
Carbonio: stati fisici, chimici e proprietà
Carbonio: stati fisici, chimici e proprietà
L'atomo di carbonio è capace di legarsi a sé stesso e ad altri atomi, producendo un elevato numero di composti. Lo studio di questi composti è l'argomento di una specifica disciplina, chiamata chimica organica. Il carbonio era noto fin dall'Antichità: Plinio il Vecchio descrive l'impiego del nerofumo nella preparazione di inchiostri, ma spetta a Antoine-Laurent Lavoisier il merito di averlo riconosciuto come elemento. Jöns Jakob Berzelius ne determinò il peso atomico, Friedrich August Kekulé ne stabilì la tetravalenza e Jakobus Henricus van't Hoff la proprietà di dare origine a composti esistenti in forme enantiomorfe, otticamente attive, saturando le sue valenze con atomi o gruppi atomici differenti.
Tutto il carbonio degli organismi viventi proviene direttamente o indirettamente da quello dell'anidride carbonica atmosferica; i vegetali verdi mediante la funzione clorofilliana utilizzano l'anidride carbonica dell'atmosfera per sintetizzare, con l'intervento dell'energia solare, una serie di composti organici che sono poi utilizzati dalle piante stesse e dagli animali come materiale plastico da costruzione e per il loro metabolismo. Insieme ai due isotopi stabili, anche quello radioattivo, 146C, viene così a essere presente nella sostanza organica, la quale può essere perciò usata come materiale per un metodo di datazioni assolute.
Attualmente, attraverso processi di sintesi chimica, è possibile produrre una quantità impressionante di molecole, stimata in oltre 6 milioni. Tra le sostanze nelle quali il carbonio è legato solo a se stesso, alcune possiedono proprietà molto differenti tra loro, come nel caso della grafite e del diamante, definite allotropi del carbonio (la proprietà di una stessa sostanza semplice di avere strutture diverse è detta allotropia). A queste due sostanze, alla fine del XX sec., se ne sono aggiunte altre: i fullereni (1985) e i nanotubi (1991), che possiedono proprietà affascinanti, alcune delle quali ancora da scoprire e da valorizzare. Questo perché il carbonio avendo la capacità di legare sé stesso può formare strutture tra loro molto diverse e attribuire a ciascuna di esse proprietà differenti: piani paralleli facilmente sfaldabili nella grafite, rigide e compatte strutture tridimensionali nel diamante, oppure forme sferiche (a pallone) nel fullerene e tubolari nei nanotubi.
A tale proposito è opportuno ricordare che un atomo di carbonio isolato nel suo stato fondamentale ha una configurazione elettronica con due elettroni a spin accoppiati nello stato 2s e due elettroni disaccoppiati in due stati 2p. La formazione di legami chimici è associata, come originariamente proposto da Linus Pauling, alla promozione di uno dei due elettroni nello stato s al rimanente orbitale 2p e conseguente ibridazione degli orbitali, ciascuno occupato da un elettrone. Si possono ottenere quattro orbitali sp3, orientati verso i vertici di un tetraedro posto l'atomo di carbonio al suo centro, che intervengono nella formazione del diamante e degli idrocarburi alifatici saturi. Un'altra possibilità consiste nella combinazione di due orbitali p con l'orbitale s con formazione di tre orbitali ibridi sp2 che giacciono su un piano e sono orientati lungo tre linee rette che formano fra di loro angoli di 120°. Questi orbitali sono coinvolti nella formazione dello scheletro dei legami σ presenti negli idrocarburi etilenici, nel benzene e suoi derivati e nella grafite. Infine, dalla combinazione dell'orbitale s con uno degli orbitali p si formano due orbitali ibridi sp che giacciono su una linea retta e sono orientati in direzione opposta. Essi sono coinvolti nella formazione degli idrocarburi acetilenici.
Grafite
Fu Abraham G. Werner, nel 1789, a coniare il termine grafite, derivandolo dal verbo greco grafèin, che significa scrivere e rimanda all'uso della grafite come punta per le matite. In realtà, la grafite è troppo tenera per essere usata a questo scopo e pertanto viene mescolata con minerali argillosi per conferirle la necessaria durezza.
Proprietà
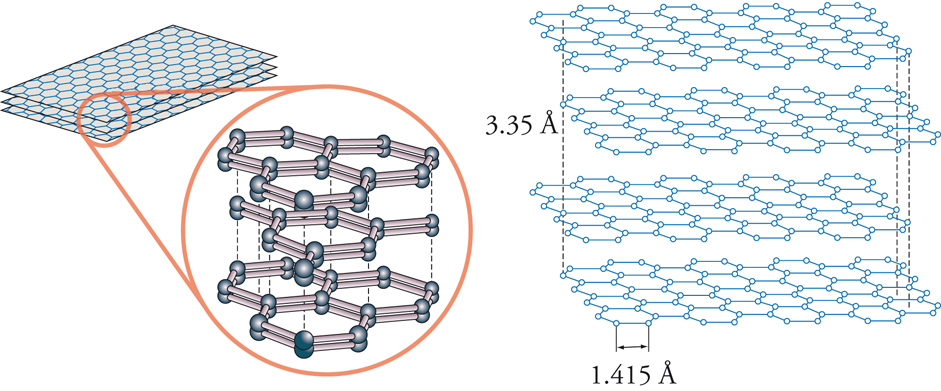
In natura la grafite si trova nelle rocce metamorfiche, come lo gneiss, il marmo e le scisti, ma può essere preparata anche artificialmente. Viene classificata come grafite cristallina a foglia, grafite cristallina a massa e grafite a massa amorfa. Dal punto di vista commerciale, la più conveniente è quella che si trova in Madagascar: da 13 tonnellate di scisti è possibile recuperare 1 tonnellata di grafite. Ha una durezza di 1÷2 sulla scala Mohs e una gravità specifica che, a seconda della purezza, varia tra 2,20 e 2,30 (per la grafite pura, 2,23). Essa è di colore grigio-nero, opaca, e ha una brillantezza metallica; è flessibile ma non elastica e possiede un punto di fusione di 3927 °C, cioè è altamente refrattaria. Tra i non-metalli, la grafite è quello con la più alta conducibilità elettrica e termica ed è chimicamente inerte. Tutte queste proprietà la rendono interessante per molte applicazioni industriali. La grafite è costituita da atomi di carbonio disposti ai vertici di esagoni regolari, planari; ciascun atomo di carbonio è legato ad altri 3 atomi di carbonio (fig. 1). La distanza del legame carbonio-carbonio è di 1,415 Å, valore intermedio tra il legame semplice (1,54) e quello doppio (1,33); i vari piani sono tenuti insieme da deboli forze di van der Waals e sono separati da una distanza di 3,35 Å. Il legame di 1 atomo di carbonio con soli 3 altri atomi (il carbonio è tetravalente) deriva da un mescolamento (ibridizzazione) dei 4 elettroni di legame con la creazione di 3 orbitali sp2 (disposti su un piano con angoli di legame di 120°), in ognuno dei quali si colloca un elettrone, mentre il quarto elettrone rimane in un orbitale p. Questo orbitale si dispone perpendicolarmente rispetto al piano definito dagli orbitali sp2 e così ogni atomo di carbonio possiede un elettrone spaiato in un orbitale p parallelo a quello di tutti gli altri atomi di carbonio, che, invece, si legano tra loro sovrapponendo gli orbitali sp2. In questa situazione, la grafite ha la possibilità di trasmettere il movimento degli elettroni in maniera facile lungo l'asse del piano definito dagli atomi di carbonio, permettendo così l'alta conducibilità elettrica direzionale già menzionata. Le deboli forze di van der Waals che tengono uniti i piani possono essere facilmente vinte, ed è ciò che accade quando si scrive con la punta della matita su un foglio di carta.
Grafite naturale e di sintesi
La grafite può essere naturale, nelle forme a scaglie o amorfa, oppure sintetica. La grafite naturale a scaglie è la forma cristallina del carbonio. La grafite amorfa è quella che si ritrova in natura, generata dalla metamorfosi di letti di carbone o di rocce sedimentarie carboniose. In realtà, essa possiede una struttura microcristallina, ma tende a essere più nera della grafite a scaglie. La grafite esiste in due forme: α (esagonale) e β (romboedrica) ed entrambe hanno identiche proprietà fisiche, tranne per la differente forma cristallina. La grafite naturale contiene fino al 30% di grafite β, quella prodotta sinteticamente è costituita solo dalla forma α: la grafite α può essere trasformata nella β attraverso trattamento meccanico, mentre la β torna alla forma α quando è riscaldata al di sopra di 1000 °C. La grafite è l'allotropo più stabile del carbonio, tuttavia è solo 0,69 kcal/mol più stabile del diamante a 26,85 °C. Conseguentemente, sarebbe logico attendersi che la interconversione tra le due strutture sia relativamente facile e che il diamante decomponga a grafite. In pratica, la grafite è stata convertita direttamente in diamante, ma solo alle estreme condizioni di 268,5 °C e 123,4 atm. Il decadimento del diamante in grafite ha un tempo di semivita di milioni di anni. Tutto ciò dipende dalla termodinamica: in realtà, esiste una barriera tra i due allotropi, un'elevata energia di attivazione che il diamante deve superare per dare luogo alla trasformazione. In più, a pressioni alte, il diamante diventa più stabile della grafite, ed è per questa ragione che a estreme condizioni avviene la trasformazione da grafite a diamante.
La grafite ha sia proprietà metalliche sia non metalliche: le prime includono la conducibilità termica e quella elettrica; le seconde, l'alta resistenza termica, l'inerzia e il potere lubrificante. Grazie alla combinazione di conducibilità e alta stabilità termica, la grafite viene utilizzata nelle batterie, nelle pile idrogeno-aria (celle a combustibile) e nei refrattari, nonché come spazzole dei motori elettrici. In quest'ultimo caso la grafite fornisce sia un'efficace lubrificazione al sito di frizione, sia una matrice capace di rimuovere contemporaneamente il calore dalla stessa zona. Nel 2004 la grafite naturale aveva un prezzo variabile tra i 230 e i 750 dollari a tonnellata, in funzione della purezza (contenuto in carbonio e ceneri), della forma cristallina e della distribuzione delle particelle. Nello stesso anno la produzione della grafite naturale era stimata in 982.000 t. La Cina ne produce circa il 70%, seguita dall'India (12%) e dal Brasile, dalla Repubblica popolare e democratica di Corea, dal Canada, dallo Zimbabwe, dalla Repubblica Ceca, dal Messico e dall'Ucraina.
La grafite sintetica viene preparata riscaldando a oltre 2500 °C il coke di petrolio. A queste temperature gli atomi di carbonio si ordinano in strati paralleli. Il coke è spezzettato e ridotto a particelle di dimensioni appropriate per l'uso finale. La polvere è quindi mescolata col bitume residuo della distillazione del carbone e con altri additivi che agiscono da collanti. Questo miscuglio può essere estruso o modellato in blocchi e cilindri. Una volta che il verde o i blocchi di carbone grezzo sono formati, vengono sottoposti a un esteso ciclo di cottura per convertire la massa in carbone solido. Questo processo può durare fino a 60 giorni e vi si presta molta cura per prevenire fratture nel materiale. Una volta completato il ciclo di cottura, il carbone cotto è pronto per il processo finale di grafitizzazione, che richiede di sottoporre il carbone a temperature di 2650÷2850 °C (raggiunte in fornaci con controllo dell'atmosfera). Un ulteriore beneficio dell'alta temperatura è quello di espellere tutte le impurezze volatili, in modo tale che la grafite (pura al 99,9%) contenga solo piccole quantità di elementi in traccia. Negli Stati Uniti, durante il 2004, la vendita di grafite sintetica, usata in particolare per innalzare il tenore di carbonio negli acciai, per supporti di catalizzatori, in metallurgia e per altri scopi, è stata pari a 249.000 t per un valore di circa 774 milioni di dollari, anche se la stima media era intorno a 1800 dollari a tonnellata.
La grafite pirolitica, una forma di grafite sintetica, viene preparata attraverso un processo denominato deposizione di vapore chimico o CVD (Chemical vapor deposition). Il metano, a bassa pressione (circa 133,3 Pa), viene scaldato a 2000 °C e quindi, sebbene molto lentamente, alla velocità di circa 2,4 cm/h, cresce uno strato di grafite. Preparata in questo modo, essa è altamente ordinata e gli strati di carbonio formano cristalli a strati, ordinati uno sull'altro come quelli di mica. La grafite pirolitica è più diamagnetica del bismuto nella direzione perpendicolare agli strati di carbonio. Siccome la densità della grafite pirolitica è più bassa di quella del bismuto (la gravità specifica è 2,1), essa è abbastanza leggera da gravitare al di sopra di un magnete sufficientemente potente. Ovviamente, uno strato può essere troppo pesante, ma, se lo si rende sottile al di sotto di 0,5 mm, resta semplicemente al di sopra di un magnete senza appoggiarvisi e, quando è spinto con un dito verso la superficie del magnete, torna indietro. In passato, la grafite si utilizzava soltanto per le matite e per i colori; attualmente, si usa per la preparazione di crogiuoli e di ceramica refrattaria, per lubrificanti, nell'industria elettrica, nell'industria siderurgica in varie leghe per batterie, negli impianti di raffreddamento delle centrali nucleari, nei freni automobilistici, per le spazzole per i motori elettrici, eccetera.
Nero di carbone, carta copiativa, inchiostro
Il nero di carbone è grafite colloidale preparata bruciando il metano in presenza di una quantità limitata di aria (cioè di ossigeno). Esso viene adoperato specialmente nella produzione della gomma, poiché, essendo questa molto suscettibile all'abrasione, se non fosse mescolata con grandi quantità di nero di carbone durerebbe pochissimo tempo (si pensi ai pneumatici delle automobili). Due famose applicazioni della grafite sono dovute a Thomas Edison; la prima riguarda la proprietà di resistenza dei granuli di carbone alla pressione a cui sono sottoposti. Edison, infatti, trovò che connettendo meccanicamente un diaframma acustico a un bottone di granuli di carbone, la resistenza può essere fatta variare in relazione alla pressione acustica, così che quando si passa una corrente attraverso il bottone, si produce una replica amplificata del segnale acustico. Questo permetteva di avere un segnale ben più forte di quello avuto dai diaframmi metallici usati da Alexander G. Bell e permise lo sviluppo delle comunicazioni telefoniche a lunga distanza. Edison ha anche usato filamenti al carbonio per le prime lampade a incandescenza.
La carta carbone (carta copiativa) è la seconda applicazione dovuta a Edison, sebbene oggi sia quasi completamente in disuso. Viene usato un film di nero di carbone spalmato su un foglio di carta sottile. La carta copiativa è intercalata tra vari fogli di carta, sul primo dei quali si esercita una pressione con il tasto di una macchina da scrivere (altra invenzione ormai in disuso) o di una penna: su tutti i fogli di carta successivi viene impressa la stessa immagine. Infine, una curiosità: una via per preparare la grafite colloidale è quella di bruciare una sostanza che produce una fiamma fumosa e di catturare il fumo su una superficie fredda. In latino questa sostanza era chiamata fuligo ed era usata per preparare la pittura nera o l'importante sostanza chiamata atramentum librarium, o inchiostro. Per preparare l'inchiostro, secondo quanto riporta Vitruvio, si frammenta la fuliggine in un mortaio e la si mescola con una gomma, per esempio d'acacia, e un po' d'acqua. La grafite colloidale è quindi peptidizzata dalla gomma, formando un sol stabile, l'inchiostro. Questa ricetta è stata scoperta in Egitto, dove fu inventata la scrittura su carta; il risultato è ora noto come inchiostro d'India o inchiostro di china. Infatti, benché la scrittura con inchiostro sia arrivata dall'Egitto, esso è stato contemporaneamente scoperto in Cina, dove però veniva usata la gelatina anziché la gomma.
Diamante
La parola diamante deriva dal latino adamas, a sua volta calco del greco, con il significato di indomabile e, per estensione, di acciaio temperato, cioè scaldato al calor rosso e poi raffreddato in acqua per dare una superficie indurita. Il diamante era una gemma rara nel Mediterraneo, venendo dall'esotica India. Plinio il Vecchio si riferisce al diamante come adamas per esprimerne la durezza, ossia la proprietà che gli conferisce importanza industriale. Fin dal IV sec. a.C. il diamante è stato riconosciuto come materiale prezioso. Il più antico riferimento conosciuto al diamante appare in un manoscritto in sanscrito prodotto da un ministro della dinastia regnante nel Nord dell'India (320-298 a.C.). In Europa appare come gioiello regale nel XIII sec., quando Luigi IX di Francia stabilì una legge che riservava i diamanti al re. Fino al 1725 l'India rimase la maggiore fornitrice di diamanti; poi si aggiunse il Brasile, vicino a Tejuca (ora Diamantina) nello Stato di Minas Gerais, e, nel 1867, la Repubblica Sudafricana (vicino all'Orange River e successivamente nella regione del Kimberley). La prima industria dedicata al taglio dei diamanti si sviluppò a Venezia, intorno agli anni Trenta del XIV sec., poi a Parigi, nel XIV sec., e successivamente a Bruges e Antwerp, nelle Fiandre. La produzione di diamanti è passata dai 50.000-100.000 carati all'anno dell'India, nel XVI sec., alla produzione mondiale (dati 2004) di circa 130×106 carati, di cui l'80% (100×106 carati o 20.000 kg) è destinato agli usi industriali e 30×106 (pari a 6000 kg) come pietra preziosa. I principali Paesi produttori sono: Repubblica Sudafricana, Botswana, Repubblica Democratica del Congo, Namibia, Federazione Russa, Australia, Angola, Cina, Canada e, in misura minore, Lesotho, Sierra Leone, Tanzania, Brasile, India.
Proprietà
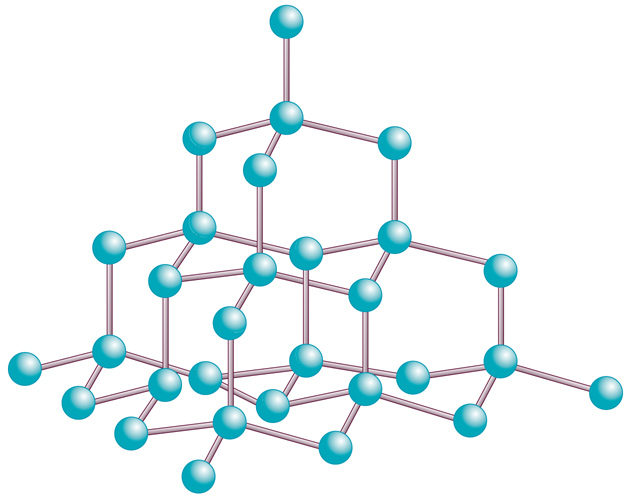
I cristalli di diamante sono costituiti di atomi di carbonio tetraedrici tetralegati (fig. 2: la lunghezza del legame tra gli atomi di carbonio è di 1,54 Å). Essi sono organizzati in anelli esagonali non planari come quelli del cicloesano, con i legami carbonio-carbonio che si estendono anche tra i piani definiti dagli anelli esagonali. La densità è di 3,51 g/cm3, un valore abbastanza alto per una sostanza non metallica. Il cristallo puro è trasparente, con indice di rifrazione che varia da 2,465 a 397 nm, da 2,427 a 527 nm, da 2,417 a 589 nm, da 2,408 a 670 nm, e da 2,402 a 763 nm. La variazione dell'indice di rifrazione dall'UV all'IR rende il diamante 3 volte più dispersivo del vetro. La durezza del diamante è il valore massimo della scala di Mohs, cioè 10; ciò lo rende massimamente abrasivo, da qui il suo principale uso industriale. Al calor rosso, cioè a circa 870 °C, il diamante brucia, fornendo anidride carbonica; in assenza di ossigeno, sublima a circa 3500 °C.
Fino al 1955 tutti i diamanti erano naturali, anche se parecchi tentativi di crearne di artificiali erano stati fatti fin dall'inizio del secolo. In quell'anno, infatti, piccole quantità di diamanti, di qualità industriale, furono preparati dalla grafite ad alta temperatura e alta pressione. Nel 1970 vennero preparati artificialmente diamanti di qualità pari alle gemme. Il prezzo del diamante dipende ovviamente dalla qualità della gemma: indicativamente oscilla (novembre 2005) da un minimo di 2336 a un massimo di 16.699 dollari a carato.
Diamanti naturali
Nella Terra i diamanti naturali sono stati creati da 1 a 3 miliardi di anni fa a profondità dove la pressione era sufficiente (ca. 55.000 atm) e la temperatura non troppo alta (ca. 1000 °C): queste condizioni si possono raggiungere a circa 150 km al di sotto dei continenti o 200 km al di sotto della crosta oceanica. La roccia era la peridotite, composta dal 60÷90% di olivina e dal 10÷40% di ortopirossene (o eclogite). Il diamante nasce come un piccolo cristallo su cui ‒ durante milioni di anni ‒ cresce a strati monomolecolari il carbonio, generando così forme cristalline (ottaedri, cubi, dodecaedri, ecc.). I diamanti sono stati portati alla superficie attraverso violente eruzioni causate dal rilascio di fluidi attivi, probabilmente acqua, nelle rocce calde del mantello superiore. La combinazione di questi fluidi con le rocce peridotitiche ed eclogitiche ha causato la corrosione della superficie delle regioni di più bassa pressione, creando dighe e prorompendo in getti caldi e turbolenti che hanno raggiunto la superficie in tubi a forma di carota chiamati diatremi, portando con loro alla superficie frammenti di roccia. Molte di queste eruzioni sono avvenute negli ultimi miliardi di anni, tipicamente nel basso Paleozoico e apparentemente nessuno dal Cretaceo in poi. Il caotico riempimento dei diatremi costituisce la roccia, facilmente erosa, chiamata kimberlite o lamproite, tipicamente arricchita in potassio, altrimenti abbastanza raro in queste rocce. I diatremi appaiono in regioni di rocce antiche e possono avere un diametro fino a 800 m.
I diamanti vengono estratti dalla kimberlite attraverso vari processi: (a) frammentazione e liberazione (si tratta di ridurre la kimberlite a piccoli frammenti, in modo da liberare i diamanti inclusi); (b) concentrazione, processo definito anche separazione di fase densa o DMS (Dense media separation); questo metodo utilizza il relativo differenziale di densità tra i diamanti e la roccia in cui erano inclusi. Il materiale da separare viene mescolato con una sospensione di ferrosilicato finemente diviso avente una densità di circa 2,65 g/cm3. La miscela viene pompata o spinta per gravità in un ciclone (un separatore dinamico che usa la forza centrifuga per effettuare la separazione), che produce un concentrato contenente diamanti e altro materiale di alta densità. Il ferrosilicato si recupera come surnatante; (c) recupero dei diamanti: il concentrato del precedente processo viene trattato in un'apposita sezione, in cui si sfruttano le proprietà specifiche del diamante per recuperarlo dal concentrato. Queste proprietà consistono nella suscettibilità magnetica, nella luminescenza ai raggi X e nella fluorescenza laser cristallografica. Il concentrato viene seccato o trattato umido attraverso un processo di separazione magnetica. La frazione non magnetica viene sottoposta a un processo di recupero con i raggi X: sotto questa irradiazione i diamanti assorbono energia, poi restituita sotto forma di luce, la quale viene riconosciuta in modo tale da consentire il recupero del diamante dal resto del campione. Dopo il recupero con i raggi X, il materiale subisce un ultimo ciclo di recupero e di concentrazione. Un separatore laser a effetto Raman viene adoperato per concentrare i diamanti fino a un livello del 95% in peso. Il separatore Raman utilizza il caratteristico segnale molecolare del diamante per distinguerlo dal restante materiale luminescente. Il più grande diamante mai trovato è il Cullinan (1905) di 3106 carati; tagliato in 105 diamanti più piccoli, tra cui la Stella d'Africa (530 carati), fu donato nel 1907 al re Edoardo VII e posto nello scettro reale conservato nella Torre di Londra.
Il taglio dei diamanti
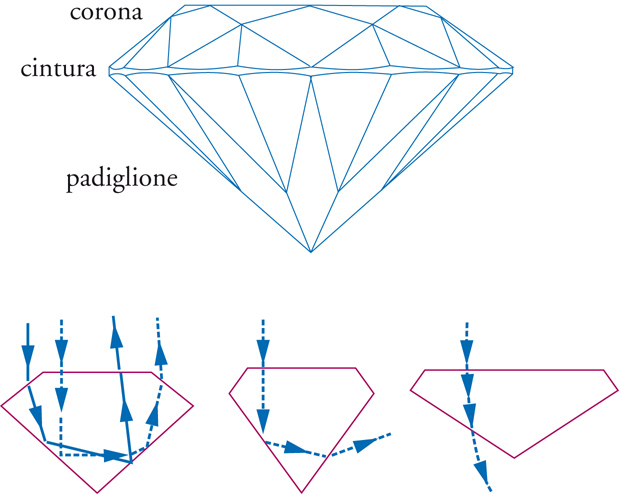
Per rendere più brillanti le pietre di diamante è necessario provvedere al loro taglio. Dal momento che esse sono le più dure fra tutti i materiali esistenti, sembrerebbe impossibile tagliare e pulire le facce di una gemma. In realtà, si può sfruttare il fatto che la durezza di un diamante dipende dalla direzione: esistono infatti direzioni più dure e più morbide. Benché non vi sia molta differenza, la direzione morbida può essere abrasa da una direzione dura. È questa proprietà che viene sfruttata nel taglio e nella pulizia di un diamante: si orienta la gemma nella direzione morbida, utilizzando un altro diamante, orientato nella direzione dura, per tagliarla. Il taglio, quindi, avviene seguendo alcuni punti specifici (fig. 3A): si identifica prima la tavola come la grande faccetta superiore. Il fuoco e la brillantezza del diamante diminuiscono se la tavola è troppo grande o troppo piccola (la tavola %, critica per lo scintillio e il fuoco del diamante, è uguale alla larghezza del diamante divisa per il diametro totale). La cintura è la stretta striscia intorno alla circonferenza esterna del diamante; il castone è la faccetta nella parte inferiore del diamante. La profondità del diamante si misura dalla tavola (faccia superiore) al castone (faccia inferiore), mentre la profondità % è data dalla misura dal castone alla tavola, diviso per la larghezza del diamante. Una profondità % troppo bassa o troppo alta provocherà uno sparpagliamento della luce uscente dalla pietra, con conseguente diminuzione dello scintillio. Il padiglione è la metà inferiore del diamante, dalla cintura inferiore al castone sulla punta inferiore. Se il padiglione è troppo alto o troppo basso, si avrà sparpagliamento della luce con conseguente diminuzione del fuoco e della brillantezza. I diamanti sono caratterizzati e valutati sulla base del peso in carati (carat weight), colore (colour), taglio (cut), purezza (clarity). Delle quattro caratteristiche del diamante, il taglio è quella più influenzata dall'uomo, poiché ne determina lo scintillio e il fuoco, perciò è dall'abilità del tagliatore che si rivela (o meno) la bellezza della gemma. Il taglio è ciò che permette al diamante di sprigionare la quantità massima di luce. Infatti, in un diamante tagliato correttamente la luce viene riflessa dalle faccette del padiglione in modo che fuoriesca dalla parte superiore della pietra. Inoltre, se un diamante viene tagliato con un padiglione troppo profondo, una frazione della luce si perde, uscendo dalla parte opposta del padiglione stesso. Infine, se il padiglione è troppo piatto, la luce esce prima che possa essere riflessa (fig. 3B). Un brillante ha tra 57 e 58 facce, a seconda che il padiglione sia completato da un punto o da una faccia (castone).
Peso in carati
Il peso di un diamante è espresso in carati. Il carato in origine era un'unità di peso naturale formata dai semi del carrubo. I diamanti erano per tradizione pesati con questi semi, fino a quando il sistema fu unificato e un carato fissato pari a 0,2 g. Un carato è diviso in 100 punti.
Colore
I diamanti possono coprire l'intero spettro dei colori. La maggior parte delle gemme, comunque, appare appena tinta di giallo o marrone, ma ci sono anche i diamanti molto rari definiti incolori. Altri, ancora più rari in natura, sono di colore ben definito e spesso vengono denominati fancies, fantasia. Tali diamanti si trovano solo occasionalmente e maggiormente in tinte come il verde, il rosso, il blu o l'ambra.
Purezza
Quasi tutti i diamanti contengono minute tracce di carbonio non cristallizzato, cioè dell'elemento dal quale sono stati originati. Molte tracce non sono visibili a occhio nudo e hanno bisogno di essere ingrandite per diventarlo. Sono chiamate inclusioni e sono le impronte digitali naturali che fanno di ogni diamante un pezzo unico. In ogni caso, meno inclusioni ci sono, più rara è la gemma.
I livelli di purezza sono classificati in modo diverso:
(a) FL (Flawless): nessuna inclusione interna o esterna di qualsiasi genere visibile a dieci ingrandimenti da un occhio esperto. È il livello massimo di purezza, corrispondente alle pietre più rare e costose;
(b) IF (Internally flawless): nessuna inclusione interna visibile a dieci ingrandimenti da un occhio esperto, ma potrebbero esservi alcune piccolissime imperfezioni esterne nella finitura;
(c) VVS-1 (Very very small inclusions-1): solitamente si tratta di un'unica piccolissima inclusione visibile a dieci ingrandimenti solo da un occhio esperto;
(d) VVS-2 (Very very small inclusions-2): piccolissime inclusioni visibili a dieci ingrandimenti solo da un occhio esperto;
(e) VS-1 (Very small inclusions-1): molto piccole inclusioni visibili a dieci ingrandimenti;
(f) VS-2 (Very small inclusions-2): diverse molto piccole inclusioni visibili a dieci ingrandimenti;
(g) SI-1 (Small inclusions-1): piccole inclusioni visibili a dieci ingrandimenti;
(h) SI-2 (Small inclusions-2): diverse piccole inclusioni visibili a dieci ingrandimenti;
(i) SI-3 (Slightly inclusions-3): inclusioni visibili a occhio nudo solo da un osservatore esperto;
(l) I-1 (Included-1): inclusioni visibili a occhio nudo;
(m) I-2 (Included-2): molte inclusioni distintamente visibili a occhio nudo che diminuiscono la brillantezza;
(n) I-3 (Included-3): molte inclusioni distintamente visibili a occhio nudo che diminuiscono la brillantezza e compromettono la struttura del diamante, rendendolo più fragile.
Diamanti artificiali
Per le loro proprietà di durezza, i diamanti hanno trovato anche larga applicazione industriale: infatti, circa l'80% della produzione totale trova impiego nelle costruzioni (seghe per materiali vari), nelle prospezioni minerarie (punte delle trivelle nelle perforazioni di pozzi petroliferi), nel taglio delle pietre preziose, nel taglio dei vetri (si stima che l'industria automobilistica americana consumi circa 1,5 carati per ogni automobile costruita). Il primo diamante sintetico venne prodotto nei primi anni Cinquanta del Novecento da ricercatori della Alemanna Svenska Elektriska Aktlebolager a Stoccolma, i quali non pubblicarono i loro risultati. Subito dopo ricercatori della General Electric pubblicarono su "Nature" il loro lavoro sulla sintesi di un diamante. Entrambi i gruppi usarono, come condizioni di pressione e temperatura, quelle ipotizzate per la produzione in natura. Una compagnia statunitense, la Gemesis di Saratoga (Florida), produce diamanti di notevoli dimensioni, usando camere di crescita. Un piccolo strato di diamanti naturali viene posto in un bagno di una soluzione di grafite fusa e di catalizzatori a base di metalli, a una temperatura di circa 1500 °C e una pressione di 58.000 atm. In questo processo, lentamente il carbonio precipita sui cristallini di diamante e una gemma di 2,8 carati di diamante giallo (il colore giallo è dovuto alla presenza di tracce di azoto nel cristallino, circa 5 atomi di azoto per ogni 100.000 di carbonio) cresce in meno di tre giorni e mezzo.
In effetti il metodo usato da Gemesis non permette il controllo delle eventuali impurezze che possono essere introdotte nel diamante sintetico. La nuova tecnica, CVD, appare in grado di controllare meglio la presenza (o l'assenza) di impurezze, anche in condizioni di lavoro meno spinte. Infatti la CVD funziona nel modo seguente: in una camera di reazione contenente uno strato di piccoli diamanti (spesso sintetici) viene fatta passare una corrente di idrogeno e metano gassosi. L'idrogeno è scisso, per azione di un filamento incandescente o di un plasma generato da microonde, in idrogeno atomico, il quale reagisce a sua volta con il metano per produrre idrogeno molecolare e il radicale metile. Quest'ultimo, infine, si deposita sui cristallini di diamante formando altri legami carbonio-carbonio, facendo così crescere il diamante sintetico. La produzione mondiale di questi ultimi è stata di circa 6×108 carati nel 2004, di cui circa 2,5×108 negli Stati Uniti.
Fullereni e nanotubi
Il 4 settembre 1985, nei laboratori della Rice University (Houston, Texas), Robert F. Curl jr., Harold W. Kroto e Richard E. Smalley, assieme ad altri due giovani ricercatori, formarono, durante lo studio della reazione di un arco elettrico tra due poli di grafite, una molecola costituita da 60 atomi di carbonio. Gli atomi erano organizzati in modo da formare un poliedro complesso contenente 12 facce pentagonali e 20 facce esagonali, con ogni pentagono circondato da sei esagoni. La simmetria era icosaedrica. A questo composto venne attribuito il nome di buckminster fullerene in ricordo delle case geodetiche realizzate dall'architetto Buckminster Fuller (1895-1983) per il padiglione degli Stati Uniti alla fiera internazionale EXPO67 a Montreal (1967), di forma simile a quella attribuita alla molecola in studio.
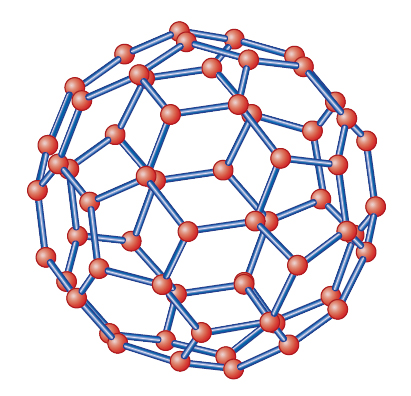
Essendo la struttura del fullerene, in verità, più immediatamente assimilabile a quella di un pallone da calcio (fig. 4), il composto è stato anche chiamato buckyball per associare il riferimento alle due strutture: finalmente il nome fullerene è rimasto a indicare tutti i composti del carbonio di forma sferica o sferoidale. In realtà, la figura del dodecaedro troncato, identica a quella della molecola di fullerene, fu disegnata da Piero della Francesca attorno al 1480 (Biblioteca Vaticana), e in maniera più importante e dettagliata riportata e discussa da Luca Pacioli nel trattato De divina proportione (1498), le cui illustrazioni furono realizzate da Leonardo da Vinci.
La struttura dei fullereni
Una molecola planare può essere costituita solo da esagoni (come nel caso della grafite), mentre per costruire una sfera occorre aggiungere anche pentagoni. Ma quanti? Nel 1770 Leonhard Euler ha dimostrato il teorema per cui in un solido vale la relazione V(ertici)− S(pigoli)+F(acce)=2. Se il solido è costituito da P(entagoni) ed E(sagoni), ovviamente F=P+E. Gli spigoli sono formati dall'accostamento delle due figure e quindi S=(5P+6E)/2. Più complesso è ricavare il numero di vertici: ogni vertice deve trovarsi all'incrocio tra pentagoni ed esagoni; la somma degli angoli interni del pentagono è di 108° e quella degli angoli interni dell'esagono di 120°, per cui non possono convergere più di tre figure geometriche: ne discende che V=(5P+6E)/3. Sostituendo nella prima relazione si ottiene P=12. Quindi è necessario che siano presenti almeno 12 pentagoni per creare una figura come il fullerene; di conseguenza, se il numero di vertici è 60, come gli atomi di carbonio, il fullerene contiene, oltre ai 12 pentagoni, 20 esagoni per 60 vertici, 90 spigoli e 32 facce. Il numero minimo di atomi di carbonio per un fullerene è quindi di 20 e il numero deve comunque essere pari anche per tutti i fullereni con più atomi di carbonio.
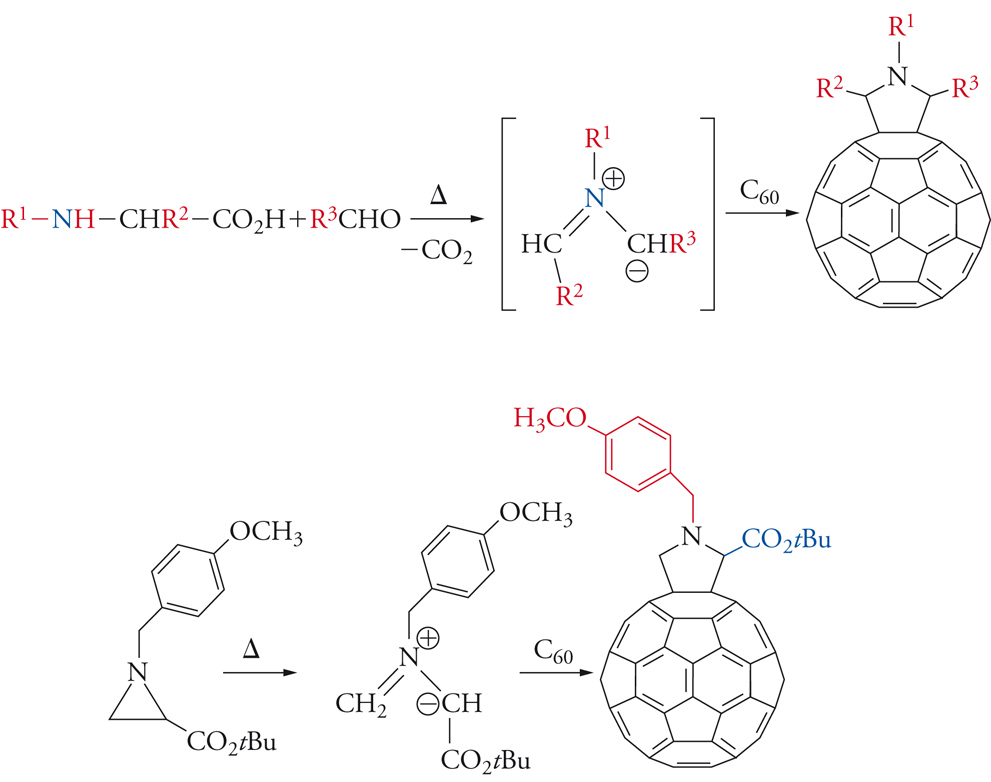
Il fullerene più abbondante, il C60, è stato studiato in dettaglio per quanto riguarda le proprietà reattive. In questa molecola vi sono due tipi di legami carbonio-carbonio: 30 congiungono i pentagoni agli esagoni e 60 formano i pentagoni. I primi possiedono una lunghezza di 1,386 Å e i secondi di 1,434 Å, vicino cioè ai valori per un legame doppio e singolo, rispettivamente (la media tra i due corrisponde alla lunghezza del legame nella grafite, dove tutte le lunghezze di legame sono equivalenti). Il diametro del fullerene è di circa 7 Å: se assumiamo che il raggio di un atomo di carbonio è di 0,7 Å, risulta che il 54% del volume del fullerene è vuoto. Gli atomi di carbonio ibridizzati sp2 (quelli del legame quasi doppio nel fullerene) impartiscono alla molecola una forte energia tensionale, che viene rilasciata in una reazione di addizione, abbastanza facile. Sono infatti state realizzate molte reazioni di somma, sia di monoaddizione che di poliaddizione. In particolare da notare la reazione 1,3-dipolare di ilidi azometiniche, che conduce alla formazione di fulleropirrolidine (fig. 5) legando l'1,3 dipolo a uno dei doppi legami dell'anello a 6 atomi.
È stato possibile addizionare al fullerene molti altri gruppi, creando molecole interessanti per le proprietà ottiche (limitatori), fotovoltaiche e anche per alcune proprietà biologiche (per es., come inibitore dell'HIV proteasi). È stato anche possibile introdurre, all'interno del fullerene, alcuni atomi, come quello dell'azoto. Oltre al fullerene C60 esistono molti altri fullereni: C70, C76, C78, C80, C84, fino a C960 e oltre. Sono stati creati fullereni all'interno di altri fullereni, in modo che siano concentrici e che possano somigliare a cipolle con un numero di strati fino a 70. Tuttavia, nonostante un elevato numero di brevetti, non vi è, al momento, alcuna applicazione pratica di queste molecole e l'interesse della chimica di base si sta spostando verso lo studio dei nanotubi.
Nanotubi
Se il fullerene viene rappresentato col simbolo C60, poiché è formato da 60 atomi di carbonio, un nanotubo potrebbe essere rappresentato da C1000000 (106 atomi di carbonio). Esso può essere definito come un cristallo singolo in una direzione con una cella unitaria che si propaga e ripete creando una struttura a elica. I nanotubi sono stati scoperti nel 1991 da Sumio Iijima della NEC Corporation, scaricando un arco a corrente diretta tra due elettrodi di carbonio immersi in un'atmosfera di un gas nobile. La presenza di un campo elettrico nella scarica ad arco sembra promuovere la formazione di nanotubi, infatti essi si formano, solo quando passa la corrente, sul più grande elettrodo negativo. La velocità di deposizione è di circa 1 mm/minuto con una corrente di 100 A di intensità e una differenza di potenziale di 20 V, che mantiene una temperatura di circa 2000÷3000 °C. L'aggiunta di piccole quantità di polvere di metalli di transizione (cobalto, nickel o ferro) favorisce la crescita dei cosiddetti nanotubi a parete singola. Recenti esperimenti di vaporizzazione laser di miscele carbonio-catalizzatore hanno permesso di abbassare la temperatura a 1200 °C e produrre nanotubi a parete singola con rese fino al 70%.
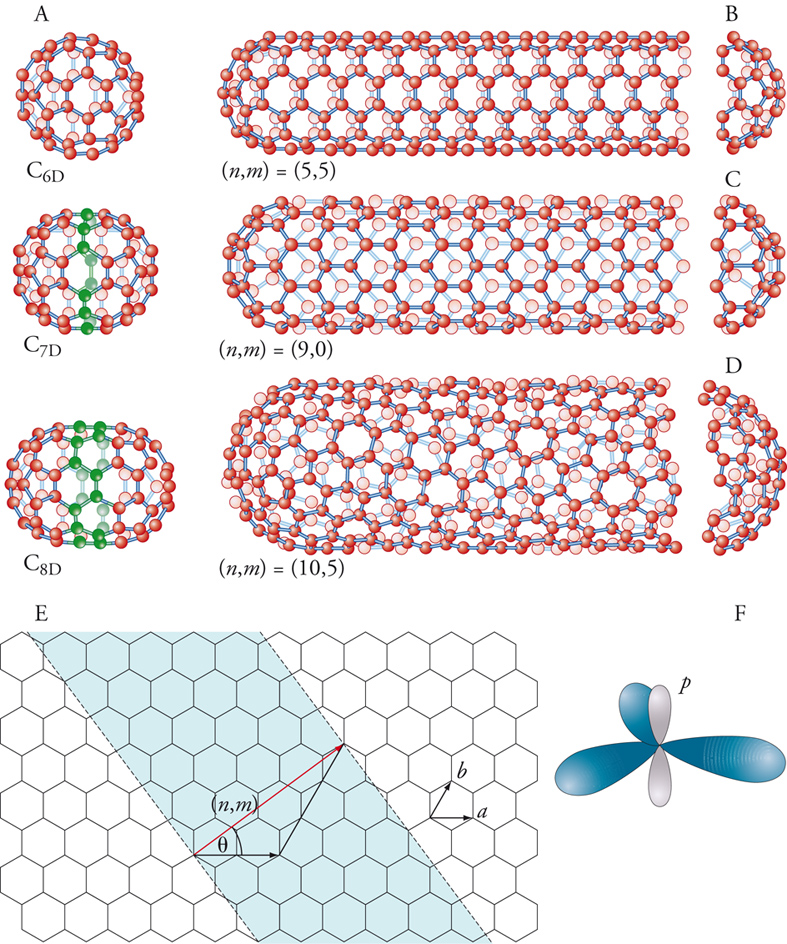
Si può pensare di costruire un nanotubo a partire da un fullerene (fig. 6). Per esempio, si taglia un C60 a metà e si inserisce una stringa di 10 atomi di carbonio per ottenere un C70 e poi altre stringhe, fino a creare un nanotubo. In effetti, il modo con cui vengono aggiunte le stringhe di atomi può condurre a tubi di struttura diversa: tubi a zig-zag, in cui gli anelli a 6 sono in file allineate e parallele, una seconda struttura a poltrona, una terza chirale. Il tutto dipende dall'angolo θ di rotazione della fila di anelli a 6 atomi: 0 gradi per la struttura a zig-zag; 30 gradi per la struttura a poltrona, e intermedia per la chirale. Vi è una precisa convenzione per distinguere i nanotubi arrotolati in modo differente. Bisogna specificare il numero di vettori unitari necessari per connettere 2 atomi nel lattice planare esagonale per formare il tubo. Questi numeri specificano un vettore espresso come (m, n), dove m e n sono interi: questi numeri costituiscono un nome unico per il tubo. Ogni tubo chiamato (n, 0) ha legami carbonio-carbonio che sono paralleli all'asse del tubo, e formano, dal lato aperto, un pattern a zig-zag: infatti, questi sono chiamati tubi a zig-zag. I tubi (n,n), con i due interi uguali, hanno legami carbonio-carbonio che sono perpendicolari all'asse del tubo e sono chiamati tubi a poltrona. Questi due tipi di nanotubi sono achirali, cioè non hanno due immagini speculari diverse (come la mano destra e quella sinistra). Tutti gli altri tubi, chiamati (m, n) sono chirali e hanno le varianti destrogira e levogira.
Per molti aspetti le proprietà dei tubi di tipo differente sono essenzialmente le stesse. L'eccezione risiede nella loro conducibilità elettrica, dove queste sottili differenze possono produrre effetti macroscopici. Per esempio, tutti i tubi a poltrona, cioè quelli dove m=n, hanno conducibilità elettrica di tipo metallico. Essi trasportano elettroni lungo l'asse del tubo, proprio come fanno i metalli, anche se non hanno alcun atomo di metallo nella loro struttura. Per contrasto, gli altri tubi sono intrinsecamente semiconduttori. Dal momento che i tubi (m, n) sono molecolarmente distinti, esiste la possibilità di separare chimicamente i vari tipi o anche di produrli selettivamente, anche se al momento queste sono solo ipotesi di ricerca. Un altro aspetto strutturale è che i tubi possono organizzarsi in corde, nelle quali, tipicamente, da dieci a 100 tubi rimangono uniti, tenuti insieme da forze di van der Waals, per la loro lunghezza. Anzi, le corde sono molto più lunghe di un singolo nanotubo: i nanotubi, infatti, sono lunghi da 100 a 1000 nm e possiedono un diametro di circa 1 nm. Le corde, invece, sono illimitate, agganciandosi nanotubo a nanotubo. Esse sono molto utili per produrre vie di conducibilità molto lunghe.
Non è ancora possibile misurare direttamente la forza necessaria per rompere un nanotubo: tuttavia, calcoli teorici hanno mostrato che un fascio di nanotubi del diametro di circa 2,4 cm (1014 nanotubi paralleli uniti assieme) potrebbe avere una forza pari a 130 GPa (cioè, ca. 1.300.000 atm), ovvero circa 100 volte più forte dell'acciaio ma con meno di 1/6 del suo peso. È stato possibile ridurre le dimensioni (lunghezza) dei nanotubi, mediante robuste ossidazioni, e funzionalizzare le estremità, creando nanotubi di dimensioni più ridotte ma solubili nei normali solventi organici. Sono state anche funzionalizzate le pareti laterali dei nanotubi e le molecole risultanti sono oggetto di studio per le loro proprietà. Tra le possibili applicazioni dei nanotubi vi è quella di collegare una serie di essi per formare fili e utilizzarli come materiali per costruire veicoli di leggerezza e forza imbattibili. Potrebbero anche essere usati, se il loro costo lo permettesse, per costruire ponti o alti edifici resistenti ai terremoti: tutte le possibili applicazioni derivano dalle proprietà meccaniche di queste sostanze, ma richiedono una loro produzione di massa, che sia anche economica.
Altre numerose applicazioni si possono immaginare nella microelettronica, dove sarebbe possibile utilizzare la combinazione della relativamente bassa velocità di trasporto di calore nella direzione perpendicolare, per utilizzarli come centri di raffreddamento (heath sink), essendo la produzione di computer della nuova generazione bloccata dal problema di sovrariscaldamento interno. Un'altra applicazione consiste nell'utilizzazione dei nanotubi come nanocannucce, in modo da poter iniettare all'interno delle strutture cellulari alcune molecole senza interessare le zone adiacenti. Queste applicazioni saranno possibili dopo aver risolto il problema dell'inserimento dei nanotubi all'interno di apparecchiature e di circuiti, in modo pratico e riproducibile, insieme alla necessità di produrre nanotubi di alta qualità in grandi volumi.
Bibliografia
Iijima, Sumio, Helical microtubules of graphitic carbon, "Nature", 354, 1991, pp. 56-58.
Maggini, Michele - Prato, Maurizio - Scorrano, Gianfranco, Addition of azomethine ylides to C60: syn-thesis, characterization and functionalization of fullerene pyrrolidines, "Journal of the American Chemical Society", 115, 1993, pp. 9798-9799.
Olson, Donald W., Diamond (industrial), "U.S. geological survey, mineral commodity summaries", 2003, pp. 58-59.
Olson, Donald W., Graphite, "Metals and minerals: U.S. geological survey, minerals yearbook", 2004, pp. 34.1-34.13.
Pauling, Linus - Hayward, Roger, The architecture of molecules, San Francisco, Freeman, 1964.
Prato, Maurizio - Maggini, Michele, Fulleropyrrolidines: a family of full-fledged fullerene deriv-atives, "Accounts of chemical research", 31, 1998, pp. 519-526.
Scorrano, Gianfranco, Fullereni, in: Enciclopedia del Novecento, Roma, Istituto della Enciclopedia Ita-liana, 2004, Suppl. III, pp. 510-517.
Smalley, Richard E., Discovering the fullerenes, "Angewandte Chemie. International edition", 36, 1997, pp. 1595-1601.
Yarnell, Amanda, The many facets of man-made diamonds, "Chemical and engineering news", 82, 2004, pp. 26-31.