'interno del genoma in esame, cioè la mappa genetica e la mappa fisica, rispettivamente (v. Collins e Galas, 1993).
Per dare un'idea dello sforzo scientifico necessario, basti pensare che il genoma umano è circa 200 volte più grande del genoma del lievito di birra, che rappresenta l'eucariote più semplice. Il genoma umano si pensa sia costituito da una sequenza di almeno 3 miliardi di coppie di basi. Se, a titolo esemplificativo, tale sequenza venisse stampata su una pagina come quella dell'elenco telefonico avremmo più di 1.200 volumi di mille pagine ciascuno. Un primo passo per cercare di leggere questa collezione di libri consiste nell'allestire una mappa genetica, cioè nell'assegnare ad alcuni geni la rispettiva posizione sul cromosoma dove sono localizzati. Ciò, riprendendo l'esempio precedente, equivale ad allestire un indice parziale dei libri di cui si è detto. La mappa genetica rappresenta quindi uno strumento per indirizzare il ‛lettore' in una direzione o in un'altra, il che è indispensabile per il riconoscimento delle sequenze specifiche.
Ovviamente, come per le carte geografiche, queste mappe possono essere a bassa o ad alta risoluzione e ciò dipende dalla quantità di informazioni che si ha a disposizione. È chiaro che si tende a ottenere delle carte geografiche sempre più dettagliate, dapprima con una visione a bassa risoluzione, per passare poi gradualmente a valori di discriminazione sempre più elevati, fino a scorgere i dettagli reali del paesaggio, vale a dire a identificare i nucleotidi che fanno parte della sequenza di quel particolare gene. Le mappe genetiche costituiscono il sistema storicamente più vecchio di cartografia dei genomi, in quanto il loro allestimento può essere effettuato anche con tecniche di genetica classica. Gli elementi primari di una mappa genetica sono infatti i difetti genetici che possono essere studiati sul fenotipo osservando la frequenza di ricombinazione del gene in cui la mutazione si manifesta o di geni che non sono responsabili della malattia ma che co-segregano con il gene-malattia.
A questi marcatori può aggiungersi una serie di altri elementi che incrementano il potere di risoluzione della mappa. Questi elementi possono essere i cosiddetti RFLP (Restriction Fragment Length Polymorphism) o, secondo metodi più recenti, delle brevi sequenze caratterizzate da ripetizioni di citosina e adenina (CA) o anche delle sequenze ripetitive di tre o quattro nucleotidi che sono sparse lungo tutto il genoma. Pur non essendo assegnabili ad alcun gene, queste sequenze costituiscono degli ottimi punti di riferimento per la mappatura (v. genetica).
Il limite a cui tende una mappa genetica è la cosiddetta mappa fisica, che consiste nell'ordinare nella giusta sequenza la ‛libreria genomica' dell'organismo di interesse. Sulla base della mappa genetica, che ne costituisce l'ossatura, si allestisce una mappa fisica. Uno strumento recentemente messo a punto per facilitare questo lavoro è certamente l'uso di STS (Sequence Tagged Sites), vale a dire piccoli tratti di DNA a sequenza nota, associati o meno a geni, che possono essere identificati mediante PCR. Il vantaggio è che questi tratti possono essere rilevati sui vari pezzi di DNA genomico che compongono la libreria, consentendo quindi di ordinare i pezzi contigui con il corretto orientamento.
Le biotecnologie hanno senz'altro permesso di aumentare la velocità di mappatura dei geni umani, tanto che nel catalogo delle malattie ereditarie umane compilato da Victor McKusick, uno dei padri della genetica umana, dagli iniziali 1.478 difetti genetici elencati nel 1966 si è giunti a 4.937 nell'edizione del 1990 (v. McKusick, 19909). Nel 1989, dopo almeno 20 anni di ricerche di genetica sia classica che molecolare, venne identificato e sequenziato il gene per la fibrosi cistica: dapprima esaminando passo per passo l'intero cromosoma e poi passando al setaccio mediante sonde a DNA la zona che si riteneva contenesse il gene, si raggiunse finalmente l'obiettivo (v. Kerem e altri, 1989; v. Riordan e altri, 1989). Se all'epoca fosse stata disponibile la sequenza del genoma umano, il processo di identificazione sarebbe stato di almeno dieci volte più veloce. La mappatura e la sequenza del genoma umano possono in effetti essere considerate, come disse lo stesso McKusick (v., 1992), ‟il vero cuore della biologia e della medicina".
b) Sequenziamento del genoma di lievito
Il primo cromosoma completamente sequenziato di un organismo eucariote è stato il cromosoma III di Saccharomyces cerevisiae, un ceppo di lievito che contiene 16 cromosomi per complessivi 15 milioni di paia di basi nucleotidiche. Questo risultato è stato pubblicato nel maggio del 1992 sulla rivista ‟Nature" (v. Oliver e altri, 1992), la stessa in cui era comparso nel 1953 il famoso articolo di Watson e Crick (v., 1953) sulla struttura a doppia elica del DNA. Al di là del risultato scientifico veramente notevole, è interessante notare come, dal punto di vista organizzativo, abbia prevalso l'approccio strategico al problema del sequenziamento che prevedeva la creazione di un network di laboratori con uno spettro di competenze integrate di elevata qualità. Verso la fine degli anni ottanta, infatti, esisteva una controversia sulla strategia complessiva da adottare e sembrava che l'ipotesi di utilizzare centri di grandi dimensioni con strutture accentrate rappresentasse l'unica strada percorribile, sia per contenere i costi, sia per la necessità di sviluppare alte tecnologie per il sequenziamento automatico ad alta velocità. Viceversa, la strategia che ha permesso di giungere al sequenziamento del cromosoma di lievito ha utilizzato un approccio diametralmente opposto: infatti, il lavoro in centri di dimensioni ridotte, in cui il sequenziamento è stato eseguito anche manualmente, si è rivelato di gran lunga più valido, dimostrando anche in questo caso quanto il fattore umano possa rivelarsi decisivo.
Dei 16 cromosomi che compongono il patrimonio genetico del lievito era già stata definita la mappa genetica e fisica: nonostante le ridotte dimensioni, questi cromosomi possiedono notevoli somiglianze con quelli degli eucarioti complessi, sia per quanto riguarda la struttura che per i meccanismi di replicazione, ricombinazione e segregazione. La facilità con cui possono essere manipolati utilizzando tecniche di genetica sia classica che molecolare ha permesso di identificarne tutte le regioni funzionali, vale a dire telomeri, centromeri e origine di replicazione (v. Petes e altri, 1991). L'impresa ha riguardato 35 laboratori (di cui 4 italiani) finanziati dall'Unione Europea, con un contratto di ricerca che vincolava ogni laboratorio a sequenziare almeno 10.000 basi (10 kb) del cromosoma in questione. Un gruppo così numeroso andava coordinato in modo da uniformare le finalità scientifiche dei laboratori coinvolti, molti dei quali lavoravano su differenti geni del lievito sulla base di differenti interessi di ricerca (v. Vassarotti e Goffeau, 1992). Questo modo di procedere - che, pur non prevedendo l'allestimento di ‛megacentri' per sequenziare, è risultato vincente per quel che riguarda la strategia - si basa principalmente sul coordinamento di due momenti essenziali del processo di sequenziamento: il coordinamento delle librerie genomiche, garantito da due cosiddetti DNA coordinators che distribuiscono ai laboratori i pezzi di cromosoma da sequenziare e che possiedono la mappa fisica relativa a tali frammenti, e la raccolta e l'immagazzinamento delle sequenze, garantiti da un centro di bioinformatica, cioè da un sequence coordinator.
Tra i molti vantaggi offerti da questo tipo di approccio ricordiamo ad esempio: la grandissima elasticità, in quanto non esiste una struttura ‛pesante', ma a ogni laboratorio è lasciata la facoltà di sequenziare il frammento che è ritenuto più interessante e più adatto al tipo di indagine in corso in quel laboratorio; la ricerca su base contrattuale, che permette di richiedere al laboratorio un impegno a consegnare la sequenza in questione entro una data stabilita; la disponibilità nel laboratorio in cui si esegue la sequenza di informazioni circa le funzioni associabili a quella sequenza.
È in questo ambito che risulta di vitale importanza lo sviluppo della bioinformatica, che nella mappatura e nel sequenziamento dei genomi trova una delle applicazioni più proficue. Nel 1991 fu coniato il termine in silico per definire un terzo modo di studiare i meccanismi biologici, che integra quelli in vivo e in vitro ed è a essi complementare, specialmente nei campi della genetica e della biologia molecolare. Mai come oggi la tecnologia dell'informazione basata sui chips al silicio è entrata a far parte in modo così importante della ricerca biologica e in particolare delle biotecnologie. Questo nuovo approccio permette l'acquisizione e il trattamento dei dati in differenti banche di sequenze specializzate, e può inoltre costituire l'interfaccia di collegamento con banche dati che immagazzinano differenti tipi di informazioni (fisiologia, metabolismo, biochimica, ecc.).
Quando le sequenze possono essere immagazzinate ed elaborate in una banca dati elettronica, codoni e proteine vengono ad assumere una veste nuova. Sono stati pubblicati interessanti lavori riguardo a metodi predittivi per la localizzazione delle proteine a partire da dati di sequenze nucleotidiche e per determinare la struttura tridimensionale delle possibili proteine prodotte da geni ‛orfani', così da poterle poi paragonare con strutture di cui si conosce la funzione. Il DNA, infatti, può essere analizzato in termini di successione di mono-, di-, tri- e oligo-nucleotidi tali da identificare particolari combinazioni a cui assegnare una funzione. Da questo tipo di analisi ci si aspetta di ottenere correlazioni tra l'organizzazione del DNA e la struttura-funzione del cromosoma in cui esso si trova.
Un aspetto interessante del progetto è che la libreria genomica era costituita da vettori chiamati YAC (Yeast Artificial Chromosome): si tratta di un sistema di trasporto e di replicazione del DNA che permette l'inserimento di DNA eterologo fino all'ordine di grandezza delle megabasi, il che rappresenta un notevole vantaggio rispetto alle librerie fagiche o allestite in cosmidi (ibridi sintetici costruiti con plasmidi e fagi) in cui possono essere inseriti solo tratti corti di DNA. Ciò consente di avere librerie di dimensioni più ridotte e quindi più facilmente analizzabili, dal punto di vista sia dello screening che dell'ordinamento dei tratti contigui.
Nella sequenza del cromosoma III sono state identificate 182 fasi aperte di lettura (Open Reading Frames, o ORF) che concordano con una mappa di trascrizione che assegnava al cromosoma non meno di 160 geni. Solo il 20% di questi 182 ipotetici geni era già stato identificato nel lievito con metodi di genetica classica o molecolare; un ulteriore 10% mostra omologia con geni di altri organismi. Le proteine identificate in questo modo sono coinvolte in disparati processi cellulari; per esempio è stato identificato un omologo del gene che, in Drosophila, controlla la sintesi del pigmento bianco dell'occhio. Un altro gene presenta un'alta omologia con un gene che controlla la fissazione dell'azoto nei batteri azotofissatori. Altre ORF presentano omologia con geni coinvolti nelle diverse fasi di sviluppo degli organismi pluricellulari (v. Oliver e altri, 1992). La presenza di questi geni appare ancora misteriosa, come pure la loro funzione. Queste ‛stranezze' starebbero a indicare che le funzioni più specializzate degli organismi complessi si sono evolute da analoghe funzioni di base presenti in organismi unicellulari. Ai restanti due terzi del cromosoma III, cioè oltre 100 geni, devono ancora essere assegnate delle funzioni.
Un modo per studiare le funzioni di un gene neosequenziato consiste nel danneggiarlo con mutazioni (per esempio delezioni) e analizzarne l'effetto a livello fenotipico (gene disrupting). Sono stati esaminati una cinquantina di questi geni ‛orfani' e solo tre sono risultati essenziali per la sopravvivenza della cellula. Negli altri casi gli effetti delle mutazioni erano indistinguibili rispetto alla situazione fenotipica del ceppo selvatico, per lo meno sulla base dei dati preliminari.
Un approccio sistematico che consente di organizzare in un secondo tempo delle indagini più accurate può consistere nell'impiego dell'elettroforesi bidimensionale, una tecnica che si sta rivelando particolarmente utile per l'identificazione di funzioni associate a nuovi geni. Per quel che riguarda il lievito, è attualmente possibile identificare su un tracciato derivante da una corsa elettroforetica bidimensionale circa 3.000 macchie, ognuna delle quali corrisponde a una specifica proteina. Una mutazione può provocare la scomparsa di una macchia o incrementarne l'estensione.
Confrontando tramite analisi computerizzata i gel derivati da ceppi mutanti di S. cerevisiae con i tracciati ottenuti da ceppi selvatici, si dovrebbe riuscire a ottenere le informazioni necessarie all'identificazione di proteine funzionali che successive analisi potrebbero correlare al gene orfano. Il problema è riuscire a mettere a punto una tecnologia talmente standardizzata da permettere a ogni laboratorio coinvolto in questa identificazione di funzioni di paragonare direttamente il proprio tracciato con quello di altri laboratori o con quelli memorizzati in una banca dati contenente tracciati a due dimensioni. Successivamente, dovrebbe essere possibile microsequenziare le proteine che si possono eluire dai gel a due dimensioni. Un problema non da poco, in quanto attualmente sono necessari almeno 40 identici gel a due dimensioni per recuperare la quantità di proteina necessaria al microsequenziamento.
Un approccio complementare è quello computerizzato, mediante il quale è possibile prevedere la struttura tridimensionale e le funzioni associate a un'ipotetica proteina codificata da un nuovo gene. Attualmente, tuttavia, con tali mezzi è possibile determinare la struttura di proteine neosequenziate nel 15% dei casi e predirne la funzione nel 40% circa. È evidente come questo tasso di determinazione debba considerevolmente aumentare per far fronte alle migliaia di nuove sequenze proteiche derivanti dal sequenziamento del genoma di lievito.
Abbiamo in precedenza accennato all'analisi del tracciato elettroforetico bidimensionale di ceppi mutanti. In questo ambito vi è la necessità di allestire due diverse collezioni di mutanti di lievito, la prima che riguarda geni non essenziali e la seconda geni essenziali per la sua crescita. La prima collezione dovrebbe essere composta da ceppi con un gran numero di delezioni per geni non essenziali, almeno 20 delezioni per ceppo. In questo modo con una collezione di 200-250 di tali ceppi si possono coprire dalle 4.000 alle 5.000 ORF per geni non essenziali, stima abbastanza vicina alla situazione reale. La collezione di mutanti per geni essenziali potrebbe essere allestita utilizzando mutanti contenenti una delezione del gene essenziale per ceppo e lo stesso gene inserito in un plasmide sotto il controllo di un promotore inducibile.
Tra i geni neosequenziati, il numero di quelli essenziali dovrebbe essere molto piccolo (circa il 5%), visto che il gran numero di studi di genetica classica condotti sul lievito era necessariamente focalizzato sugli effetti letali per il fenotipo. Quindi, circa altri 250 ceppi mutanti dovrebbero essere sufficienti a completare la collezione. Questo vuol dire che una rete di sei laboratori con due-tre persone ciascuno potrebbe allestire questa collezione in otto anni: un tempo giusto per poterla confrontare con i dati di sequenziamento del genoma che saranno disponibili nel frattempo. I ceppi, inoltre, potrebbero essere distribuiti a qualunque laboratorio interessato allo studio di uno o più particolari fenotipi, ampliando il numero di informazioni disponibili e creando un'enorme rete costituita da tanti piccoli gruppi di laboratori operanti su differenti fenotipi.
I risultati finora ottenuti permettono di avanzare due ipotesi: 1) l'informazione genica è ridondante (ma si sa che nel lievito questo è valido solo per una minoranza di casi); 2) esiste una grande varietà di funzioni biologiche di cui non si sa praticamente nulla e il cui ruolo è da ricollegare con la messa a punto più fine della meccanica cellulare. Si tratta di funzioni fondamentali la cui mancanza, però, non necessariamente risulta in un fenotipo evidente.
Dobbiamo adesso affrontare la fase più difficile del progetto, che dovrà coinvolgere ricercatori con interessi differenti ed esperienze scientifiche di alto livello, in modo da riuscire a ridisegnare le funzionalità fisiologiche codificate dalle 315.356 paia di basi contenute nel cromosoma III, che rappresenta solo il 2,5% dell'intero genoma di lievito costituito da circa 14 milioni di basi. Si pensa che entro l'anno 2000 i 16 cromosomi di lievito dovrebbero essere completamente sequenziati. Attualmente è in corso il sequenziamento dei cromosomi VII, X, XIV e XV.
Oltre al lievito vi sono altri organismi il cui genoma è in fase di mappatura o sequenziamento (v. tab. 5; v. Vassarotti e Goffeau, 1992); tali studi sono finanziati da programmi specifici dell'Unione Europea.

c) Mappatura del genoma di topo
Nell'ottobre 1993 venne pubblicata sulla rivista ‟Science" la mappa genetica del topo, la prima relativa a un mammifero nella storia della biologia moderna (v. Copeland e altri, 1993). Storicamente il topo, dato il breve periodo di gestazione e le grandi possibilità di incrocio, è stato l'animale di laboratorio d'eccellenza anche per gli studi di genetica classica.
Recentemente sono state sviluppate tecnologie che consentono di inserire in cellule embrionali di topo geni umani, i quali possono in seguito essere espressi nel corso dello sviluppo e del differenziamento dell'animale; sono stati in tal modo creati animali transgenici in cui possono essere studiate le funzioni di tali geni mediante mutazioni che possono inattivarli o aumentarne il grado di attività. Gli effetti di tali modificazioni sono particolarmente utili per studiare numerose patologie genetiche umane, l'autoimmunità e altri disturbi del sistema immunitario, alcune patologie neurologiche, le malattie congenite, alcuni tipi di tumore, varie anomalie della riproduzione e malattie metaboliche come il diabete e l'aterosclerosi.
Come per la mappa genetica umana, la mappa genetica del topo serve per due scopi distinti. Innanzitutto essa fornisce uno strumento per l'analisi genetica, e quindi - date le possibilità di manipolazione - permette la mappatura di mutazioni che causano caratteristiche fenotipiche particolarmente interessanti, la localizzazione cromosomica di geni già clonati e sequenziati e la creazione di animali genotipicamente ben definiti. In secondo luogo, l'allestimento delle mappe genetiche - che è un prerequisito essenziale per poter poi sviluppare le mappe fisiche - fornisce le indicazioni necessarie per ordinare gli YAC contenenti i vari segmenti contigui del genoma del topo.
Per la costruzione delle mappe dei Mammiferi si considerano generalmente quattro tipi di marcatori genetici, detti anche loci: loci che codificano per isoenzimi, loci mutanti, geni clonati e segmenti anonimi di DNA altamente polimorfici a sequenza conosciuta. Ovviamente, in alcuni casi queste categorie di marcatori possono parzialmente sovrapporsi, ma questo rappresenta un vantaggio ai fini di stabilire in seguito l'ordine corretto in cui questi marcatori si susseguono lungo il genoma. Ogni categoria di locus apporta quindi un pezzo di informazione e svolge un differente ruolo nella creazione della mappa genetica.
I loci mutanti sono ovviamente quelli conosciuti da più tempo, in quanto più semplici da osservare e studiare, spesso applicando le leggi della genetica mendeliana. Già centinaia di anni fa in Giappone, allo scopo di divertire la corte imperiale, venivano allevati ceppi di topi che presentavano colorazioni strane del pelo e alterazioni neurologiche che ne modificavano il comportamento. In genere questi loci riguardano geni importanti dal punto di vista biologico ma poco caratterizzati sul piano biochimico.
I geni clonati e sequenziati forniscono invece importanti informazioni, specialmente allorché vengono posti a confronto geni omologhi di topo e di altri mammiferi, uomo compreso.
I segmenti di DNA altamente polimorfici, nonostante il basso contenuto informativo sul piano biologico, si prestano molto bene a essere identificati mediante PCR e monitorati nel corso di esperimenti di incrocio tra varie specie di topo, permettendo quindi di identificare i vari segmenti di ricombinazione e la distanza tra un locus e un altro.
Con questo approccio è stata quindi preparata una mappa genetica dei 21 cromosomi di topo basata su 2.616 loci, portando il potere di definizione della mappa a circa 0,6 cM (il centimorgan dà una stima della distanza tra un gene e un altro ed è basato sulla frequenza di ricombinazione cromosomica che si instaura al momento della meiosi). Dei 2.616 loci, 917 mostrano omologia con geni umani.
Questo grande numero di segmenti conservati permette di stimare il grado di divergenza delle linee ancestrali verificatosi nel corso dell'evoluzione umana e murina. Sulla base di questi dati si pensa che si siano verificati almeno 150 riassetti genici da quando le due specie si sono separate e conseguentemente geni molto simili hanno assunto differenti posizioni sui rispettivi genomi.
Paragonando le mappe genetiche è possibile trasferire le informazioni per geni omologhi da mappe ricche di dettagli, come quella murina, in mappe a basso livello di definizione, come sono tuttora le mappe genetiche di bovini, suini e ovini (v. Hino e altri, 1993; v. O'Brien e altri, 1993).
La disponibilità di una mappa genetica così ricca di informazioni apre la strada ad approcci impensabili solo due o tre anni fa. Innanzitutto la cosiddetta dissezione genetica di caratteri poligenici: finora, infatti, era possibile determinare la base genetica di una certa caratteristica fenotipica solo se questa era sotto il controllo di un singolo gene, mentre in futuro sarà possibile seguire le basi molecolari di malattie il cui decorso è controllato da vari fattori e quindi da differenti geni.
Esistono diversi ceppi di topo che presentano vario grado di suscettibilità a malattie quali il diabete, l'epilessia, il cancro, le infezioni virali o batteriche, l'obesità, ecc.; altri ceppi presentano invece caratteristiche ereditabili che influenzano la morfologia scheletrica, il pH sanguigno, la durata della vita, il comportamento, ecc. (v. Festing, 1979). Nella maggior parte dei casi queste diversità sono il risultato di effetti combinati di geni differenti che interagiscono tra loro. Sezionare geneticamente questi caratteri equivale a seguire simultaneamente sull'intero genoma i marcatori che influenzano un dato fenotipo. Questo è particolarmente vantaggioso negli studi di fisiologia dei Mammiferi, anche perché risulterà molto più facile trovare delle variazioni piuttosto che identificare un singolo gene la cui mutazione ha spesso effetti catastrofici e letali. Un altro settore che può trarre vantaggio da questi studi è sicuramente la ricerca sul cancro, in quanto da tempo è noto che una degenerazione genetica a carico di determinate regioni cromosomiche è responsabile dell'insorgere e della progressione della malattia tumorale. Queste regioni possono essere riconosciute in quanto i marcatori genetici nei tessuti normali risultano eterozigoti, mentre nei tessuti cancerosi si osserva l'instaurarsi dello stato omozigotico. Ciò rende il topo un modello ideale per studiare geneticamente il fenomeno dell'oncogenesi.
d) Mappatura del genoma umano
Per quanto riguarda il genoma umano, nel settembre 1994 è stata pubblicata sulla rivista ‟Science" una mappa genetica ad alta risoluzione della specie umana (v. CHLC, 1994). L'importanza di questo risultato è enorme, in quanto, per la prima volta nella sua storia, la specie umana ha la capacità di capire il proprio assetto genetico e come questo contribuisca alla patologia individuale e della specie stessa. I tentativi di associare le malattie umane al patrimonio genetico sono iniziati negli anni trenta, con i primi studi di linkage genetico tra il cromosoma X e il daltonismo e l'emofilia. Un serio ostacolo a questi studi di linkage è rappresentato dalla poco numerosa progenie degli uomini e dalla grande eterogeneità dei loro accoppiamenti.
Nel 1959 venne messo a punto un metodo, chiamato LOD score (Logarithm of the Odds Ratio for linkage), che aggirava questa difficoltà e rendeva l'analisi del linkage per la specie umana molto più agevole. Questo metodo statistico venne impiegato con successo su un numero limitato di caratteri riguardanti polimorfismi dei gruppi sanguigni e di altre proteine plasmatiche, consentendo l'allestimento dei primi raggruppamenti di linkage.
Negli anni settanta, con lo sviluppo e l'utilizzazione degli elaboratori elettronici, questo metodo divenne più rapido ed efficiente, anche se la scarsità di marcatori genetici ne limitava grandemente l'impiego. Fu negli anni ottanta, grazie ai sempre più potenti elaboratori e all'avvento delle moderne biotecnologie, che furono evidenziati molti RFLP nel DNA umano (v. Keates, 1981). Vennero poi evidenziati altri tipi di marcatori, come i VNTR (Variable Number of Tandem Repeats), chiamati anche minisatelliti, e sequenze ripetute di dinucleotidi, chiamati microsatelliti. In pratica, questi vari tipi di marcatori - geni, RFLP, VNTR - possono essere considerati alla stregua di ‛segnalibri' che danno un'idea di come sono posizionati gli uni rispetto agli altri e in quale direzione. Ovviamente, la distanza tra un marcatore e l'altro può essere più o meno grande e questo dà un'idea del grado di risoluzione della mappa. Più sono numerosi i loci, maggiore è la definizione della mappa.
Nel 1994 l'uso sempre più frequente delle tecniche di PCR ha ulteriormente aumentato il numero di sequenze che etichettano una certa zona del DNA umano, facendolo quasi triplicare nel giro di pochi mesi (v. tab. VI), tanto che la mappa genetica umana è risultata composta da 5.840 loci con un potere di risoluzione di circa 0,7 cM, valore paragonabile a quello ottenuto nella mappa genetica del topo. Tale risultato è tra l'altro in linea con uno degli obiettivi che il Progetto Genoma Umano statunitense si era prefissato, vale a dire una mappa genetica a 2 cM completa entro il 1995. Si può quindi prevedere che nel giro di pochi anni sarà raggiunto un altro obiettivo, cioè una mappa fisica basata sull'impiego di STS situati dapprima a una distanza media di circa 300 kb l'uno dall'altro, e successivamente di 100 kb, che sembra un valore accettabile per iniziare il sequenziamento vero e proprio. A questo scopo l'obiettivo primario è quello di mettere a punto tecnologie di sequenziamento robotizzate che consentano di ottenere velocità annue di sequenziamento intorno agli 80 milioni di basi, così da prevedere che il lavoro potrà essere completato intorno all'anno 2005.

A questo stadio, come si è visto, è cruciale il supporto bioinformatico. Attualmente vi sono tre banche dati di primaria importanza per il Progetto Genoma Umano: la banca di sequenze di DNA (GenBank), la banca per le mappe cromosomiche (Genome Data Base) e la banca di sequenza e struttura delle proteine (Protein Information Resource). Si tratta di banche dati separate e indipendenti, ma in considerazione delle problematiche descritte in precedenza in relazione al lievito sembra che sarà opportuno integrarle in una sorta di banca dati ‛virtuale', unificando i protocolli di interrogazione e di inserimento delle informazioni (v. Cuticchia e altri, 1993). In questo modo le sequenze di DNA e delle proteine correlate potrebbero essere collegate tra loro insieme alle annotazioni relative alle loro funzioni e alle omologie con sequenze analoghe di altre specie. Questo faciliterebbe in seguito sia la gestione dei segmenti di DNA sequenziati, sia l'identificazione dei geni su tali segmenti e le funzioni a essi associate.
4. Terapia genica
a) Aspetti tecnologici
La tecnologia del DNA ricombinante ha fornito gli strumenti per definire il ruolo di uno specifico prodotto genico nella patogenesi di alcune malattie umane. La possibilità di caratterizzare le patologie in termini molecolari ha quindi portato alla definizione di approcci terapeutici basati sulla correzione del difetto genico responsabile della malattia (v. Mulligan, 1993). Questo è il principio su cui si fondano le tecniche di terapia genica. Una certa patologia può essere trattata trasferendo materiale genetico all'interno di specifiche cellule del paziente anziché utilizzare un trattamento farmacologico convenzionale che in molti casi non è disponibile. Ciò può avvenire in due modi: ex vivo, cioè prelevando dal paziente le cellule da trattare e reinserendole a procedimento avvenuto, oppure in vivo, vale a dire intervenendo sulle cellule di un determinato distretto corporeo direttamente nell'area in cui si trovano. In ambedue i casi è comunque necessario veicolare il DNA esogeno all'interno della cellula (v. Miller, 1990).
Attualmente esistono differenti metodi per trasferire geni in cellule bersaglio e sono in corso studi preclinici e clinici di alcune patologie umane che vanno dalle malattie del sistema ematopoietico al cancro e all'AIDS. Questi metodi possono essere raggruppati in due grandi categorie: i sistemi virali e quelli non virali (v. tab. 7).

Nei sistemi virali le sequenze di DNA sono trasferite utilizzando come vettori dei virus. I vettori retrovirali e i vettori associati ad Adenovirus, nei quali il DNA esogeno viene integrato nel DNA cromosomico delle cellule bersaglio, vengono utilizzati soprattutto nella terapia genica ex vivo. L'impiego dei vettori retrovirali, che consentono di trasferire stabilmente DNA esogeno con una resa molto vicina al 100%, è strettamente dipendente da determinate condizioni: in primo luogo l'entrata dei Retrovirus è assolutamente subordinata all'esistenza di recettori specifici per tali virus sulla superficie cellulare, e ciò può ovviamente rappresentare una limitazione; la seconda condizione necessaria è che vi sia una replicazione del DNA cellulare in modo che si verifichi l'integrazione del genoma provirale contenente i geni da trasferire, e pertanto il trasferimento è tanto più efficace quanto maggiore è il grado di proliferazione delle cellule bersaglio, almeno durante un limitato periodo di tempo.
I vettori a base di Adenovirus consentono il rilascio in vivo di geni estranei, in quanto sono in grado di infettare efficacemente anche cellule non in replicazione e di esprimere alti livelli di prodotto genico; inoltre, le particelle virali sono relativamente stabili e ben si prestano a procedure di separazione e purificazione. Al contrario dei Retrovirus, tuttavia, essi portano a lisi le cellule infettate. Infatti, nonostante che l'integrazione di sequenze adenovirali all'interno del DNA cromosomico sia senz'altro possibile, soprattutto in cellule non permissive, non sembra che questo fenomeno abbia un ruolo critico nel ciclo vitale del virus ed è relativamente inefficiente. Infatti, l'espressione dei prodotti genici codificati dalle sequenze trasferite è transitoria, al contrario di quanto succede con i Retrovirus, per i quali si ha un'espressione stabile. Il problema della lisi cellulare è stato risolto utilizzando mutanti di Adenovirus difettivi per un gene E1 competente per la replicazione. In questo modo è possibile utilizzare più efficacemente tali vettori per procedure di terapia genica in vivo. Studi preclinici hanno mostrato che si ottiene un efficace trasferimento genico nelle cellule bersaglio e che l'espressione genica viene mantenuta per significativi periodi di tempo.
Vi sono altri sistemi di vettori di sequenze di DNA esogeno, quali virus adeno-associati (VAA), virus erpetici e diversi virus a RNA. Questi sistemi rivestono tuttavia un interesse più di base che applicativo a causa di vari problemi associati alla sicurezza del loro impiego.
I VAA sono virus di piccole dimensioni, abbastanza stabili, che possono infettare le cellule umane anche non in divisione; tuttavia risultano poco efficienti in termini di integrazione e spesso presentano fenomeni di riassetto e di delezione. Una proprietà dei VAA è che l'integrazione nel genoma dell'ospite avviene frequentemente in una piccola regione del cromosoma 19, una regione implicata nel riassetto cromosomico associato alla leucemia cronica; risulta quindi discutibile il vantaggio di poter usufruire di un unico punto di integrazione piuttosto che di un'integrazione casuale.
I vettori a virus erpetici - che sono abbastanza interessanti in quanto rappresenterebbero gli unici strumenti per trasferire geni in cellule del sistema nervoso centrale (v. Andersen e altri, 1992) - presentano però una notevole complessità nella regolazione della replicazione virale che rende difficile l'allestimento di virus ricombinanti completamente incapaci di replicazione. Inoltre, va rilevato che anche i ceppi incompetenti per quanto riguarda la replicazione sono tossici per le cellule bersaglio, in quanto esprimono alti livelli di specifiche proteine virali che sono dannose per il metabolismo cellulare (v. anche virus).
La maggior parte dei metodi non virali di trasferimento genico sfrutta i meccanismi che le cellule di mammifero utilizzano per incorporare le macromolecole. Attualmente l'interesse è focalizzato su metodi che utilizzano i processi di endocitosi mediati da recettori, in quanto permettono l'entrata del DNA nelle cellule bersaglio in vivo (v. Wu e altri, 1991); in questo modo il DNA esogeno viene coniugato a un polipeptide riconosciuto da uno specifico recettore di membrana che lega il complesso DNA-polipeptide portandolo all'interno della cellula. Il problema principale di questo approccio è che le vescicole endocitiche contenenti il coniugato vengono dirette verso i lisosomi dove avviene la degradazione. Questo fenomeno è prevenibile se, una volta portato all'interno, il complesso DNA-peptide può fuoriuscire dall'endosoma; in effetti, coniugando il DNA di interesse con peptidi fusogeni controllati dal gene HA del virus influenzale viene indotta la distruzione della vescicola endocitica con conseguente fuoriuscita dal DNA esogeno (v. Wagner e altri, 1992): questi peptidi sono infatti fattori proteici virali presenti sull'involucro pericapsidico in grado di facilitare il processo di fusione mediante il quale il virus penetra nella cellula. I risultati ottenuti con questo sistema si sono rivelati promettenti, anche se è possibile ottenere solo un'espressione transitoria dei prodotti controllati dal gene trasferito.
b) Applicazioni in medicina umana
Solo pochi anni fa la maggior parte dei biotecnologi considerava la terapia genica come una possibilità sperimentale di difficile applicazione, almeno per questo millennio. Tuttavia, nel 1990 alcuni ricercatori dei National Institutes of Health negli Stati Uniti realizzarono il primo intervento di terapia genica su un essere umano: nel caso specifico si trattava di una bimba di quattro anni affetta da deficienza di adenosindeaminasi (ADA), una patologia ereditaria causata da una mutazione del gene preposto alla codificazione di detto enzima, il cui risultato finale è un'immunodeficienza congenita che mina le difese immunitarie dell'individuo rendendolo suscettibile di severe e mortali forme di infezione (v. The ADA..., 1990). In questo caso l'inserimento del gene corretto venne allestito ex vivo, prelevando le cellule del sistema ematopoietico dal midollo osseo e ‛correggendole' in vitro, con successivo reimpianto nel paziente. Da allora altri pazienti in età pediatrica hanno beneficiato di questo trattamento che ha restituito loro la capacità di vivere una vita normale.
In effetti, molti degli sforzi e delle conoscenze finora acquisite nell'ambito dell'applicazione clinica della terapia genica si sono focalizzati sulle cellule del sistema ematopoietico, che meglio si prestano a essere geneticamente curate al di fuori del corpo del paziente per poi esservi reintrodotte (v. Anderson, 1992). La difficoltà maggiore è quella relativa al trasferimento genico in cellule staminali midollari - le uniche che possono garantire la correzione permanente del difetto genetico - che sono presenti in scarso numero nel midollo osseo e non appaiono facilmente riconoscibili con gli attuali metodi analitici. Vi è inoltre una difficoltà connessa all'efficienza di trasfezione mediante vettori retrovirali che, come si è detto poc'anzi, richiedono cellule in proliferazione, mentre le cellule staminali midollari sono in gran parte quiescenti (v. Ohashi e altri, 1992; v. Correll e altri, 1992).
Per quanto riguarda il trattamento genico di epatociti, vi sono evidenze sperimentali relative a una malattia ereditaria, l'ipercolesterolemia familiare, causata da un difetto del gene che codifica per il recettore delle lipoproteine a bassa densità (LDL). In questo caso sono state utilizzate sia tecniche ex vivo che in vivo e sono stati sperimentati vettori retrovirali e adenovirali per il trasferimento dei geni negli epatociti (v. Wilson e altri, 1992).
Relativamente al polmone, sono da segnalare le sperimentazioni in atto per la cura genica della fibrosi cistica, una delle malattie ereditarie a più alta incidenza nella popolazione caucasica, causata da difetti del gene che codifica per la proteina CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) responsabile del controllo del flusso di ioni cloro attraverso la membrana cellulare (v. Yoshimura e altri, 1992). La malattia si manifesta con un'ipersecrezione di muco a livello polmonare e pertanto le cellule bersaglio della terapia genica risultano essere le cellule dell'epitelio polmonare. In questo caso, data la difficoltà di prelievo e reimpianto di queste cellule, la metodologia di elezione risulta essere quella in vivo. Studi su modelli animali hanno dimostrato l'utilità di vettori adenovirali e di liposomi per trasferire il gene corretto nelle cellule polmonari. Tuttavia, sono in corso ulteriori studi, in quanto l'espressione della proteina è transitoria, con una durata che va da una settimana fino a quaranta giorni circa (v. Rosenfeld e altri, 1992).
Vi sono poi studi volti a cercare di trapiantare cellule modificate in modo che queste mettano in circolo i loro prodotti genici: questo potrebbe rappresentare uno strumento per la cura dei disturbi della coagulazione, dato che è possibile trapiantare stipiti cellulari come cheratinociti, mioblasti o fibroblasti modificati in modo da esprimere le proteine di interesse e immetterle in circolo (v. Dhawan e altri, 1991).
Malattie della coagulazione determinate dalla mancanza del fattore VIII o del fattore IX potrebbero essere definitivamente curate, evitando l'uso di frequenti trasfusioni del fattore mancante prodotto con metodi estrattivi o con tecniche di DNA ricombinante. Gli esperimenti finora effettuati con cheratinociti dimostrano che, a fronte di una persistente espressione in vivo del prodotto genico dopo il trapianto, vi è però una transitoria dismissione in circolo della sostanza di interesse. Si pensa che questo dipenda sia dal tipo di prodotto da secernere che dalla linea cellulare in cui viene espresso. Risultati migliori sono stati infatti ottenuti con mioblasti in cui il fattore IX veniva efficacemente sintetizzato e secreto per più di sei mesi dopo il loro trapianto nel muscolo (v. Dai e altri, 1992). Lo stesso vale per l'ormone della crescita. Nonostante questi risultati positivi, vi sono ancora dei fattori critici che dovrebbero essere indagati, come il sito di impianto, l'età delle cellule, i vari passaggi in coltura che queste hanno subito, ecc. Da non dimenticare poi il problema di una possibile reazione immunitaria nei confronti di queste cellule trapiantate.
La possibilità che la terapia genica venga utilizzata anche per il trattamento di patologie acquisite è oggetto di assidua indagine. Riuscire a modificare sia linfociti tumore-infiltranti (LTI) che cellule tumorali stesse rappresenterebbe sicuramente un progresso in questa direzione (v. Anderson, 1992). Gli LTI possono essere usati come veicoli per portare nei pressi della massa tumorale grandi quantità di citochine e per esprimere molecole ad attività antitumorale. In questo caso devono essere chiariti i meccanismi che stanno alla base sia del riconoscimento delle cellule tumorali da parte degli LTI, sia della regolazione dell'espressione dei geni in essi trasferiti.
Altri studi sono orientati all'utilizzazione di cellule tumorali per stimolare una risposta immunitaria dell'ospite. Protocolli clinici dimostrano che cellule tumorali irradiate esprimenti il fattore GM-CSF (Granulocyte-Macrophage Colony Stimulating Factor) - la più interessante tra le molecole studiate - possono essere utilizzate come una sorta di vaccino per il trattamento di differenti forme di cancro, come è stato messo in evidenza nel 1993 nel corso di un incontro organizzato dall'NIH Recombinant DNA Advisory Committee.
Altri tentativi di terapia genica del cancro prevedono l'introduzione nelle cellule cancerose di geni per proteine tumore-soppressore, in modo da inibire l'espressione degli oncogeni; altri ancora sono diretti a ottenere una maggior resistenza delle cellule del midollo osseo nei confronti dei trattamenti chemioterapici (v. Podda e altri, 1992).
Un certo numero di studi si è focalizzato anche sullo sviluppo di trattamenti genici nei confronti dell'AIDS, di malattie cardiovascolari e del sistema nervoso centrale. Per quanto riguarda l'AIDS, gli sforzi sono tesi a rendere le cellule, in modo particolare linfociti T e cellule staminali totipotenti, resistenti nei confronti del virus HIV (Human Immunodeficiency Virus) inibendo sia l'infezione che il rilascio di particelle virali dalle cellule (v. Malim e altri, 1992).
5. Biotecnologie e risorse genetiche
a) Aspetti della biodiversità
Le caratteristiche individuali degli esseri viventi, siano essi animali, piante o microrganismi, rappresentano la base delle biotecnologie. Sono gli individui delle suddette specie, infatti, a produrre le molecole da cui dipendono le biotecnologie, non essendovi nessun'altra fonte alternativa. Questo è un concetto estremamente importante per capire a fondo l'origine delle biotecnologie (v. Biotechnology and..., 1992).
Nonostante ci siano molte controversie in merito, vi è un certo consenso sul fatto che esistano sul nostro pianeta circa 10 milioni di specie di Eucarioti, 1 milione delle quali vive nei mari, mentre le altre sono terrestri. Per quanto riguarda invece i Procarioti, come i Batteri, e i Virus, è quasi impossibile formulare una ragionevole ipotesi circa la quantità delle specie viventi sul pianeta. Sono state classificate circa 3.000 specie di Batteri, anche in base al fatto che possono essere coltivate in laboratorio; tuttavia ve n'è un numero imprecisato di cui non si sa praticamente nulla. Usando sonde al DNA, alcuni ricercatori norvegesi hanno recentemente stimato che esistano 4.000-5.000 specie di Batteri in un grammo di terra proveniente da una faggeta. Da ciò si possono trarre delle ovvie conclusioni circa la nostra conoscenza del numero di specie che popola la Terra.
Degli 8-10 milioni di specie di Eucarioti, solo 1,4 milioni sono stati classificati; delle restanti specie, come abbiamo già detto, non si conosce praticamente nulla, a parte il nome, la località di rinvenimento e alcuni commenti descrittivi (v. May, 1992). Probabilmente, solo poche decine di migliaia di specie di Eucarioti sono state accuratamente descritte e solo su alcune di esse si è concentrato lo studio per eventuali applicazioni biotecnologiche. Vi è inoltre un grande squilibrio tra le specie conosciute nella parte temperata del pianeta, che ammontano a circa 1 milione, e quelle delle zone tropicali, delle quali si hanno notizie relativamente a forse 400 mila specie su un totale di quasi 8 milioni.
Alcuni gruppi di organismi sono più conosciuti di altri, e se quasi tutte le specie di Vertebrati sono classificate, dell'1,5 milioni di specie di Funghi se ne conoscono solo 69 mila e del milione di specie di Nematodi si arriva appena a 13 mila specie riconosciute.
Dall'insieme di piante, animali e microrganismi derivano cibo, farmaci, fibre di vario genere, biomassa a scopo energetico; essi contribuiscono anche a mantenere aria e acque pulite, impediscono l'erosione del suolo, determinano le condizioni climatiche caratteristiche delle varie zone del pianeta.
Sfortunatamente, mentre la popolazione mondiale dal 1950 a oggi è cresciuta da 2,5 a 5,5 miliardi di individui, da allora è stato perso il 5% dell'ozono stratosferico e distrutto un terzo delle foreste. Un miliardo e mezzo di persone vive al di sotto dei livelli di sussistenza e, nonostante siano state intraprese campagne di pianificazione familiare, in tutti i paesi in via di sviluppo la proporzione di individui in giovane età rimane estremamente elevata rispetto alla media dei paesi industrializzati.
Per queste e altre ragioni, la velocità di estinzione delle specie viventi è dalle mille alle diecimila volte superiore rispetto alla media degli ultimi 65 milioni di anni, e un quinto o più delle specie esistenti, milioni delle quali non saranno mai conosciute, è minacciato di estinzione nei prossimi trent'anni.
Si ritiene che circa l'80% della biodiversità si trovi nei paesi in via di sviluppo, nei quali, sfortunatamente, è presente solo il 6% degli scienziati, insieme a 4,3 miliardi di persone. Ciò significa che queste nazioni non hanno il supporto scientifico per utilizzare la biodiversità in modo efficiente, per studiarla e per preservarla, mentre si è diffusa l'opinione che i paesi industrializzati possano risultare gli unici beneficiari di questo patrimonio: bisogna ammettere che ciò è almeno in parte vero, in quanto solo questi hanno le conoscenze biotecnologiche in grado di sostenere questa attività.
b) Applicazioni biotecnologiche nel settore della biodiversità
Le relazioni che intercorrono tra biotecnologie e diversità genetica sono molteplici (v. Beese, 1990). Le biotecnologie possono fornire potenti strumenti per metodi di identificazione genetica che permettano di analizzare l'assetto genetico di un certo organismo e di paragonarlo con altri, in modo da evidenziarne le differenze e il possibile impiego. Tecniche per il mantenimento a lungo termine di embrioni, cellule o geni aiutano a preservare le risorse genetiche in modo da riuscire a capirne le potenzialità di utilizzazione. Questo si è rivelato particolarmente importante per i Batteri, che spesso possono presentare caratteristiche fisiologiche sorprendenti.
Attualmente vi sono almeno quattro aree in cui le biotecnologie possono grandemente influenzare la conoscenza e la conservazione della diversità genetica. Lo sviluppo di metodi di screening molecolare persegue lo scopo di valutare la diversità genetica sia in popolazioni libere che in collezioni di cellule e ceppi di microrganismi: l'obiettivo è quello di mettere a punto procedure per acquisire dati genetici attraverso l'analisi di un gran numero di campioni. Questi dati potrebbero essere utilizzati sia a livello tassonomico e di classificazione, permettendo di stabilire la distanza genetica tra le varie specie, sia per identificare i tratti genetici associati a proprietà utili ai fini biotecnologici. Tali informazioni genetiche dovrebbero progressivamente integrare e sostituire i dati fenotipici, in modo da rendere più razionale la gestione delle collezioni di materiale vivente e consentire una migliore analisi delle variazioni genetiche in natura (v. Anderson e Fairbanks, 1990).
Le tecnologie utilizzabili in questa sede non si discostano da quelle che vengono impiegate per la mappatura e il sequenziamento di genomi, includendo la determinazione di RFLP, VNTR e l'uso della PCR. Gli studi finora compiuti indicherebbero che RFLP possono essere usati con successo per verificare le differenze genetiche complessive della popolazione in esame, in quanto consentono di prendere in considerazione differenti marcatori contemporaneamente. La PCR si è rivelata utile per studi filogenetici e di classificazione, mentre i VNTR possono essere utilizzati per identificare uno specifico locus. Questi metodi vengono impiegati dagli allevatori per individuare e selezionare i portatori di determinati geni.
Una delle applicazioni della bioinformatica nel settore della protezione della diversità genetica implica l'utilizzazione di banche dati a scopo tassonomico (v. Fortuner, 1993). Gli elenchi in cui sono riportate le caratteristiche delle specie viventi non classificate, siano esse piante, animali, microrganismi, ecc., sono in gran parte ancora su supporto cartaceo e l'integrazione e coordinazione tra i vari elenchi è minima; sono inoltre di difficile consultazione, non aggiornabili in tempo reale e del tutto privi di annotazioni di tipo genetico-molecolare. Attraverso l'uso della bioinformatica questo settore, considerato a torto poco interessante, subirebbe una drastica rivitalizzazione e potrebbe divenire il punto di partenza di qualsiasi iniziativa volta alla conservazione della diversità biologica.
Un settore economicamente importante è rappresentato dalla diversità genetica riscontrabile negli animali domestici. La specie umana attualmente utilizza circa diciotto specie tra Mammiferi e Uccelli: di queste, sette servono a produrre la maggior parte degli alimenti di origine animale, mentre le altre sono importanti solo per piccoli e spesso isolati gruppi di esseri umani e raramente sono utilizzate altrove. All'interno delle prime sette specie considerate, nel corso di diecimila anni sono state sviluppate circa tremila razze. Questo dato indica quanto sia grande il potenziale di creazione di diversità genetica.
Le biotecnologie hanno grandemente contribuito, e ancor più contribuiranno, sia alla conservazione che al miglioramento delle ‛risorse' genetiche degli animali domestici, in particolare relativamente alla creazione degli animali transgenici e alla mappatura dei genomi di specie quali la mucca e il maiale (v. Biotechnology and ..., 1992). Gli animali transgenici si ottengono modificando, attraverso l'inserimento di geni esogeni, il patrimonio genetico dell'animale in modo stabile e trasmissibile. A questo scopo si utilizzano tecniche di trasferimento genico impiegando Retrovirus, analogamente a quanto descritto nella parte dedicata alla terapia genica, oppure il costrutto genico viene iniettato nel nucleo dell'ovulo fecondato allo stadio unicellulare. Un'alternativa interessante viene fornita dall'utilizzazione di cellule staminali embrionali derivanti dalla massa interna della blastocisti; una volta isolate, tali cellule possono essere coltivate e mantenute in vitro, modificate con i due sistemi sopra descritti e reimpiantate nuovamente nella blastocisti, ove sono incorporate andando poi incontro ai vari processi di differenziamento che portano all'animale completo, in genere chimerico. Se la linea cellulare transgenica viene coinvolta anche nello sviluppo delle gonadi, allora anche la prole sarà transgenica.
Avvalendosi di questo approccio sono state recentemente prodotte pecore transgeniche che secernono nel latte una proteina umana, l'α1-antitripsina, da utilizzare come farmaco per il trattamento dell'enfisema polmonare e della fibrosi cistica. Questo dato è estremamente importante, in quanto sembra che ciò consentirebbe di allestire linee di animali deputati alla produzione di farmaci che non potrebbero essere prodotti con le tecnologie farmaceutiche attuali.
BIBLIOGRAFIA
Albertini, A., Bombardieri, E., Bonini, P., Salvatore, F. (a cura di), Applicazioni diagnostiche della PCR, Milano 1993.
Andersen, J. K., Garber, D. A., Meaney, C. A., Breakefield, X. O., Gene transfer into mammalian central nervous system using herpes virus vectors: extended expression of bacterial lacZ in neurons using the neuron-specific enolase promoter, in ‟Human gene therapy", 1992, III, pp. 487-499.
Anderson, W. F., Human gene therapy, in ‟Science", 1992, CCLVI, pp. 808-813.
Anderson, W. R., Fairbanks, D. J., Molecular markers: important tools for plant genetic resource characterization, in ‟Diversity", 1990, VI, pp. 51-53.
Beese, K. (a cura di), Proceedings of the European workshop on biotechnology for improvements in the assessment, conservation and utilization of biological diversity, Brussels 1990.
Biotechnology and genetic resources. Proceedings of the workshop, a cura di US-EC Task Force on Biotechnology Research, Airlie, Va., 1992.
CHLC (Cooperative Human Linkage Center), A comprehensive human linkage map with centimorgan density, in ‟Science", 1994, CCLXV, pp. 2049-2054.
Collins, F., Galas, D., A new five-year plan for the U.S. Human Genome Project, in ‟Science", 1993, CCLXII, pp. 43-46.
Copeland, N. G. e altri, A genetic linkage map of the mouse: current applications and future prospects, in ‟Science", 1993, CCLXII, pp. 57-66.
Correll, P. H., Colilla, S., Dave, H. P., Karlsson, S., High levels of human glucocerebrosidase activity in macrophages of long-term reconstituted mice after retroviral infection of hematopoietic stem cells, in ‟Blood", 1992, LXXX, pp. 331-336.
Cuticchia, A. J., Chipperfield, M. A., Porter, C. J., Kearns, W., Pearson, P. L., Managing all those bytes: the Human Genome Project, in ‟Science", 1993, CCLXII, pp. 47-48.
Dai, Y., Roman, M., Naviaux, R. K., Verma, I. M., Gene therapy via primary myoblasts: long-term expression of factor IX protein following transplantation in vivo, in ‟Proceedings of the National Academy of Sciences", 1992, LXXXIX, pp. 10892-10895.
Dhawan, J., Pan, L. C., Pavlath, G. K., Travis, M. A., Lanctot, A. M., Blau, H. M., Systemic delivery of human growth hormone by injection of genetically engineered myoblasts, in ‟Science", 1991, CCLIV, pp. 1509-1512.
Eisenstein, B. I., The polymerase chain reaction, a new method of using molecular genetics for medical diagnosis, in ‟New England journal of medicine", 1990, CCCXXII, pp. 178-183.
Erlich, H. A. (a cura di), PCR technology, New York 1989.
Erlich, H. A., Gelfand, D., Sninsky, J. J., Recent advances in polymerase chain reaction, in ‟Science", 1991, CCLII, pp. 1643-1651.
Festing, M. F. W., Inbred strains in biomedical research, New York 1979.
Fortuner, R., Advances in computer methods for systematic biology: artificial intelligence, databases, computer vision, Baltimore, Md., 1993.
Hino, O., Testa, J. R., Buetow, K. H., Taguchi, T., Zhou, J. Y., Bremer, M., Bruzel, A., Yeung, R., Levan, G., Levan, K. K. e altri, Universal mapping probes and the origin of human chromosome 3, in ‟Proceedings of the National Academy of Sciences", 1993, XC, pp. 730-734.
Keates, B. J. B., Linkage and chromosome mapping in man, Honolulu 1981.
Kerem, B., Rommens, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A., Buchwald, M., Tsui, L. C., Identification of the cystic fibrosis gene: genetic analysis, in ‟Science", 1989, CCXLV, pp. 1073-1080.
Kornberg, A., DNA synthesis, New York 1974.
Kwok, S., Higuchi, R., Avoiding false positives with PCR, in ‟Nature", 1989, CCCXXXIX, pp. 237-238.
McKusick, V. A., Mendelian inheritance in man. Catalogue of autosomal dominant, autosomal recessive and X-linked phenotypes, Baltimore, Md., 19909.
McKusick, V. A., Human genetics: the last 35 years, the present and the future. Presidential address, Eighth international congress of human genetics, in ‟American journal of human genetics", 1992, L, pp. 663-670.
Malim, M. H., Freimuth, W. W., Liu, J., Boyle, T. J., Lyerly, H. K., Cullen, B. R., Nabel, G. J., Stable expression of transdominant Rev protein in human T cells inhibits human immunodeficiency virus replication, in ‟Journal of experimental medicine", 1992, CLXXVI, pp. 1197-1201.
Mantero, G., Zonaro, A., Albertini, A., Bertolo, P., Primi, D., DNA enzyme immunoassay: general method for detecting products of polymerase chain reaction, in ‟Clinical chemistry", 1991, XXXVII, pp. 422-429.
May, R., How many species inhabit the Earth?, in ‟Scientific American", 1992, CCLXVII, 4, pp. 42-48 (tr. it.: Quante sono le specie che vivono sulla Terra?, in ‟Le scienze", 1992, XXV, 292, pp. 16-23).
Miller, A. D., Retrovirus packaging cells, in ‟Human gene therapy", 1990, I, pp. 5-14.
Mulligan, R. C., The basic science of gene therapy, in ‟Science", 1993, CCLX, pp. 926-932.
Mullis, K. B., Faloona, F. A., Specific synthesis of DNA in vitro via a polymerase catalyzed chain reaction, in ‟Methods in enzymology", 1987, CCLV, pp. 335-350.
Narayanan, S., Overview of principles and current uses of DNA probes in clinical and laboratory medicine, in ‟Annals of clinical and laboratory science", 1992, XXII, pp. 353-376.
O'Brien, S. J., Womack, J. E., Lyons, L. A., Moore, K. J., Jenkins, N. A., Copeland, N. G., Anchored reference loci for comparative genome mapping in Mammals, in ‟Nature genetics", 1993, III, pp. 103-112.
Ohashi, T., Boggs, S., Robbins, P., Bahnson, A., Patrene, K., Wei, F. S., Wei, J. F., Li, J., Lucht, L., Fei, Y. e altri, Efficient transfer and sustained high expression of the human glucocerebrosidase gene in mice and their functional macrophages following transplantation of bone marrow transduced by a retroviral vector, in ‟Proceedings of the National Academy of Sciences", 1992, LXXXIX, pp. 11332-11336.
Oliver, S. G. e altri, The complete DNA sequence of yeast chromosome III, in ‟Nature", 1992, CCCLVII, pp. 38-46.
Petes, T. D., Malone, R. E., Symington, L. S., Recombination in yeast, in The molecular and cellular biology of the yeast Saccharomyces cereviesiae, vol. I, Genome dynamics, protein synthesis and energetics (a cura di J. R. Broach, J. R. Pringle e E. W. Jones), Cold Spring Harbor, N. Y., 1991, pp. 407-521.
Podda, S., Ward, M., Himelstein, A., Richardson, C., de la Flor-Weiss, E., Smith, L., Gottesman, M., Pastan, I., Bank, A., Transfer and expression of the human multiple drug resistance gene into live mice, in ‟Proceedings of the National Academy of Sciences", 1992, LXXXIX, pp. 9676-9680.
Riordan, J. R., Rommens, J. M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., Zielenski, J., Lok, S., Plavsic, N., Chou, J. L. e altri, Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA, in ‟Science", 1989, CCXLV, pp. 1066-1073.
Rosenfeld, M. A., Yoshimura, K., Trapnell, B. C., Yoneyama, K., Rosenthal, E. R., Dalemans, W., Fukayama, M., Bargon, J., Stier, L. E., Stratford-Perricaudet, L. e altri, In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the airway epithelium, in ‟Cell", 1992, LXVIII, pp. 143-155.
Sanger, F., Nicklen, S., Coulson, A. R., DNA sequencing with chain terminating inhibitors, in ‟Proceedings of the National Academy of Sciences", 1987, LXXIV, pp. 5463-5467.
Sanger, F., Nicklen, S., Coulson, A. R., The ADA human gene therapy clinical protocol, in ‟Human gene therapy", 1990, I, pp. 327-362.
U.S. Department of Health and Human Services - U.S. Department of Energy, Understanding our genetic inheritance. The U.S. Human Genome Project: the first five years, Bethesda, Md., 1990.
Vassarotti, A., Goffeau, A., Sequencing the yeast genome: the European effort, in ‟Trends in biotechnology", 1992, X, pp. 15-18.
Wagner, E., Zatloukal, K., Cotten, M., Kirlappos, H., Mechtler, K., Curiel, D. T., Birnstiel, M. L., Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes, in ‟Proceedings of the National Academy of Sciences", 1992, LXXXIX, pp. 6099-6103.
Watson, J. D., Crick, F. H. C., Molecular structure of nucleic acids, in ‟Nature", 1953, CLXXI, pp. 737-738.
Wilson, J. M., Grossman, M., Wu, C.H., Chowdhury, N. R., Wu, G. Y., Chowdhury, J. R., Hepatocyte-directed gene transfer in vivo leads to transient improvement of hypercholesterolemia in low density lipoprotein receptor-deficient rabbits, in ‟Journal of biological chemistry", 1992, CCLXVII, pp. 963-967.
Wu, G. Y., Wilson, J. M., Shalaby, F., Grossman, M., Shafritz, D. A., Wu, C. H., Receptor-mediated gene delivery in vivo. Partial correction of genetic analbuminemia in Nagase rats, in ‟Journal of biological chemistry", 1991, CCLXVI, pp. 14338-14342.
Yoshimura, K., Rosenfeld, M. A., Nakamura, H., Scherer, E. M., Pavirani, A., Lecocq, J. P., Crystal, R. G., Expression of the human cystic fibrosis transmembrane conductance regulator gene in the mouse lung after in vivo intratracheal plasmid-mediated gene transfer, in ‟Nucleic acids research", 1992, XX, pp. 3233-3240.