
Gene
Gene
Come per tutti gli altri elementi, anche nel caso del gene non è semplice darne una definizione. Il primo passo verso la comprensione di qualsiasi tipo di unità consiste nel darne una definizione soddisfacente. Appellandoci al buon senso, potremmo dire che i criteri principali per decidere della bontà o meno di una definizione di un'unità elementare sono, da una parte, il grado della sua univocità, cioè l'essere i suoi confini netti piuttosto che sfumati; e dall'altra, la quantità di eccezioni. E qui ci si accorge subito che, anche limitandoci solo al campo della genetica, non tutte le unità sono definibili in modo altrettanto convincente. Per esempio, mentre 'genoma aploide', 'cromosoma', 'coppia di desossiribonucleotidi' (cioè un'unità di mutazione e di ricombinazione), 'cellula' e 'individuo' sono unità genetiche discrete, e quindi definibili in modo soddisfacente, al contrario 'popolazione', 'specie' e 'gene' sono unità a confini fortemente vaghi e sfumati. In particolare, gene è un'unità la cui definizione ha subito numerosi e radicali modificazioni in rapporto al grado di conoscenza raggiunto nel campo della genetica (soprattutto molecolare). Occorre però aggiungere che le definizioni che via via sono state create si sono aggiunte (più che andate a sostituire) alle precedenti. In altre parole, molte delle definizioni più antiche sono tuttora valide e utili in determinati contesti.
Il gene è un'unità genetica molto sui generis perché la più piccola quantità di materiale genetico di un organismo dotata di tutte le sue proprietà informazionali è il suo genoma aploide, non un suo gene. Un gene rappresenta, invece, la più piccola parte di un genoma in grado, almeno potenzialmente, di dare origine a una catena polipeptidica. L'approccio storico è il modo più conveniente per rendere maggiormente comprensibile il concetto di gene così come lo si è concepito in tre periodi che si sono succeduti ma al tempo stesso si sono anche ampiamente sovrapposti, ciò causando una certa arbitrarietà nella loro definizione precisa: (a) quello della genetica mendeliana-morganiana o premolecolare, che va dalla riscoperta delle leggi di Mendel, all'inizio del Novecento, fino agli anni Cinquanta, e che ha portato al concetto di gene come unità puramente formale; (b) quello della genetica molecolare nella sua prima fase prevalentemente 'proteica', in cui cioè gran parte delle informazioni sui geni erano ottenute per inferenze che partivano dalle conoscenze delle catene polipeptidiche da essi codificate; e in una fase successiva caratterizzata soprattutto dalla scoperta degli introni nel 1976. È in questo periodo che si è arrivati al concetto classico di gene strutturale che codifica per una catena polipeptidica, perfettamente valido tuttora e a cui è stato qui assegnato l'aggettivo di 'ortodosso'; questo periodo arriva fino a circa la metà degli anni Ottanta; (c) quello attuale, in cui le conoscenze sui geni, anzi sul materiale genetico in generale, sono state ottenute direttamente, invece che per inferenza dalle proteine: la genetica ‒ per quella sua parte tesa a chiarire la struttura chimica dettagliata del materiale ereditario ‒ è così sempre più diventata una scienza descrittiva. È evidente che questa, come qualsiasi altra suddivisione storica, è in larga misura arbitraria, anche perché i tre periodi appena elencati si sovrappongono ampiamente fra loro.
La più semplice definizione dei geni e la loro suddivisione in due classi
Sono geni tutti i segmenti del genoma suscettibili di essere trascritti. I geni vengono suddivisi in due categorie: i geni 'destinati' a essere trascritti e tradotti costituiscono la classe dei geni strutturali che si esprimono nelle catene polipeptidiche; mentre i geni che sono solo trascritti (RNA) costituiscono la classe dei geni i cui 'esecutori fenotipici' sono gli RNA come tali. I primi sono quelli di gran lunga più studiati: a tal punto che, a meno di precisazioni, quando si dice 'gene' comunemente si intende gene che esprime una catena polipeptidica. E anche qui si farà questa scelta, addirittura non si parlerà affatto dei geni degli RNA 'non destinati' a essere tradotti. Questi ultimi comprendono i geni per gli RNA transfer e per gli RNA ribosomali, alcuni dei quali sono ribozimi, cioè molecole di RNA capaci di catalizzare reazioni chimiche; per queste loro proprietà sono ritenuti i discendenti attuali delle prime macromolecole della vita (in quanto forniti tanto di proprietà informazionali che catalitiche, a differenza del DNA e delle proteine).
Concetto 'premolecolare' di gene
Nel periodo premolecolare la genetica si era sviluppata attraverso due approcci soltanto, quello formale (che comprendeva la genetica delle popolazioni) e quello citogenetico.
L'approccio formale. Basato solo su inferenze statistiche (come quelle delle leggi di Mendel sulle frequenze relative dei vari fenotipi nella progenie di determinati incroci, esso ha portato alla individuazione (soprattutto in Drosophila melanogaster) di alcune centinaia di caratteri ereditari che, in base alle modalità di trasmissione, potevano essere attribuiti con certezza a entità singole, di cui non si sapeva nulla salvo che erano singole e che erano situate nei cromosomi. Queste conoscenze, solo apparentemente limitate, in realtà hanno costituito la base ‒ solidissima, e mai smentita in seguito ‒ di tutta la genetica. Tali entità, inizialmente denominate 'fattori mendeliani' (caratteri unifattoriali), sono state poi chiamate geni. Per molto tempo ne sono stati del tutto ignorati la natura chimica (fino al punto di ritenere che fossero proteine), la forma, le dimensioni e il modo di funzionare. Un ulteriore, gravissimo, limite derivava dal modo ‒ l'unico disponibile ‒ con cui si potevano individuare i geni: un gene era scopribile solo in base alle modalità di trasmissione degli eventuali caratteri fenotipici alternativi da esso dipendenti (per es., occhio rosso oppure bianco; piselli bianchi oppure verdi; statura normale oppure nanismo acondroplasico). Quindi si potevano scoprire solo geni variabili, cioè con almeno due alleli e, in questo ambito già molto limitato, solo se le variazioni fenotipiche associate ai genotipi per questi alleli erano percepibili con i metodi di indagine di quel tempo, che erano ancora rudimentali.
Le variazioni fenotipiche potenzialmente utili erano perciò solo una frazione molto piccola di quelle reali. Poiché quello che si scopriva era in genere un fenotipo molto evidente causato dall'assenza di funzione di un determinato gene (della cui esistenza si sarebbe rimasti all'oscuro se non si fosse scoperto quel fenotipo), ne era risultata una nomenclatura molto peculiare ma pressoché inevitabile, che rispecchiava l'ignoranza riguardo alla funzione perfino dei pochi geni allora noti: un gene non veniva denominato per quello che faceva normalmente, perché ciò restava sconosciuto anche dopo la sua scoperta, ma per ciò che succedeva quando la sua funzione veniva a mancare; come se chiamassimo il rene l'organo del coma uremico, invece che l'organo della escrezione. Per esempio, il gene che ora chiamiamo 'gene del fattore VIII della coagulazione' era stato denominato 'gene della emofilia', perché lo si era scoperto chiarendo le modalità di trasmissione di questa malattia. Del resto ciò è esattamente quello che era accaduto per le vitamine, a cui per decenni è stato assegnato il nome riferendosi alle malattie che insorgevano in seguito alla loro carenza: per esempio, la vitamina B1 era denominata 'vitamina antiberiberica', invece di precocarbossilasi (cioè precursore del coenzima della carbossilasi).
L'approccio citogenetico. Ha permesso la descrizione, in certi casi molto dettagliata (cromosomi giganti, o salivari, di D. melanogaster), ma comunque limitata dal livello di risoluzione consentito dal microscopio ottico, dei cromosomi di alcune specie (dalle quali è rimasta esclusa per molto tempo la nostra). I tre progressi più significativi nella caratterizzazione dei geni, compiuti nel periodo premolecolare, sono stati la loro localizzazione, il test di allelismo e il test cis-trans.
La localizzazione dei geni
Il primo e fondamentale risultato è stato la dimostrazione che i geni sono situati nei cromosomi, donde la concezione, piuttosto naïf considerando le conoscenze della genetica attuale, che i cromosomi fossero semplicemente una collana di geni. In seguito si è raggiunta una localizzazione molto più dettagliata soprattutto nei batteriofagi, nei batteri e in D. melanogaster, ovviamente con approcci molto diversi da un caso all'altro.
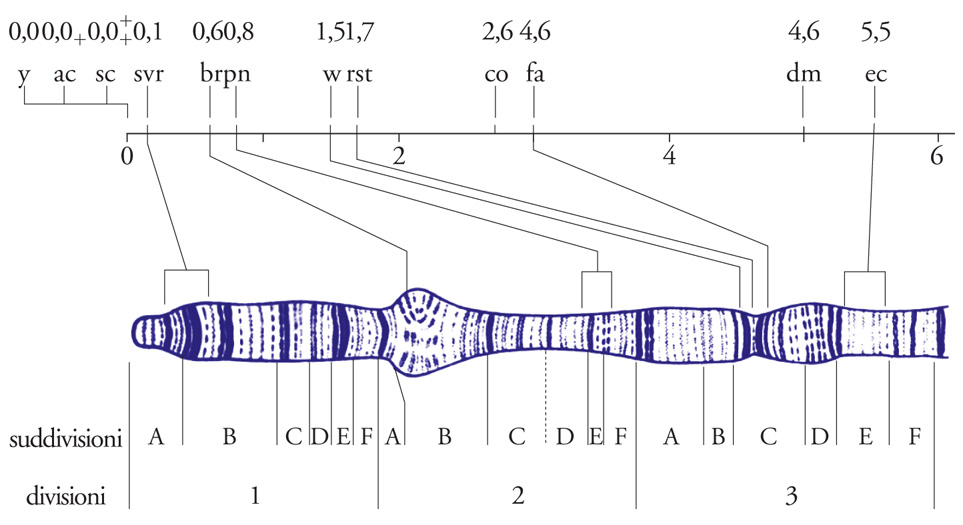
In Escherichia coli, il metodo più noto per localizzare i suoi geni lungo il cromosoma circolare che costituisce il suo genoma ha sfruttato il fenomeno della coniugazione. In particolari condizioni e con specifici ceppi di E. coli, un batterio 'donatore', dopo aver formato un ponte citoplasmatico con un batterio 'ricevente', sintetizza una copia del proprio cromosoma e la trasferisce in modo sequenziale a partire da un punto di inizio fino ad arrivare, a processo ultimato, a trasferire l'estremità opposta; viene trasferito cioè l'intero cromosoma. Questo è quello che si verifica se il processo viene lasciato proseguire indisturbato fino al suo completamento. Però è sperimentalmente possibile interromperlo in tempi diversi, accertando così quali geni sono passati dal donatore all'accettore e quali invece 'non hanno fatto in tempo' a essere trasferiti; in questo modo, si possono determinare le posizioni relative dei geni lungo il cromosoma batterico in base al tempo necessario perché ognuno di essi venga trasferito. In D. melanogaster si è arrivati a una conoscenza molto avanzata combinando le informazioni ottenute con entrambi gli approcci: mediante l'approccio formale si sono ottenute mappe di ricombinazione, mentre attraverso quello citogenetico si sono potute costruire mappe fisiche (cioè, i geni sono stati inseriti in posizioni precise su uno dei cromosomi giganti). Il confronto tra queste due mappe (fig. 3) ha dimostrato che sono perfettamente colineari, cioè che i geni vi si dispongono nello stesso ordine, sebbene le distanze relative che li separano varino molto da una mappa all'altra.
Il test di allelismo (o test di complementazione)
In molti casi di fenotipi anormali su base genetica, come per esempio microrganismi normalmente autotrofi che invece presentavano richieste nutrizionali (istidina−, cioè richiedenti l'istidina) o individui con deficit funzionali (per es., sordità), questo test ha permesso di accertare se tali alterazioni erano dovute a un'anomalia di un solo gene o di più geni. La base logica di questo test è addirittura elementare. La si può esporre con un esempio molto semplice: se la progenie di due individui affetti da sordità autosomica recessiva (cioè omozigoti per un allele silente di un gene autosomico necessario per un udito normale) è costituita da figli con udito normale, è certo che il gene compromesso in un partner è diverso da quello compromesso nell'altro, per cui si può concludere che i geni coinvolti nell'udito sono almeno due, cioè che costituiscono almeno due gruppi di complementazione (infatti l'incrocio sarebbe interpretabile come A0A0;B+B+× A+A+;B0B0 e i figli, essendo tutti A0A+/B+B0 e quindi possedendo tutti un allele + tanto per il gene A che per il B, avrebbero tutti un udito normale); se, invece, tutti i loro figli sono anch'essi sordi, è segno che il gene compromesso in un genitore è lo stesso di quello compromesso nell'altro (ciò, naturalmente, non escluderebbe che i geni coinvolti nell'udito siano più di uno, ma che i due genitori possano essere omozigoti per un allele silente dello stesso gene).
Il test cis-trans e i cistroni
Questo test, insieme a quello precedente, ha portato all'individuazione dei geni come 'unità di funzione', pur in assenza di qualsiasi informazione sulla loro natura chimica e sul loro modo di funzionare, cioè con un approccio esclusivamente formale. Esso permette di accertare se due siti genetici che influenzano lo stesso carattere e di cui si è a conoscenza che sono molto vicini (per cui non sarebbe da escludere che siano situati nello stesso gene) facciano o meno parte della stessa unità genetica funzionale che, se definita con questo test, viene denominata 'cistrone'. Il principio su cui si basa questo approccio è concettualmente molto semplice, ma esso non è stato mai di facile attuazione, perché lo si può applicare solo a materiali biologici che soddisfino requisiti molto stringenti, soprattutto in termini del grado di conoscenza già disponibile su di essi (che deve essere molto avanzato) e della 'maneggevolezza' come materiale sperimentale (incroci facilmente realizzabili, tempi di generazione brevi e progenie molto numerosa).
Si supponga di avere individuato due siti genetici A e B, ciascuno con un allele normale e uno silente (A+ e A0; B+ e B0) e che l'omozigosi, anche per uno solo degli alleli silenti, abolisca una certa funzione riconoscibile fenotipicamente, e che la funzione compromessa sia la stessa per i due siti. Si pone allora la domanda: i due siti sono situati nella stessa, oppure in due distinte unità funzionali (comunque entrambe necessarie per la comparsa del fenotipo normale)? Per poter rispondere mediante il test cis-trans si devono avere due tipi di individui entrambi eterozigoti tanto per il sito A che per quello B, ma che differiscono fra loro per il modo in cui questi alleli sono assortiti: più precisamente, si deve avere almeno un individuo A+B+/A0B0 e almeno uno A+B0/A0B+ (nel primo caso i due alleli 0 e i due alleli + sono stati ricevuti dallo stesso genitore, e si dice che sono in cis; nel secondo caso sono stati ricevuti da due genitori diversi e si dice che sono in trans). Se i due tipi di individui presentano lo stesso fenotipo (normale), cioè se l'assortimento dei 4 alleli è irrilevante per il fenotipo, si conclude che i due siti fanno parte di cistroni diversi; se, invece, i due tipi di individui differiscono e, in particolare, il fenotipo del primo è normale e quello dell'altro corrisponde alla mancanza della funzione, si conclude che A e B fanno parte dello stesso cistrone. Il perché di queste conclusioni è ovvio. Infatti, se i due siti sono situati nella stessa unità funzionale, solo il genotipo A+B+/A0B0 ha un fenotipo normale (per il fatto di possedere, sia pure in singola dose, un'unità funzionante, in quanto normale tanto nel sito A che nel B), mentre nel genotipo A+B0/A0B+ non funzionerebbe nessuna delle due copie dell'unità funzionale (essendo una alterata nel sito A e l'altra nel B).
È fondamentale chiarire che il tipo di cistrone, cioè di unità funzionale, identificabile con il test cis-trans, dipende dal fenotipo che si prende in esame. Tornando all'esempio appena presentato e ragionando con quello che sappiamo attualmente, se supponiamo che A e B siano risultati localizzati in cistroni diversi sulla base di un fenotipo accertato a livello proteico qualitativo (cioè in sequenze che codificano per catene polipeptidiche diverse), ciò non escluderebbe affatto che essi appartengano alla stessa unità funzionale responsabile di un altro fenotipo: un caso classico tra i moltissimi esistenti è quello di sequenze che, pur codificando per catene polipeptidiche diverse, dipendono tutte da una sequenza regolatrice unica, come l'operatore di operoni del tipo della β-galattosidasi e dell'istidina (in casi come questo il cistrone si identifica, invece che con una sequenza codificante, con la regione genetica detta 'operone', che comprende tutte le sequenze codificanti di quell'operone più un operatore condiviso da tutte, che cioè le coregola tutte assieme). In altre parole, l'operone è un singolo cistrone regolativo che comprende cistroni qualitativi (o 'codificoni') diversi. Si deve notare che il test cis-trans non porta alla descrizione delle unità genetiche funzionali, bensì solo ad accertare se siti genetici diversi, ma che controllano lo stesso carattere e sono molto vicini, fanno parte della stessa unità funzionale, che resta però elusiva. Se si confrontano i mezzi di indagine dell'epoca premolecolare con quelli attuali, si resta sbalorditi ‒ può sembrare un'esagerazione, ma non lo è ‒ da quanto fosse avanzata la genetica: nessuna delle inferenze fondamentali a cui si era giunti sui geni senza conoscerne né la natura chimica, né il modo di funzionare è stata in seguito dimostrata falsa, cioè, usando la terminologia di Karl Popper, 'falsificata'. Questo è probabilmente uno degli esempi che meglio illustrano l'efficacia conoscitiva raggiungibile con inferenze basate su una buona logica e su fatti bene accertati.
Concetto 'molecolare' di gene: i geni strutturali 'ortodossi' delle catene polipeptidiche
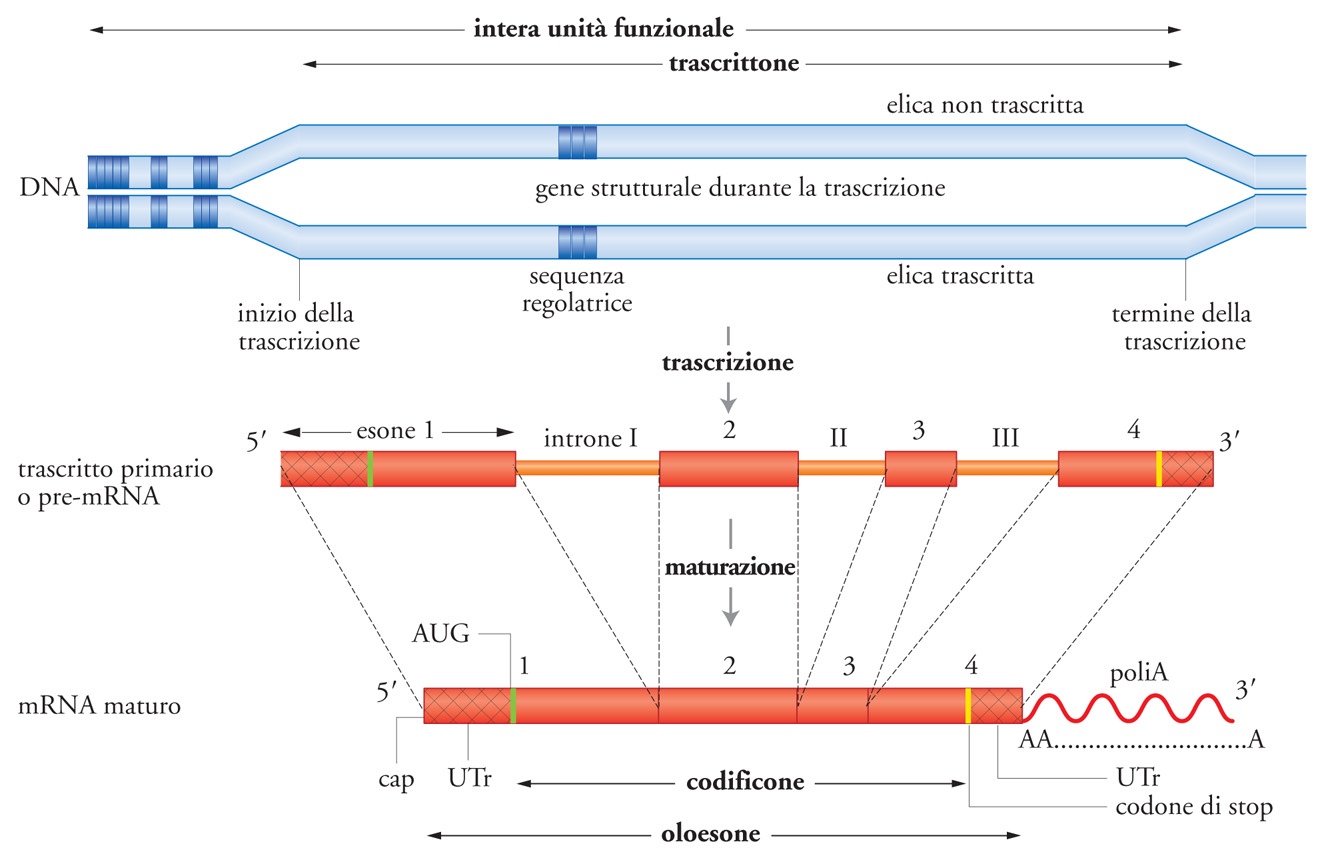
In questa sezione sono descritti i geni strutturali (gs) delle catene polipeptidiche (cp) secondo la visione a cui si era arrivati verso la metà degli anni Settanta del Novecento, cioè dopo i primi 20-25 anni di biologia molecolare: conoscenze raggiunte a livello molecolare, ma quasi esclusivamente proteico, cioè in genere non ancora direttamente a livello del DNA. Gran parte di quello che si sapeva sui gs si basava su inferenze ‒ solidissime, ma pur sempre inferenze ‒ derivate da ciò che si era effettivamente constatato a livello delle proteine. Quando è diventato possibile guardare direttamente il DNA, queste inferenze nel complesso si sono dimostrate giuste a eccezione ‒ ma si trattava di un'eccezione cospicua ‒ dell'esistenza degli introni: l'mRNA e la sua traduzione erano in sostanza come si era creduto che fossero, ma si è scoperto che i gs sono ben diversi da come essi erano stati immaginati. Infatti negli eucarioti questi geni non sono costituiti semplicemente da una sequenza continua complementare a quella dell'mRNA corrispondente, ma da una molto più lunga, formata da esoni e da introni (destinati a essere rimossi per splicing), e la sequenza dell'mRNA deriva solo dagli esoni (fig. 4). Come risultato di questi progressi il gene ‒ nato come fattore mendeliano puramente formale e così rimasto fino alla nascita della genetica molecolare ‒ è diventato per gli eucarioti il gene strutturale 'ortodosso', di cui si presentano varie possibili definizioni.
Possibili definizioni, ristrette o allargate, di gs ortodossi e di cistroni regolativi
Per ogni tipo di cp sintetizzata in un organismo esiste nel genoma di quell'organismo (almeno) una sequenza di DNA che codifica la sequenza dei suoi amminoacidi (da cui dipende non solo la struttura tridimensionale della cp, ma anche il suo destino topologico, cioè dove andrà una volta sintetizzata) e che può quindi essere denominata 'codificone' di quella cp. Il 'codificone' è la forma più ridotta possibile di gs, la sua accezione minima, nel senso che qualsiasi definizione si decida di adottare per un gs essa deve in ogni caso comprendere il codificone in esso contenuto. Una definizione più ampia di gs ortodosso comprende invece tutta la sequenza destinata a essere trascritta, e in questo caso la sua denominazione potrebbe essere 'trascrittone'. Ma di ogni cp sintetizzata in una cellula vengono specificate nel suo genoma non solo la sua sequenza amminoacidica, ma anche dove (cioè in quali cellule e tessuti), quando e quanta ne viene prodotta. La regolazione di queste proprietà non qualitative (cioè non attinenti al tipo di cp) è il risultato di interazioni che coinvolgono due tipi di elementi.

I fattori diffusibili agiscono quindi sia in cis che in trans (tipicamente fattori di trascrizione), estranei al gene o addirittura alla cellula dove la cp viene sintetizzata, e che sono spesso impropriamente denominati trans-factors, come se agissero solo in trans. I cis-acting elements (promoters, silencers, enhancers ecc.) recepiscono i segnali trasmessi con i fattori diffusibili e, su questa base, influenzano l'espressione del trascrittone sotto il loro controllo. Esso ‒ come dice il nome ‒ è solo quello in cis. Alcuni dei cis-acting elements fanno parte del trascrittone (per es., gli enhancers situati in un introne), altri, invece, sono situati al di fuori di esso, pur influenzandone in cis la trascrizione. Comunque la proprietà che li accomuna è che agiscono tutti solo in cis. Su questa base può quindi essere concettualmente opportuno in certi casi utilizzare, come unità genetiche di funzione, unità ancora più espanse del trascrittone, che comprendano cioè cis-acting elements non trascritti, ma coinvolti nella regolazione della sua trascrizione (cistroni regolativi). La tab. 1 elenca in forma schematica diversi tipi possibili di definizione di gs ortodossi e di cistroni regolativi.
Confrontando i gs dei procarioti e i gs ortodossi degli eucarioti si evince che la più ovvia delle differenze tra i gs dei procarioti e i gs eucariotici è l'esistenza, in questi ultimi, degli introni. Queste sequenze trascritte, ma destinate a non far parte degli mRNA perché vengono rimosse (per splicing), sono in genere più lunghe ‒ e spesso molto più lunghe ‒ degli esoni, per cui i trascrittoni eucariotici sono di regola molto più lunghi di quelli procariotici, talora addirittura di due ordini di grandezza (centinaia, rispetto a poche Kb), sebbene i loro codificoni in genere codifichino per cp di lunghezze simili. Questa, comunque, non rappresenta l'unica differenza tra le due classi di geni ma, oltre a essere la più cospicua, bisogna sottolineare che essa non era stata affatto inferita da un punto di vista logico, mentre il fatto che solo i gs eucariotici devono comprendere sequenze cis-acting coinvolte nella loro regolazione differenziativa era evidentemente del tutto atteso.
Il processo che porta all'espressione fenotipica dei gs ortodossi è suddiviso in due segmenti: il primo passo consiste nella successione di eventi che, a partire dal gene, conducono alla sintesi della cp corrispondente. Una proprietà fondamentale degli iter genotipo-fenotipo dei gs ortodossi è che, fino alla conclusione del primo passo, cioè fino all'espletamento della sintesi proteica, essi consistono in sostanza negli stessi passaggi. Questo insieme di eventi, che costituisce ciò che si chiama 'espressione dei gs delle cp a livello proteico', rappresenta quindi il loro denominatore comune anatomo-funzionale. Quindi, se si scopre un fenotipo a livello di una catena polipeptidica, si può ‒ se si tratta di un fenotipo qualitativo come, per esempio, una singola sostituzione amminoacidica ‒ inferire in che cosa consiste la variazione verificatasi nel DNA che la codifica e ‒ se si conosce la sequenza amminoacidica di quella cp ‒ anche dove è situata quella variazione. Da quella fase in poi, invece, ogni espressione fenotipica procede per la sua propria strada, tanto che si può ben dire che le strade che conducono a fenotipi distanti dalla espressione primaria (la produzione delle cp) dei gs sono infinite. Questo è il motivo per cui, se i fenotipi studiati riguardano proprietà molto a valle della cp, spesso tutto quello che si può dire del/i gene/i responsabile/i (ammesso, e non concesso, che il fenotipo in esame abbia una base, almeno in parte, genetica) riguarda solamente le sue/loro proprietà formali (uno oppure più geni; autosomici o X-linked; alleli dominanti o codominanti o recessivi, ecc.); e, in condizioni citogenetiche estremamente favorevoli (per es., i cromosomi giganti della drosofila), è possibile determinare anche la sua/loro sede in un certo cromosoma. Questa era la situazione della genetica in epoca premolecolare.
Classificazioni dei gs ortodossi
Ne esistono diverse, ciascuna basata su un certo criterio e utile in un determinato contesto. Si può, per esempio, parlare di geni lunghi o lunghissimi (come, rispettivamente, del gene CFTR che è lungo ca. 250 Kb, e del gene della distrofina che è lungo più di 2Mb) e di geni corti o cortissimi (come i geni delle globine, lunghi ca. 1 Kb); di geni del cromosoma X, di geni autosomici e di geni mitocondriali; di geni di recettori, o di enzimi o di ormoni oppure di proteine trasportatrici o di immunoglobuline, di oncogeni o di geni soppressori di tumori, ecc.; di geni adibiti a funzioni indispensabili o non indispensabili; di geni singoli o di geni duplicati, e così via. Inoltre, è importante la distinzione tra geni costitutivi e geni adattativi (nei procarioti) e quella tra geni house-keeping e geni differenziativi (negli eucarioti pluricellulari). I geni costitutivi sono quelli che si esprimono in qualsiasi circostanza, mentre i geni adattativi (inducibili oppure repressibili) producono la cp corrispondente solo quando essa è utile alla cellula. I geni house-keeping sono i gs, che si esprimono in tutte le cellule dell'organismo in quanto necessari per le funzioni che ogni cellula effettua per suo conto (duplicazione del DNA, sintesi proteica, produzione dell'ATP, ecc.), mentre si denominano 'geni differenziativi' i geni che si esprimono solo in certe cellule dell'organismo (pur essendo presenti anche in tutte le altre) come, per esempio, i geni dell'emoglobina, che si esprimono solo negli eritroblasti, il gene dell'insulina, che funziona solo nelle cellule β delle isole di Langerhans del pancreas, ecc.
Come si scoprivano prima, e come si scoprono ora, i gs ortodossi
Si è già visto che per molto tempo gli unici geni che si potevano scoprire erano quelli che possono causare variazioni fenotipiche cospicue. La scoperta dei gs, e del fatto che per ogni tipo di cp che si trova deve esistere almeno un gs che la codifichi, ha finalmente permesso di scoprire i geni indipendentemente dalla loro eventuale variabilità, semplicemente inferendone l'esistenza dal numero di tipi di cp. In questo primo periodo di studi era quindi dalla biochimica, più che dalla genetica, che ci si poteva aspettare di giungere a una stima ragionevolmente attendibile del numero di geni per organismo. Ma verso la fine degli anni Ottanta la parola è tornata alla genetica (molecolare), perché è diventato possibile esaminare il DNA direttamente, invece di inferirne le proprietà da quelle delle cp da esso codificate. Rendendo breve il resoconto di una storia piuttosto lunga relativamente agli studi sulla genetica, il sequenziamento di segmenti molto estesi di DNA ha portato alla scoperta di gs del tutto nuovi e di cui non si sospettava nemmeno l'esistenza, ogni volta che si individuava, in una sequenza di DNA, una ORF (Open reading frame), cioè un insieme di sequenze identificabili come esoni (soprattutto in quanto in grado di codificare per una sequenza amminoacidica di lunghezza ragionevole): si è così passati dalla fase in cui l'esistenza dei gs veniva inferita dalle cp da essi codificate, alla fase opposta (reverse genetics), in cui cioè l'esistenza di una cp era inferita dalla scoperta di una sequenza di DNA con tutte le proprietà caratteristiche dei gs; non solo, ma per il modo stesso con cui essa era stata scoperta, si sapeva fin dall'inizio esattamente la sua sequenza amminoacidica. Molti geni legati a malattie gravi, inoltre, sono stati scoperti sequenziando una regione in cui li si era localizzati (positional cloning) con metodi di genetica formale (analisi degli aplotipi); un esempio molto noto è quello del gene CFTR della fibrosi cistica.
Caratteri quantitativi individuali uni- o poligenici e caratteri poligenici, ma occasionalmente unigenici
I caratteri sono proprietà non condivise da tutti i membri di un insieme. In altre parole, una proprietà è un carattere solo se è distintiva. Se questo insieme è l'insieme delle specie, avere o non avere le ali è un carattere, mentre non lo è l'essere costituiti da atomi. Nell'ambito di una popolazione o di una specie, sono caratteri le proprietà non condivise da tutti gli individui (caratteri qualitativi, come il gruppo sanguigno Rh) o condivise da tutti, ma in misura variabile (caratteri quantitativi, come la statura, il peso, la pressione sanguigna e l'intelligenza).
I caratteri individuali qualitativi sono in genere dovuti a una sola causa (caratteri unifattoriali), che può essere ambientale oppure genetica (caratteri unigenici). È su questi ultimi che, essendo i più semplici, la genetica ha ottenuto i massimi successi (gli esempi più noti riguardano le variazioni individuali di sequenze amminoacidiche). Quindi è di questi che ci si occupa di solito anche a livello didattico, malgrado che i caratteri quantitativi polifattoriali siano quelli più rilevanti per la qualità della vita. Della determinazione genetica di questi ultimi si sa però molto poco, salvo che per i fenotipi estremi in senso negativo (per es., per i nani e per gli idioti). In questi casi, alla base della variazione estrema di un certo carattere c'è quasi sempre una causa unica ed estrema che, per il fatto di essere unica piuttosto che multipla, rende quel particolare carattere, in quel caso specifico, unifattoriale invece che polifattoriale, e quindi relativamente facile da identificare. Nel caso in cui questa causa unica sia genetica, cioè nel caso di fenotipi negativi estremi unigenici, alla base del fenotipo c'è l'assenza di funzione di un gene indispensabile alla comparsa di un fenotipo normale. È vero che nella grandissima maggioranza dei casi il carattere in questione richiede l'espressione di tutta una serie di geni, e che in tutti gli individui normali effettivamente tutti questi geni funzionano, anche se in misura variabile da uno all'altro (per cui il carattere è poligenico); ma se in un individuo anche uno solo di questi geni non funziona affatto, questo unico deficit è sufficiente a causare un fenotipo estremo, il quale si comporta da unigenico. Esistono infiniti esempi di situazioni di questo tipo, che in genere sono caratterizzate dalla familiarità (per es., due fratelli con il fenotipo estremo): il caso più comune è quello di coppie in cui entrambi i genitori sono eterozigoti per un allele silente dello stesso gene, il cui corretto funzionamento sia richiesto affinché si verifichi la normalità di un certo carattere fenotipico come l'intelligenza e l'udito. È evidente che la progenie di queste coppie spesso comprende due o più fratelli/sorelle che presentano fenotipo negativo estremo (per es., idiozia o sordità).
È facile allora spiegarsi perché, mentre la familiarità per fenotipi estremi negativi (che sono rari, ma non rarissimi) è relativamente comune, essa non esiste per fenotipi estremi positivi: non si conosce nessun esempio di un Leonardo con un figlio del livello di suo padre. Mentre è sufficiente un deficit funzionale grave di un solo gene per essere idioti, per essere potenzialmente (cioè 'ambiente permettendo') un genio o un campione olimpionico è necessario un genotipo che, oltre a essere molto speciale per un'intera costellazione di geni, sia diploide (e quindi questa situazione, oltre che eccezionalmente rara, non è trasmissibile da un singolo individuo). È un po' come per le automobili: una sola alterazione del motore di una Cinquecento può renderla incapace di muoversi, ma non esiste nessuna modifica in grado di trasformarla in una Ferrari. In conclusione, di regola l'ereditarietà dei caratteri fenotipici complessi può essere interpretata in termini genetici soltanto per i minus varianti estremi. Per quanto ne sappiamo, fanno eccezione a questa regola solo quei plus varianti estremi in cui il vantaggio diventa operante solo in presenza di uno specifico fattore ambientale avverso. Questo è il caso dell'allele fy, che allo stato omozigote conferisce una resistenza completa a Plasmodium vivax, e dell'allele βS, che allo stato eterozigote protegge dalle forme più gravi di malaria da Plasmodium falciparum, per cui queste resistenze si comportano come caratteri unigenici, invece che poligenici, come di solito accade per la resistenza alle malattie.
Il concetto attuale di gene e i geni 'eretici'
Il concetto di gene è rimasto in gran parte immodificato rispetto a quello del periodo precedente, malgrado l'enorme aumento delle conoscenze. Si è trattato infatti di un progresso soprattutto quantitativo, nel senso che ora si conosce la sequenza di molte decine di migliaia di geni, cioè di un numero a cui non si era sperato di arrivare nemmeno con le previsioni più ottimistiche; ma il concetto che abbiamo ora dei geni e del loro modo di funzionare non è cambiato molto, almeno per quanto riguarda i gs ortodossi, che si ritiene costituiscano la grande maggioranza dei gs. Sono emerse però due importanti eccezioni, rappresentate dai geni 'eretici'.
Geni eretici, perché contraddicono il dogma della genetica molecolare
È noto che cellule somatiche diverse di uno stesso organismo sintetizzano cp diverse, ma la spiegazione classica ‒ e mai contestata fino agli anni Settanta ‒ di questo fenomeno di differenziazione biochimica (alla base degli altri tipi di differenziazione di ordine gerarchico superiore, come, per es., quella morfologica) era che cellule diverse di uno stesso organismo producono cp diverse, perché esprimono geni differenti, pur avendo tutte gli stessi geni. E questo è certamente vero a eccezione che per i geni IG delle immunoglobuline dei linfociti B e per i geni TCR (T-cell receptor) dei linfociti T.
Per i geni IG (e più tardi anche per i TCR) era risultato a priori molto difficile da accettare che essi fossero esistiti e fossero stati trasmessi in modo continuo nel corso delle generazioni in numero infinitamente grande (pari cioè al repertorio di anticorpi diversi che un vertebrato è in grado di produrre), dal momento che di sicuro solo pochissimi di essi erano stati utili alla specie. Ci si è accorti allora ‒ prima solo sul piano logico e in seguito constatandolo direttamente a livello del DNA ‒ che il dogma a cui si è accennato consiste in realtà di due dogmi e che solo il primo (quello che afferma che se una cellula produce una determinata cp, vuol dire che quella cellula ha il gs per quella cp) è assolutamente irrinunciabile. Al secondo (quello che affermava che se una cellula somatica di un organismo ha un gene, vuol dire che lo hanno tutte le altre cellule somatiche di quell'organismo), invece, si poteva e si doveva rinunciare affermando che i diversi cloni di linfociti B producono Ig diverse perché hanno geni IG diversi (per cui sarebbe impossibile in linea di principio ottenere una Dolly da un linfocita B).
Geni eretici perché comprendono sequenze che fanno parte di molti codificoni diversi
Era noto già dalla metà degli anni Settanta che alcuni pre-mRNA possono andare incontro a splicing diversi, dando così origine a mRNA differenti e quindi a cp diverse per certe particolari proprietà, come quella di comportarsi da recettori di membrana oppure di essere secrete. L'esempio più noto era quello delle Ig, che nei linfociti B si localizzano nella membrana cellulare con un dominio idrofobico che la attraversa, mentre nelle plasmacellule, essendo prive di questo dominio, hanno un destino topologico diverso: esse sono secrete e diventano anticorpi circolanti. Sono stati scoperti in seguito molti geni i cui pre-mRNA subiscono splicing alternativi, dando così origine ad alcuni tipi diversi di codificoni e quindi di cp. Ma è solo molto più recentemente che si sono scoperti geni che, per splicing alternativi, danno origine non a 'pochi' tipi diversi di mRNA, ma addirittura a molte migliaia di differenti mRNA. Questo fenomeno riguarda alcuni dei geni che si esprimono nel sistema nervoso ed è di sicuro implicato nel determinare l'estremo grado di plasticità di questo sistema. Non c'è dubbio che il suo studio riserverà molte sorprese ma, a parte questo, è evidente che dal punto di vista genetico, per i geni di questa classe è caduta completamente la relazione in genere stretta tra numero di geni e numero di tipi di cp sintetizzate in un organismo. L'osservazione che negli eucarioti molti geni codificano per più di una cp spiega, almeno in parte, la sorprendente nozione che il numero di geni del genoma di un eucariote superiore (per es., l'uomo) è maggiore rispetto a quello di un batterio come E. coli soltanto per un fattore 10 (ca. 30.000 vs ca. 3000).
Quanti sono i geni preposti alla sintesi di ognuno dei differenti tipi di prodotti genici (RNA o cp)?
Al principio dell'era molecolare la risposta a questo genere di domanda era data per scontata: ogni prodotto genico è dovuto all'espressione di un gene per genoma aploide, il quale gene è responsabile della sintesi di quel tipo di prodotto solamente. Oggi si sa bene che ciò è vero spesso, ma non sempre. Considerati sotto questo aspetto, infatti, i geni possono essere suddivisi in tre classi (di cui solo quella dei geni ortodossi rispetta questa regola): (a) i geni per gli RNA ribosomali. Il prodotto di ognuno di questi geni è richiesto in tutte le cellule (tipici geni house-keeping) e in quantità considerevoli (una molecola per ribosoma). Inoltre la produzione degli RNA ribosomali, come quella di tutti gli RNA che costituiscono il prodotto genico terminale, passa attraverso una sola amplificazione anziché due, come nel caso dei geni per le cp (compresi quelli delle proteine ribosomali), che sono amplificati anche con la traduzione. Ognuno di questi geni è presente in molte decine di copie per genoma aploide; (b) i gs ortodossi delle cp. Ognuno di essi è destinato a esprimersi prima o poi almeno in un tipo di cellule, se non in tutte (se si tratta di geni house-keeping) e a passare attraverso entrambi i tipi di amplificazione. Di regola ognuno di questi geni è presente in singola copia per genoma aploide. Questi geni costituiscono la maggioranza dei gs; (c) i gs per le Ig e per i TCR. Si tratta di una delle due categorie di geni eretici dei Vertebrati. Sia i geni IG che i TCR sono costituiti da un insieme pressoché infinito di geni diversi, simili fra loro per alcuni aspetti comuni a tutti, ma ciascuno dotato di una propria struttura e specificità. Molti di essi si esprimono in ogni organismo (per es., quelli degli anticorpi verso antigeni ubiquitari), ma sono infinitamente più numerosi quelli che non hanno mai avuto, e verosimilmente non avranno mai, l'occasione di esprimersi. Ebbene, questi geni quasi certamente inutili non sono presenti nemmeno in singola dose nel genoma aploide e il loro vastissimo repertorio si crea solo in una fase postzigotica e solo in certe cellule molto specializzate attraverso la continua formazione, per processi di riarrangiamento genico, di cloni somatici specifici (veri e propri piccoli eserciti di ultraspecialisti), ciascuno dei quali portatore dei geni che codificano per una determinata Ig o TCR: il quasi infinito repertorio di Ig e di TCR di un organismo è l'espressione dell'esistenza in quell'organismo di un vasto repertorio pressoché infinito di cloni, ciascuno numericamente molto ridotto, ma suscettibile di 'esplodere' in determinate circostanze (tipicamente in presenza dell'antigene riconosciuto dall'anticorpo codificato nel clone corrispondente).
Alcune considerazioni sull'evoluzione dei gs
Nell'evoluzione dei gs si distinguono due tipi di eventi, quelli su 'larga scala' e quelli 'puntiformi'.
Eventi evolutivi su larga scala. Sono stati scoperti studiando le relazioni tra esoni e domini proteici. Ci si è accorti che nei geni il cui codificone è costituito da più di un esone, e si tratta della maggioranza dei geni degli eucarioti, spesso ognuno di questi esoni codifica per un dominio della cp, cioè per una regione dell'intera molecola con individualità anatomo-funzionale propria. Per esempio, se la cp è un recettore di membrana, spesso un esone codifica per la sua sezione extracellulare, l'esone successivo per il suo segmento idrofobico transmembrana e quello ancora successivo per la sua parte citoplasmatica. In altri termini, i gs hanno in genere una struttura modulare in cui ogni modulo è un esone che codifica per una parte deputata a una certa funzione tra quelle svolte dalla molecola proteica nel suo insieme. In molti casi, inoltre, è dato osservare che uno dei 'moduli' (o esoni) di un gene presenta una omologia significativa con un esone di un altro gene e che i segmenti di sequenza polipeptidica codificati da questi esoni svolgono funzioni simili nelle cp rispettive, proprio come se alcuni geni si fossero formati combinando assieme un certo numero di moduli, ciascuno dei quali sia in grado di impartire una, o più, proprietà funzionali alla molecola di cui fa parte. Gli introni che separano gli esoni che codificano per questi moduli marcherebbero i punti in cui essi sarebbero stati inclusi per trasposizione nella sequenza finale del gene. Questa è la forma ridotta ai minimi termini dell'ipotesi evoluzionistica della formazione di alcuni geni per exon-shuffling (dal verbo inglese to shuffle, mescolare). Un altro ben noto evento evolutivo su larga scala è rappresentato dalla duplicazione genica.
Eventi puntiformi. Consistono quasi sempre in quelle singole sostituzioni nucleotidiche di una sequenza codificante, che risultano in una singola sostituzione amminoacidica nella cp corrispondente. Questi eventi si scoprono sia in confronti interspecie sia intraspecie, cioè tra individui della stessa specie o della stessa popolazione. Le variazioni prendono in questi ultimi casi il nome di 'variazioni alleliche' che, se non sono rare (cioè se l'allele meno comune ha una frequenza di almeno circa l'1%), vengono dette 'polimorfiche'. Si conoscono più di un milione di variazioni alleliche nell'uomo (la specie più studiata), ma questo numero, grande in senso assoluto, non lo è rispetto ai circa tre miliardi di basi del nostro genoma aploide. L'esistenza di variazioni alleliche dei gs costituisce la conditio sine qua non di qualsiasi loro processo evolutivo, perché si può passare da una situazione in cui in un certo sito di un gs c'è lo stesso nucleotide in tutti i genomi di una specie, a un'altra situazione in cui in tutti i genomi c'è invece un altro nucleotide solo attraversando una fase intermedia più o meno lunga in termini di numero di generazioni, durante la quale i due nucleotidi, il vecchio e il nuovo, coesistono (cioè in certi genomi in quella posizione c'è un nucleotide e negli altri genomi ce ne è un altro); questo è ciò che viene definito 'sito polimorfico'.
Se lasciamo da parte i casi in cui non solo la mutazione che ha prodotto l'allele nuovo ma anche il suo destino successivo si sono svolti per puro caso (in gergo, per 'deriva genetica'), che avrebbe agito cioè su alleli perfettamente equivalenti (alleli neutrali), restano quelli in cui il processo di sostituzione è stato adattativo, ossia 'guidato' dalla selezione che, un passo alla volta, può portare alla sostituzione di un dato gs con un altro migliore e diverso dal precedente per tutta una serie di singole sostituzioni amminoacidiche. Tutto sarebbe concettualmente semplice, vale a dire tipicamente darwiniano, se ciascuno degli n cambiamenti risultanti nell'adattamento finale procedesse lungo un gradiente, causando cioè un effetto progressivo sequenziale, un miglioramento parziale rispetto a quelli già avvenuti e che inoltre fosse totalmente indipendente dall'ordine in cui i singoli cambiamenti si sono effettivamente susseguiti (ossia tre cambiamenti meglio di due e due meglio di uno, e così via). Ma in genere alcuni dei cambiamenti intermedi non sono affatto vantaggiosi, anzi addirittura compromettono gravemente l'espressione del gene. E, siccome la selezione non può prevedere il risultato finale, ma solo guardare a quello parziale momento per momento, essa non può fare altro che opporsi a percorsi controcorrente di questo genere. Potrebbe sembrare una difficoltà insuperabile (che, del resto, riguarda l'evoluzione nel suo insieme, cioè anche ad altri livelli, come quello dell'anatomia comparata), ma che invece è risolvibile almeno per i geni duplicati e/o per gli pseudogeni (lo pseudogene di un gs è una sequenza a esso molto simile, ma non in grado di produrre la corrispondente cp a causa di una o più mutazioni che lo rendono silente). Questa situazione, infatti, dà l'opportunità, a uno dei due geni duplicati e/o allo pseudogene, di accumulare mutazioni non soggette alla selezione; queste possono poi essere trasferite, eventualmente anche a blocchi di più di uno per volta, al gene funzionante, purché in tandem (e in genere succede proprio questo per il modo stesso in cui si originano le duplicazioni geniche) con meccanismi ricombinatori del tipo della gene conversion: la ridondanza è in questi casi 'la carta vincente del successo evolutivo'.
Bibliografia
De Carli 1997: De Carli, Luigi e altri, Genetica generale e umana, Padova, Piccin, 1997.
Lewin 2005: Lewin, Benjamin, Genes VIII, Oxford-New York, Oxford University Press, 2005 (trad. it.: Il gene VIII, Bologna, Zanichelli, 2006).
Montalenti 1979: Montalenti, Giuseppe, Introduzione alla genetica, Torino, UTET, 1979.
Siti internet
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi
(search: Unigene)
Tavola I
La topografia dei genomi: geografia o autonomia?
La successione dei nucleotidi lungo un genoma è perfettamente paragonabile a quella delle lettere in un’enciclopedia, anche perché in entrambi i casi si individuano facilmente diverse gerarchie di successione. Semplificando al massimo, un gene corrisponde a una voce di un’enciclopedia e l’intero genoma aploide all’intera enciclopedia. È evidente che l’ordine delle lettere di una voce è altrettanto indispensabile per la sua comprensibilità quanto lo è l’ordine dei nucleotidi del gene per la sua corretta espressione. Ma, al di là di questo livello gerarchico elementare (cioè intra-voce e intra-gene), non è più chiaro – salvo che per pochi genomi – se questa similitudine continui a essere valida anche ai livelli gerarchici superiori. Infatti, mentre nel caso dell’enciclopedia è evidente che il suo funzionamento richiede il rispetto rigoroso anche di un ordine gerarchico superiore, che consiste nel disporre in un certo ordine (in genere alfabetico) le varie voci, l’esistenza di un significato funzionale nella disposizione dei geni in un genoma è accertata solo per pochi genomi, come quelli di alcuni batteriofagi in cui i geni che si esprimono per primi, cioè gli early genes, sono situati all’estremità opposta del genoma rispetto ai late genes.
Di regola, invece, pur conoscendo perfettamente per i molti genomi completamente sequenziati l’esatta posizione dei loro geni lungo il genoma, non sappiamo rispondere a una domanda fondamentale: la disposizione dei geni nel genoma è una delle tante disposizioni possibili all’incirca equivalenti, oppure è una delle poche compatibili con un corretto funzionamento del genoma nel suo insieme? Non sappiamo cioè quasi mai se, permutando le posizioni rispettive dei vari geni, accadrebbe qualcosa di funzionalmente rilevante e non sappiamo, tanto meno, cosa accadrebbe in caso affermativo. In altre parole, non sappiamo se la topografia dei genomi è da assimilare più a una geografia o più a una anatomia, cioè a una topografia basata su relazioni talmente strette ed evidenti tra struttura e funzione da sembrare finalistiche. Comunque una cosa è certa: la trasmissione corretta dei genomi da una generazione all’altra di una specie richiede che la loro topografia sia la stessa per tutti i genomi di quella specie.
Tavola II
Il gene: un’unità sui generis
Le unità, in linea di principio, sono unità quantitative: una molecola di una sostanza rappresenta la più piccola parte di
quella sostanza che possiede tutte le sue proprietà; un nefrone, salvo che per la quantità di urina che produce, è un rene in miniatura, in quanto l’urina da esso prodotta è dal punto di vista qualitativo uguale a quella prodotta dall’intero organo.
La relazione tra un gene e il genoma aploide di cui esso fa parte è esattamente l’opposto. Nell’uomo, per esempio, il cui genoma aploide comprende circa 30.000 geni, ognuno di essi è responsabile, da solo, dell’intera produzione del ‘suo’ mRNA, mentre non contribuisce affatto alla produzione di ciascuno degli altri tipi di mRNA. Delle circa 30.000 funzioni del genoma, una è effettuata completamente da uno solo dei suoi 30.000 geni, un’altra solo da un altro di questi geni, e così via. Questa differenza tra la relazione anatomo-funzionale propria delle unità quantitative e quella delle unità qualitative nella loro globalità ha conseguenze estreme: mentre è possibile vivere perfettamente con un nefrone in meno (anzi, addirittura con un solo rene), la perdita di funzione anche di uno solo dei geni di un genoma ha spesso conseguenze gravi, se non addirittura letali.