
Neoplasie
Neoplasie
Cause e prevenzione, di Bruce N. Ames, Lois S. Gold e Walter C. Willett
Oncologia clinica, di Georges Mathé e Paolo Pontiggia
Oncologia sperimentale, di Giancarlo Vecchio
Cause e prevenzione di Bruce N. Ames, Lois S. Gold e Walter C. Willett
SOMMARIO: 1. Introduzione. 2. Andamenti epidemiologici. 3. I meccanismi della carcinogenesi: a) lesioni del DNA, mutazioni e divisione cellulare; b) punti di controllo del ciclo cellulare; c) sistemi di difesa. 4. I principali fattori di rischio: a) fattori endogeni; b) dieta; c) tabacco; d) infezioni croniche, flogosi e cancro; e) ormoni. 5. Fattori di rischio meno importanti: a) attività professionale; b) esposizione al sole; c) fattori iatrogeni; d) inquinamento. 6. Fattori ereditari. 7. Errori di valutazione; limiti di validità dei test sugli animali. 8. Conclusione. □ Bibliografia.
1. Introduzione
I dati epidemiologici indicano che è possibile diminuire l'incidenza del cancro specialmente attraverso la riduzione del fumo, un maggior consumo di frutta e verdura e il controllo delle infezioni. Altre precauzioni consistono nell'evitare un'intensa esposizione ai raggi solari, nell'intensificare l'attività fisica, nel ridurre il consumo di bevande alcoliche e, per quanto possibile, di carne rossa. Per conseguire una netta riduzione del cancro della mammella è probabile che si debbano modificare i livelli degli ormoni sessuali; lo sviluppo di metodi pratici per ottenere questo risultato dovrebbe costituire un obiettivo di ricerca prioritario. La determinazione del potenziale ruolo protettivo di specifici antiossidanti e di altri costituenti di frutta e verdura merita grande attenzione. Già ora siamo in grado di ridurre il rischio di contrarre molte forme di cancro, ma vi è ampio spazio per ulteriori progressi. Gli studi sui meccanismi della carcinogenesi hanno indicato quanto importante sia il ruolo svolto dal danno ossidativo endogeno a carico del DNA, un danno peraltro controbilanciato da elaborati processi di difesa e di riparazione. La velocità della divisione cellulare, che è influenzata dagli ormoni, dall'età, dalla citotossicità e da processi infiammatori, è anch'essa un fattore chiave, in quanto determina la probabilità che lesioni del DNA si trasformino in mutazioni. È probabile che questi meccanismi siano alla base di molti dei casi osservati; nello stesso tempo essi suggeriscono interventi pratici e aree di ulteriore ricerca.
In questo articolo verranno discusse le cause di cancro principali e secondarie, evidenziandone in particolar modo i meccanismi, la cui comprensione - grazie ai rapidi progressi che si stanno registrando - consente di attuare la prevenzione. B. E. Henderson, R. K. Ross e M. C. Pike (v., 1991) hanno esaminato le probabili cause di cancro nel 1991, a un decennio di distanza dall'esauriente rassegna di Doll e Peto (v., 1981). L'opera di Henderson e collaboratori, in seguito integrata dalla nostra (v. Ames e altri, 1995), include una tabella del numero stimato dei casi di cancro negli Stati Uniti, suddivisi in base all'area geografica, con l'indicazione delle cause note e di quelle possibili.
Non verrà invece discusso un altro tema di grande importanza, come la scoperta di un cancro nelle primissime fasi e delle lesioni preneoplastiche, la cui trattazione esula dallo scopo di questo articolo.
2. Andamenti epidemiologici
Nel 1993, negli Stati Uniti, il cancro ha provocato il 23% delle morti premature misurate in anni-persona, pari a circa 530.000 decessi (v. Miller e altri, 1993). Alle quattro principali forme di cancro (del polmone, del colon-retto, della mammella e della prostata) è imputabile il 55% dei decessi. Secondo i dati, aggiornati al 1993, raccolti nell'ambito del programma SEER (Surveillance, Epidemiology, and End Results) del National Cancer Institute, il tasso di mortalità ripartito per età per tutte le forme di cancro messe insieme (esclusi il cancro del polmone e quello dei bronchi) è diminuito dal 1950 al 1990 per tutti i gruppi di età eccettuato quello dagli 85 anni in su: la diminuzione è andata dal 71%, nel gruppo 0-4 anni, all'8% nel gruppo 74-85. ‟Se si escludesse il cancro del polmone, il tasso di mortalità complessivo dovuto al cancro risulterebbe diminuito di oltre il 14% tra il 1950 e il 1990" (v. Miller e altri, 1993). Il fumo, oltre a causare la maggior parte dei casi di cancro del polmone, contribuisce ad altre patologie maligne, quali i tumori della bocca, dell'esofago, del pancreas, della vescica, la leucemia e forse il cancro del colon; se si escludessero anche questi tumori la diminuzione risulterebbe maggiore.
Se si tiene conto anche del cancro del polmone, la mortalità complessiva per cancro è diminuita di oltre il 25% per ciascun gruppo d'età sotto i 45 anni ed è aumentata per i gruppi d'età al di sopra dei 55 anni. Le diminuzioni dei decessi per cancro durante il periodo in questione riguardano principalmente i tumori dello stomaco, del collo dell'utero, dell'utero e del retto. Gli aumenti sono dovuti soprattutto al cancro del polmone, causato dal fumo (responsabile del 30% di tutti i decessi per cancro negli Stati Uniti), e ai linfomi non-Hodgkin (NHL). Le ragioni dell'aumento dell'NHL non sono chiare, ma è possibile che il fumo vi contribuisca e l'infezione da HIV rappresenti una causa relativamente rara ma in aumento.
Per interpretare i cambiamenti del tasso di mortalità si devono considerare sia i mutamenti dei tassi di incidenza (il numero dei nuovi casi di cancro diagnosticati) sia gli effetti del trattamento. Per certi tipi di cancro, l'aumento dei tassi di incidenza è dovuto, in parte, al fatto che è divenuto possibile effettuare una diagnosi precoce. Doll e Peto (v., 1981) nel loro esauriente studio sulle cause del cancro fanno notare che i tassi di incidenza non dovrebbero essere considerati isolatamente, perché potrebbero riflettere una maggiore registrazione dei casi e miglioramenti diagnostici; ad esempio, l'apparente aumento del tasso di incidenza del cancro tra gli uomini nati negli anni quaranta rispetto a quelli nati negli anni novanta del secolo scorso potrebbe spiegarsi proprio in questo modo, e il rapido aumento - nelle diverse classi d'età - dell'incidenza del cancro della prostata, non accompagnato da un aumento significativo della mortalità, è quasi certamente dovuto in larga misura al maggior numero di indagini su vasta scala (screening) e alla scoperta casuale del tumore durante una prostatectomia per ipertrofia prostatica benigna.
3. I meccanismi della carcinogenesi
a) Lesioni del DNA, mutazioni e divisione cellulare.
Le lesioni del DNA (basi danneggiate) hanno una certa probabilità di dare origine a mutazioni quando la cellula si divide. Il danno endogeno del DNA è diffuso (v. Ames e altri, 1993): in ogni cellula di un ratto vecchio sono presenti circa un milione di lesioni ossidative. Un agente mutageno esogeno, per esempio il vinilcloruro, produce un aumento delle lesioni in aggiunta al tasso base di lesioni endogene. Il potere mutageno di una particolare lesione dipende dalla velocità con cui viene ‛tagliata' dagli enzimi di riparazione del DNA e dalla probabilità che dia luogo a una mutazione quando la cellula si divide.
Mutazioni che si verifichino in diversi geni critici possono portare alla formazione di tumori (v. Vogelstein e altri, 1989). In circa la metà dei tumori umani si riscontrano mutazioni nel gene p53 che codifica per un soppressore di trasformazione, la proteina p53, la quale svolge un ruolo di controllo sul ciclo cellulare: la sua inattivazione dà luogo a una divisione cellulare incontrollata.
La divisione cellulare costituisce quindi un fattore critico nella mutagenesi (v. Ames e Gold, 1990; v. Ames e altri, 1995 e 1996; v. Henderson e altri, 1991). Pertanto un fattore decisivo nel determinare il potere mutageno di un agente è la sua capacità di incrementare il tasso di divisione delle cellule di primaria importanza; nel caso del cancro queste sono le cellule staminali, che, a differenza delle loro cellule figlie, non vengono eliminate. Aumentando il tasso di divisione delle cellule staminali aumenta la frequenza di mutazione e quindi la probabilità che compaia un cancro. Come previsto, le cellule che non si dividono vanno raramente incontro a trasformazione neoplastica. Un'intensificazione della divisione cellulare - e quindi un maggior rischio di cancro - può essere causata da agenti disparati, come più alti livelli di particolari ormoni, eccesso di calorie, infezioni e infiammazioni croniche, o sostanze chimiche assunte in dosi che provocano la divisione cellulare (v. Cunningham e altri, 1994; v. Ames e altri, 1996). Se aumentano sia il tasso di lesioni del DNA sia quello di divisione cellulare, allora si avrà un effetto moltiplicativo sulla mutagenesi (e sulla carcinogenesi); ciò può avvenire, per esempio, per l'assunzione, in dosi elevate, di un mutageno che, uccidendo un certo numero di cellule, stimoli la divisione cellulare per favorire la sostituzione delle stesse. Anche dosaggi protratti e massicci di agenti chimici che non danneggiano il DNA possono causare l'uccisione di cellule e la conseguente divisione cellulare, e quindi incrementare il rischio che si sviluppi un cancro.
b) Punti di controllo del ciclo cellulare.
In corrispondenza di alcuni stadi del ciclo cellulare vi sono alcuni ‛punti di controllo' che prevengono la divisione di cellule con troppe lesioni del DNA, e quindi inibiscono la comparsa di mutazioni (v. Ames e altri, 1995). Questa difesa, come la riparazione del DNA, non è perfetta. La rilevazione di lesioni nei geni trascritti è effettuata dall'apparato di trascrizione che produce l'mRNA. La presenza di lesioni sembra indurre la riparazione del DNA e anche fermare la divisione cellulare a livello dei punti di controllo. Il meccanismo può essere il seguente: la proteina p53, che agisce nel punto di controllo tra lo stadio G1 e lo stadio S, si associa alla proteina di replicazione e riparazione RPA (Replication Protein A); quando si verifica un danno del DNA, la proteina RPA si lega al DNA a catena singola e libera la p53; quest'ultima determina un blocco della divisione cellulare tra gli stadi G1 e S, prevenendo così la trasformazione delle lesioni in mutazioni. Inoltre la p53 può indurre la morte cellulare programmata (apoptosi) se il DNA è troppo danneggiato per essere riparato (v. neoplasie: Oncologia sperimentale, vol. XI).
c) Sistemi di difesa.
Il DNA viene protetto dagli agenti mutageni da sistemi di difesa, quali le transferasi del glutatione, che sono quasi tutti inducibili e agiscono impedendo l'accumulo di sostanze chimiche reattive elettrofile nelle cellule (v. Ames e altri, 1990). Inoltre, gli enzimi di riparazione del DNA, anche questi quasi tutti inducibili, provvedono a sostituire le eventuali basi danneggiate. L'effetto di una particolare aggressione chimica dipende dal livello di ciascuna difesa, che, a sua volta, dipende dalle precedenti esposizioni all'agente chimico. Le difese possono essere parzialmente disattivate dalla mancanza di particolari micronutrienti nella dieta (per esempio gli antiossidanti; v. Ames e altri, 1993).
4. I principali fattori di rischio
a) Fattori endogeni.
Se vengono ridotti i principali fattori esogeni di rischio per il cancro - il fumo, le infiammazioni croniche e una dieta non bilanciata - proporzionalmente aumenterà sia l'età alla quale il cancro fa la sua comparsa, sia la quota dei tumori provocati da processi endogeni.
I sottoprodotti ossidanti del metabolismo normale provocano estesi danni al DNA, alle proteine e ai lipidi. Noi sosteniamo che questi danni (uguali a quelli prodotti dalle radiazioni) costituiscono uno dei principali fattori responsabili dell'invecchiamento e delle malattie degenerative che ne sono proprie, quali il cancro, l'infarto, la cataratta e determinate disfunzioni cerebrali (v. Ames e altri, 1993). Le difese antiossidanti contro questi danni comprendono l'ascorbato, i tocoferoli e i carotenoidi.
La degenerazione delle cellule somatiche durante l'invecchiamento sembra contribuire in buona parte alle malattie degenerative. L'incidenza complessiva del cancro in varie specie di mammiferi sembra aumentare con la quarta potenza dell'età. Peraltro, la vita media dell'uomo è andata crescendo nei circa 60 milioni di anni della sua evoluzione, per cui si è andata via via spostando l'età alla quale il rischio di cancro è maggiore; quindi, a due anni di età tale rischio è molto alto in una specie a vita breve come il ratto, ma basso nell'uomo.
Un importante fattore di longevità sembra essere il tasso metabolico basale, che è circa sette volte più alto in un ratto che in un essere umano e che potrebbe far aumentare notevolmente il livello degli ossidanti endogeni e di altri sottoprodotti mutageni del metabolismo. Il livello del danno ossidativo a carico del DNA sembra essere approssimativamente collegato al tasso metabolico in un certo numero di specie di mammiferi.
Alcuni sottoprodotti del metabolismo normale, per esempio il radicale superossido, il perossido di idrogeno e il radicale ossidrile, sono gli stessi mutageni ossidativi prodotti da radiazioni. Il danno ossidativo a carico del DNA, delle proteine e di altre macromolecole si accumula con l'età, ed è stato postulato che sia un importante, ma non l'unico, tipo di danno endogeno che conduce all'invecchiamento. La perossidazione dei lipidi dà luogo a mutageni: epossidi lipidici, idroperossidi lipidici, radicali alcossilici e perossilici lipidici ed enali (aldeidi α, β-insature). L'ossigeno monoatomico, una forma di ossigeno mutagena ad alta energia, può essere prodotto per trasferimento di energia dovuto all'assorbimento della luce, per combustione respiratoria da neutrofili o per perossidazione di lipidi, ed è efficacemente neutralizzato dai carotenoidi che dissipano l'energia in eccesso (v. radicali liberi: Biologia e patologia, vol. XI).
Nel metabolismo normale, il DNA si ossida perché le difese antiossidanti, per quanto numerose, non sono perfette. Il DNA ossidato viene riparato da enzimi, i quali tagliano i tratti lesionati che vengono quindi escreti nelle urine. Sono stati sviluppati metodi per analizzare parecchie di queste basi danneggiate escisse, presenti nelle urine di Roditori e di esseri umani, che per la maggior parte sono risultate basi libere derivanti da riparazioni da parte delle glicosilasi. È stato stimato che il numero degli eventi ossidativi del DNA per cellula, al giorno, sia pari a circa 100.000 nel ratto e approssimativamente dieci volte meno nell'uomo. Gli enzimi di riparazione del DNA rimuovono efficacemente la maggior parte delle lesioni, ma non tutte. Le lesioni ossidative del DNA si accumulano con l'età, così che un ratto vecchio (di due anni) ha circa un milione di lesioni del DNA per cellula, circa il doppio rispetto a un ratto giovane. Anche le mutazioni si accumulano con l'età: per esempio, la frequenza delle mutazioni somatiche è circa nove volte maggiore nella popolazione anziana che fra i neonati, ma è sconosciuto quale sia il contributo delle lesioni ossidative del DNA a questo fenomeno. L'importanza di questo tipo di lesioni nel cancro e nell'invecchiamento è sottolineata dall'esistenza di specifiche glicosilasi di riparazione, che le tagliano dal DNA: per esempio, nel caso di una lesione causata da un danno ossidativo a carico dei residui di guanina nel DNA, che produce 8-oxo-guanina, la soppressione dell'attività glicosilasica specifica comporta un apprezzabile incremento del tasso di mutazione spontanea, il che dimostra l'intrinseco danno mutageno di questa lesione. Probabilmente anche molte delle altre principali lesioni ossidative del DNA (circa 20) contribuiscono all'insorgenza di tumori e all'invecchiamento.
Per la sua vicinanza agli ossidanti generati durante la fosforilazione ossidativa, il DNA mitocondriale (mtDNA) subisce un danno ossidativo dieci volte maggiore rispetto a quello del DNA nucleare. La cellula si difende contro questo alto tasso di danneggiamento tramite un continuo turnover di mitocondri, quindi, presumibilmente, rimuovendo i mitocondri alterati che stanno producendo più ossidanti. Malgrado questo turnover, con l'età le lesioni ossidative si accumulano nell'mtDNA a una velocità più alta che nel DNA nucleare, e ciò può spiegare il maggior numero di mutazioni che vi si riscontrano.
Gli ossidanti danneggiano le proteine oltre che il DNA. Gli enzimi proteolitici protettivi che idrolizzano le proteine ossidate non bastano a evitarne l'accumulo col tempo. In due malattie umane associate all'invecchiamento prematuro, la sindrome di Werner e la progeria, le proteine ossidate si accumulano a una velocità molto più alta del normale. Anche le lipofuscine fluorescenti, che si pensa siano in parte originate da legami crociati tra una proteina e prodotti della perossidazione dei lipidi, si accumulano con l'età. Un'altra comune conseguenza di stress ossidativi, come la radiazione UV o il fumo, sono le cataratte, anch'esse dovute ad accumulo di una proteina ossidata o a insufficiente protezione antiossidante.
b) Dieta.
Si ritiene che circa un terzo dei casi di cancro dipenda dalla dieta, ma solo lentamente si sta chiarendo quali siano i fattori responsabili.
1. Prevenzione del cancro tramite riduzione delle calorie o delle proteine (v. Ames e altri, 1995). - Nei Roditori una dieta ipocalorica in sostituzione di un'alimentazione ad libitum determina una marcata diminuzione dell'incidenza dei tumori e un aumento della durata della vita, ma fa calare la fertilità. La riduzione delle proteine sembra avere effetti simili, ma è stata studiata meno. L'adattamento (fitness) darwiniano negli animali sembra essere incrementato da cambiamenti ormonali che differiscono la funzione riproduttiva durante periodi di scarsa disponibilità di cibo, perché le risorse alimentari vengono impiegate nel mantenimento del corpo finché non siano nuovamente sufficienti a consentire una riproduzione coronata da successo. Si stanno facendo progressi nella comprensione dei meccanismi responsabili del marcato effetto delle restrizioni alimentari sull'invecchiamento e sul cancro, effetto che può in buona parte essere dovuto a un minor danno ossidativo. Una limitazione della dieta porta a una più efficiente riparazione del DNA e a una migliore respirazione mitocondriale. Un rinvio del declino - dipendente dall'età - delle difese antiossidanti negli animali sottoposti a dieta ipocalorica suggerisce che questa potenzi le funzioni di mantenimento e riduca il danno ossidativo. Le migliori difese antiossidanti possono anche render conto della più efficiente risposta immunitaria. Ambedue le diminuzioni, calorica e proteica, rallentano l'accumulo di proteine ossidate che accompagna l'invecchiamento nei ratti. Rispetto a quelli alimentati ad libitum, i roditori soggetti a dieta ipocalorica presentano, in diversi tessuti, tassi mitotici decisamente più bassi che, con ogni probabilità, contribuiscono alla diminuzione dell'incidenza dei tumori. Pertanto, gli effetti della più elevata attività necessaria al mantenimento in condizioni di regime alimentare limitato - riduzione del danno ossidativo a carico del DNA e delle proteine, diminuzione delle lesioni del DNA e delle proteine, rallentamento della divisione cellulare - potrebbero quindi risultare in una diminuzione delle mutazioni somatiche che portano al cancro. Per quanto i dati epidemiologici sulle limitazioni della dieta negli esseri umani siano scarsi, l'ipotesi che una diminuzione della crescita staturale possa costituire una misura importante nella profilassi del cancro umano è avvalorata da studi epidemiologici che indicano tassi più elevati di cancro della mammella e di altri tumori tra le persone più alte: per esempio le donne giapponesi, che sono ora più alte, vanno incontro a un menarca anticipato e presentano tassi di cancro della mammella maggiori di prima. Inoltre, in molti casi, i differenti tassi di cancro della mammella nei vari paesi e i diversi andamenti di questa patologia nel corso del tempo all'interno dei singoli paesi, sembrano corrispondere alle variazioni dei tassi di crescita e dell'altezza media raggiunta.
2. La prevenzione del cancro tramite l'introduzione di frutta e verdura nel regime alimentare (v. tab. I; v. Ames e altri, 1995; v. Block e altri, 1992). - Il consumo di una quantità sufficiente di frutta e verdura è associato a un minor rischio di malattie degenerative quali il cancro, le malattie cardiovascolari, la cataratta e le disfunzioni cerebrali e immunitarie. Sono stati passati in rassegna circa 200 studi epidemiologici, e tutti concordano nel mettere in relazione la mancanza di un adeguato consumo di frutta e verdura con l'incidenza del cancro (v. Block e altri, 1992). Il quarto della popolazione che consuma meno frutta e verdura, rispetto a quello che ne consuma di più, presenta un tasso di incidenza circa doppio per la maggior parte dei tipi di cancro (polmone, laringe, cavo orale, esofago, stomaco, colon e retto, vescica, pancreas, collo dell'utero e ovaie). L'effetto protettivo nel caso dei tumori legati agli ormoni è più debole e meno sicuro: per il cancro della mammella l'effetto protettivo sembra essere di circa il 30%. Secondo un'altra ricerca, sembra che il consumo di frutta e verdura eserciti un effetto protettivo nei confronti non solo del cancro, ma anche dei disturbi cardiaci e di altre malattie degenerative dell'invecchiamento. Solo il 9% degli Americani consuma il quantitativo di frutta e verdura raccomandato dal National Cancer Institute e dal National Research Council: due porzioni di frutta e tre di verdura al giorno.
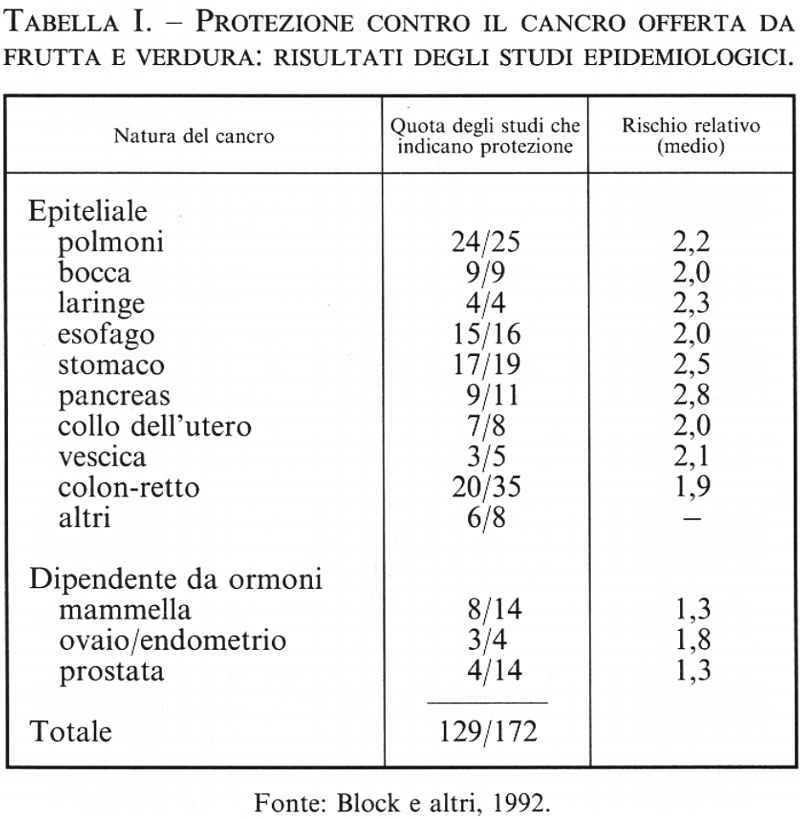
Studi sui meccanismi biochimici suggeriscono che gli antiossidanti presenti nella frutta e nella verdura possono spiegare una buona parte dell'effetto benefico di questi alimenti. Comunque è difficile distinguere, sulla base di studi epidemiologici, gli effetti dovuti all'assunzione col regime alimentare degli antiossidanti ascorbato, tocoferoli e carotenoidi da quelli dovuti ad altre importanti vitamine e ad altri costituenti della frutta e della verdura. Inoltre, è improbabile che tutti i composti che condividono proprietà antiossidanti abbiano effetti simili contro tutti i tipi di cancro, dato che ogni antiossidante ha una funzione peculiare e una distinta distribuzione nel corpo. Infine, anche se uno specifico antiossidante può svolgere un ruolo cruciale nel limitare l'incidenza del cancro, la sua concentrazione in una particolare popolazione può già essere sufficiente, talché un maggior consumo non sarebbe di giovamento.
Sono stati riportati solo pochi studi randomizzati volti a saggiare la possibilità che gli antiossidanti contribuiscano a prevenire il cancro negli esseri umani. In una ricerca condotta nella campagna cinese, una combinazione di integratori antiossidanti è risultata efficace nel ridurre l'incidenza del cancro dello stomaco, una malattia che è stata ripetutamente associata a uno scarso consumo di frutta e verdura. Comunque, le integrazioni di β-carotene non hanno ridotto il numero dei casi di cancro della pelle, così come la somministrazione delle vitamine C ed E e di β-carotene non ha fatto diminuire i casi di polipi del colon. Secondo i risultati di un ampio studio condotto di recente in Finlandia su trentenni forti fumatori, le integrazioni di β-carotene produrrebbero un leggero incremento del rischio di cancro del polmone e di malattie coronariche, nonché della mortalità complessiva, in contrasto con i risultati di molte osservazioni circa la funzione protettiva esercitata dal consumo di frutta e verdura. In questo stesso studio una modesta dose di vitamina E si è dimostrata senza relazione col rischio di cancro del polmone; va peraltro rilevato che non è stata somministrata vitamina C, la quale, come noto, è necessaria per rigenerare la vitamina E. La durata della ricerca finlandese (sei anni) forse è stata insufficiente per osservare un effetto protettivo che potrebbe operare nei primi stadi della carcinogenesi. Attualmente, quindi, i dati epidemiologici sull'efficacia di un maggior consumo di antiossidanti nella prevenzione del cancro umano non risultano coerenti. Ciononostante, dati biochimici che indicano la presenza di massicci danni ossidativi a carico del DNA, delle proteine e dei lipidi, così come un indizio indiretto - il maggior danno ossidativo a carico del DNA dello sperma umano, che si riscontra in presenza di un insufficiente apporto di ascorbato nella dieta -, suggeriscono la necessità di ulteriori indagini sugli antiossidanti potenzialmente efficaci. Oltre che sugli antiossidanti a piccola molecola introdotti nel regime alimentare, come la vitamina C, la vitamina E e vari carotenoidi, è stata richiamata l'attenzione su un certo numero di antiossidanti fisiologici precedentemente trascurati, tra cui l'urato, la bilirubina, la carnosina e l'ubichinolo. L'ubichinone (CoQ1), per esempio, è una piccola molecola cruciale per il trasporto di elettroni nei mitocondri per generare energia: la sua forma ridotta, l'ubichinolo, è un efficace antiossidante delle membrane. Livelli ottimali di ubichinone/ubichinolo nella dieta potrebbero essere importanti in molte malattie degenerative.
Oltre agli antiossidanti, molti altri composti contenuti nella frutta e nella verdura possono contribuire alla riduzione del cancro. L'acido folico può essere particolarmente importante (v. Ames e altri, 1995): una sua scarsa assunzione causa rotture di cromosomi nei Roditori e negli esseri umani e aumenta l'incidenza del tumore in alcuni tipi di roditori. L'acido folico è necessario per la sintesi dei nucleotidi del DNA e la carenza di folato provoca rotture del DNA attraverso la mancata incorporazione di uracile. L'assunzione di bassi livelli di folato è stata associata a parecchie neoplasie, compresi gli adenomi e i tumori del colon. Una carente assunzione di acido folico con la dieta sembra diffusa tra gli Americani, com'è dimostrato dagli alti livelli di omocisteina nel sangue e dall'evidente relazione con difetti del tubo neurale negli embrioni. Circa il 15% della popolazione degli Stati Uniti e circa la metà dei bambini o degli anziani di colore appartenenti a famiglie a basso reddito presentano una concentrazione di acido folico inferiore a 4ng/ml di siero, che rappresenta un valore di soglia al di sotto del quale sono state osservate rotture dei cromosomi. La presenza nella dieta di fibra ottenuta soltanto da cibi di origine vegetale può contribuire ad abbassare il rischio di cancro del colon. La vitamina A, che può derivare da alcuni carotenoidi nonché da alimenti di origine animale, regola il differenziamento cellulare e riduce l'incidenza del tumore in molti animali e forse nell'uomo. Frutta e verdura possono anche ridurre il rischio di cancro perché contengono un'ampia varietà di possibili antiossidanti, come i flavonoidi, nonché di induttori di enzimi disintossicanti, come gli indoli; inoltre, gli alimenti di origine vegetale contengono una vasta gamma di estrogeni deboli che possono agire come antiestrogeni competendo con gli estrogeni endogeni.
3. Altri aspetti della dieta. - Se, da un lato, i benefici della frutta e della verdura nella prevenzione del cancro sono comprovati molto chiaramente da studi epidemiologici, dall'altro forti analogie a livello internazionale suggeriscono che il grasso animale (ma non quello vegetale) e la carne rossa possano far aumentare l'incidenza dei tumori della mammella, del colon e della prostata. Tuttavia, studi prospettici ad ampio spettro hanno concordemente dimostrato che sussiste una associazione debole o nulla tra il consumo di grassi e l'incidenza del cancro della mammella. Il grasso animale e la carne rossa sono stati invece associati al rischio di cancro del colon in numerosi studi di casi-controllo e per coorti, ma l'associazione con il consumo di carne sembra essere quella più diffusa. Il consumo di grasso animale e di carne rossa è stato associato al rischio di cancro della prostata in molteplici studi: i meccanismi ipotizzati alla base di queste associazioni comprendono gli effetti dei grassi assunti con la dieta sui livelli degli ormoni endogeni, gli effetti proliferativi degli acidi biliari sulla mucosa del colon, gli effetti dovuti a cancerogeni prodotti nella cottura della carne e un'eccessiva assunzione di ferro. È possibile, per quanto indimostrato, che un'eccessiva assunzione di ferro (nell'intestino il ferro-eme della carne è assorbito come tale) contribuisca alla produzione di radicali dell'ossigeno; comunque, l'importanza di questi meccanismi resta ipotetica. Alcuni risultati sperimentali suggeriscono che un maggior consumo di calcio contrasti la proliferazione indotta da un eccesso di grassi nella dieta, riducendo quindi il rischio di cancro del colon; comunque, studi di casi-controllo e studi per coorti hanno portato a risultati divergenti. Per quel che riguarda la frequenza del cancro del colon, alcune delle grandi differenze geografiche che sono state attribuite a fattori alimentari sono probabilmente dovute a differenze di attività fisica, che, secondo molti studi, è inversamente correlata al rischio di contrarre il cancro del colon.
La cottura del cibo è un fattore che, probabilmente, contribuisce all'insorgenza del cancro (v. Ames e altri, 1995). Durante la cottura si formano svariate sostanze chimiche: i seguenti quattro gruppi di sostanze chimiche che provocano tumori nei Roditori hanno attirato l'attenzione a causa del loro potere mutageno e cancerogeno e della loro concentrazione.
1) Nitrosammine: queste sostanze si formano dagli ossidi d'azoto presenti nelle fiamme a gas o da altri processi di combustione, e alcune di esse, come la dimetilnitrosammina, possono essere presenti in quantità relativamente elevate rispetto al loro potere cancerogeno nei Roditori. Stranamente, considerato il potere mutageno e cancerogeno delle nitrosammine, i livelli raggiunti da queste sostanze nel pesce o nella carne cotti nei forni a gas o sul barbecue non sono stati adeguatamente studiati.
2) Ammine eterocicliche: si formano dal riscaldamento di amminoacidi o proteine, e nei Roditori sono risultate essere dei potenti mutageni e cancerogene per molti organi, benché di solito nei cibi cotti se ne formino quantità relativamente scarse.
3) Idrocarburi policiclici: si formano nel processo di carbonizzazione della carne, anch'essi in quantità modeste.
4) Furfurale e furani analoghi: si formano dal riscaldamento di zuccheri e sono presenti in dosi relativamente alte nella dieta.
Gli studi epidemiologici sulla cottura dei cibi sono ardui e finora non hanno permesso di valutare quale effetto cancerogeno essa abbia sugli esseri umani.
Le bevande alcoliche causano cirrosi epatica e infiammazione e cancro del fegato; l'alcool è inoltre una causa importante di cancro orale ed esofageo (ed esercita un'azione sinergica con quella del fumo) e forse contribuisce al cancro colon-rettale. Anche il cancro della mammella è associato al consumo di alcool.
c) Tabacco.
Il fumo, che si pone al primo posto tra i fattori responsabili di tutti i tipi di cancro, è un elemento voluttuario e come tale evitabile (v. fig. 1; v. Doll e Peto, 1981; v. Peto e altri, 1994). Negli Stati Uniti esso contribuisce a circa un terzo dei casi di cancro, a circa un quarto delle malattie di cuore e a circa 400.000 morti premature all'anno. Si è accertato che il tabacco causa tumori del polmone, della vescica, della bocca, della faringe, del pancreas, del rene, dello stomaco, della laringe, dell'esofago e probabilmente del colon, oltre a provocare un numero ancora maggiore di morti per malattie diverse: negli anni novanta sono risultati imputabili al tabacco circa tre milioni di morti all'anno su scala mondiale e, se si continuerà a fumare al ritmo attuale, fra qualche decennio si arriverà a circa dieci milioni di morti all'anno. ‟In tutta la seconda metà di questo secolo (1950-2000) il numero totale di morti causate dal fumo nei paesi sviluppati sarà di circa sessanta milioni" (v. Peto e altri, 1994). Le prove a favore dell'ipotesi che il fumo passivo provochi il cancro sono molto deboli: è stato stimato che esso causi fino a 3.000 ulteriori casi di cancro negli Stati Uniti, per quanto tale stima sia stata decisamente contestata.
I meccanismi tramite i quali il fumo di tabacco provoca il cancro non sono ben compresi. Il fumo, oltre a comportare un grave stress ossidativo, contiene una gran varietà di mutageni e di sostanze che risultano cancerogene per i Roditori. Gli ossidanti nel fumo delle sigarette (soprattutto gli ossidi di azoto) esauriscono gli antiossidanti del corpo; pertanto, per raggiungere lo stesso livello di ascorbato nel sangue, i fumatori devono ingerirne due o tre volte di più dei non fumatori, ma raramente lo fanno. Regimi alimentari inadeguati (e il fumo) possono danneggiare non soltanto il DNA somatico, ma anche quello spermatico. Quando la quantità di ascorbato assunta per via alimentare è insufficiente a mantenere l'ascorbato nel liquido seminale a un livello adeguato, allora le lesioni ossidative del DNA spermatico aumentano di due volte e mezza. Un insufficiente livello di ascorbato nel plasma è più comune tra i maschi singoli, i poveri, i fumatori. Il tasso di mutazione della linea germinale è influenzato molto di più dal padre (dato che gli spermatociti si dividono costantemente, l'età del padre costituisce un importante fattore di rischio) che dalla madre (i cui oociti si formano durante la vita intrauterina): per tale ragione il fumo paterno può far aumentare il rischio di difetti alla nascita e di cancro infantile nella prole.
d) Infezioni croniche, flogosi e cancro.
I leucociti e gli altri fagociti combattono i Batteri, i parassiti e le cellule infettate da virus distruggendoli con ossido e superperossido d'azoto, che reagiscono formando perossinitrito (un potente agente ossidante e nitrante, mutageno), ipoclorito (un agente clorante e ossidante, mutageno) e perossido di idrogeno (un agente ossidante mutageno). Questi ossidanti proteggono gli esseri umani da una morte immediata per infezione, ma inducono anche danni ossidativi a carico del DNA, mutazioni, nonché una cronica uccisione di cellule che provoca una divisione cellulare compensativa, contribuendo così al processo cancerogeno. Gli antiossidanti sembrano inibire alcune patologie flogistiche croniche.
Le infezioni croniche contribuiscono a circa un terzo dei casi di cancro nel mondo (v. Ames e altri, 1995; v. IARC, 1994). I virus delle epatiti B e C sono un'importante causa di infiammazione cronica che conduce al cancro del fegato, una delle forme di cancro più comuni in Africa e in Asia (circa la metà dei casi su scala mondiale si verifica in Cina). I virus delle epatiti B e C infettano circa 500 milioni di persone in tutto il mondo. Vaccinare i neonati alla nascita costituisce potenzialmente un metodo efficace per ridurre il rischio di contrarre il cancro del fegato; in Italia la vaccinazione dei neonati contro l'epatite B è stata resa obbligatoria dal 1991.
L'aflatossina, una tossina mutagena che si trova nei prodotti a base di arachidi e granturco contaminati da muffe, sembra agire in sinergia con l'infezione cronica da epatite nello sviluppo del cancro del fegato. Rilievi eseguiti su popolazioni dell'Africa e della Cina confermano che queste popolazioni sono cronicamente esposte ad alti livelli di aflatossina. Negli Stati Uniti il cancro del fegato è raro: benché i virus delle epatiti B e C infettino meno dell'1% della popolazione, a essi si può imputare la metà dei casi di cancro del fegato fra i non asiatici e percentuali anche maggiori tra gli asiatici.
Un'altra importante infezione cronica è la schistosomiasi, ampiamente diffusa in Asia e in Egitto. In Asia le uova di Schistosoma japonicum, deposte nella mucosa del colon, causano flogosi e conseguente cancro del colon. In Egitto le uova di Schistosoma haematobium, deposte nella vescica, causano infiammazione e cancro della vescica. Opisthorchis viverrini, un trematode epatico, infetta milioni di persone in Thailandia e in Malaysia: i trematodi si annidano nei dotti biliari e fanno aumentare il rischio di colangiocarcinoma. Le infezioni da Clonorchis sinensis, cui vanno soggetti milioni di Cinesi, determinano un aumento del rischio di cancro delle vie biliari. Il batterio Helicobacter pylori, che infetta lo stomaco e colpisce oltre un terzo della popolazione mondiale, è un'importante causa di cancro dello stomaco, ulcere e gastrite. Nei paesi ricchi, tuttavia, l'infezione è spesso asintomatica, il che suggerisce che l'infiammazione possa essere almeno parzialmente soppressa, probabilmente attraverso un consumo adeguato di antiossidanti.
Anche un'infiammazione cronica di origine non infettiva può portare al cancro: per esempio, il fatto che l'esposizione all'amianto induca un'infiammazione cronica può essere in buona parte la ragione per cui essa rappresenta un significativo fattore di rischio per il cancro del polmone.
Il virus del papilloma umano, un importante fattore di rischio per il cancro del collo dell'utero, non sembra agire attraverso un meccanismo flogistico; la sua diffusione è in rapporto alla promiscuità sessuale, che rappresenta un modo efficace per trasmettere qualsiasi virus.
e) Ormoni.
Henderson e i suoi collaboratori hanno passato in rassegna la vasta letteratura che segnala l'importante effetto cancerogeno degli ormoni sessuali, forse corresponsabili addirittura di un terzo di tutti i casi di cancro (v. Henderson e altri, 1991 e 1993). È probabile che gli ormoni agiscano causando la divisione cellulare (v. cap. 3, § c). Il rischio di cancro dell'endometrio - un cancro che sembra estremamente sensibile a un'esposizione cumulativa agli estrogeni - aumenta da 10 a 20 volte in seguito all'uso prolungato di estrogeni esogeni. Gli estrogeni aumentano la divisione delle cellule dell'endometrio, mentre i progestinici la riducono; quindi l'aggiunta di progestinici alla terapia a base di estrogeni, spesso praticata dopo la menopausa, può ridurre il rischio di cancro dell'endometrio.
Il cancro delle ovaie sembra collegato a fattori che aumentano la divisione delle cellule epiteliali di superficie. L'incidenza di questo tumore può essere diminuita riducendo il numero delle ovulazioni, ad esempio con le gravidanze, o bloccandole con l'uso di contraccettivi orali, la cui assunzione per oltre 5 anni sembra ridurre fino al 50% il rischio di questa formazione maligna.
Una maggiore esposizione cumulativa agli estrogeni, determinata da fattori come un menarca precoce, una menopausa tardiva e prolungate terapie a base di estrogeni dopo la menopausa, fa aumentare i rischi di cancro della mammella. In questo tipo di tumore, infatti, le cellule neoplastiche sembrano proliferare in presenza di estrogeni, e anche i progestinici sembrano intensificare la divisione cellulare; pertanto, la loro aggiunta a una terapia a base di estrogeni non solo non riduce, ma può forse aumentare ulteriormente il rischio di cancro della mammella. La gravidanza influisce in modo complesso sul cancro della mammella, in quanto tra le donne che hanno avuto prole il rischio aumenta (probabilmente a causa della stimolazione ormonale) per un periodo iniziale di uno o due decenni, ma l'incidenza del tumore nell'arco della vita alla fine risulta ridotta, forse in virtù di un differenziamento permanente delle cellule staminali che dà luogo a una minore proliferazione. L'allattamento riduce moderatamente l'incidenza del cancro della mammella. L'osservazione che gli ormoni influenzano l'incidenza del cancro della mammella suggerisce alcuni modi per ridurla: uno di questi è sviluppare un contraccettivo ormonale capace di imitare l'effetto di una menopausa precoce, che potrebbe diminuire della metà tale rischio, riducibile anche, nelle donne giovani, attraverso l'esercizio fisico che probabilmente influenza i livelli ormonali. Un altro metodo per ridurre questo rischio potrebbe consistere nella diminuzione del consumo di alcool, regolarmente associato al rischio di cancro della mammella in studi prospettici a largo spettro, nonché in molti studi di casi-controllo, in quanto sembra aumentare i livelli di estrogeni endogeni. Cibi come i semi di soia, i quali contengono sostanze debolmente estrogene che competono con gli estrogeni endogeni più potenti, potrebbero anch'essi ridurre il rischio di cancro della mammella.
5. Fattori di rischio meno importanti
a) Attività professionale.
La IARC (International Agency for Research on Cancer) dell'Organizzazione Mondiale della Sanità valuta i rischi di contrarre un cancro in seguito all'esposizione ad agenti chimici. Metà dei 60 elementi e composti chimici giudicati cancerogeni per gli esseri umani costituiscono fattori di rischio per piccoli gruppi di persone esposte per periodi prolungati ad alte concentrazioni di tali sostanze per motivi professionali. Risultano cancerogene le sostanze impiegate in particolari lavorazioni, come nell'industria della gomma o nella produzione di carbon coke, nonché specifiche ammine aromatiche, prodotti petrolchimici, metalli, ecc. La questione di quale quota dei casi di cancro sia attribuibile all'esposizione a sostanze cancerogene per motivi professionali è controversa, ma sembra ragionevole supporre che possa ammontare a qualche punto percentuale. Doll e Peto (v., 1981) hanno preso in esame i fattori che impediscono di eseguire correttamente tali stime, quali ad esempio la mancanza di dati accurati sull'esposizione passata e presente ai cancerogeni e l'esistenza di fattori ‛di confondimento', come lo status socioeconomico e il fumo (in patologia si intende per confondimento l'associazione, priva di carattere di causalità, tra una malattia e un determinato agente, dipendente dalla frequente correlazione tra l'esposizione a questo agente e il vero agente causale: così, ad esempio, l'incidenza di cancro del polmone, che non è in rapporto all'assunzione di alcool, è elevata tra i bevitori, che sono spesso anche fumatori). Nella stima di Doll e Peto, il cancro del polmone era di gran lunga il tipo di tumore più frequente fra le neoplasie dovute ad attività professionale. La preminenza del fumo come causa del cancro del polmone impedisce di imputare una precisa percentuale di casi di questa patologia all'esposizione a particolari sostanze: l'amianto, per esempio, sembra moltiplicare l'effetto del fumo piuttosto che limitarsi a sommarsi a esso, mentre da solo è un noto fattore di rischio di induzione del mesotelioma. All'amianto si imputava un'alta percentuale di tumori di origine professionale, mentre di recente questa stima è stata ridimensionata.
Le quantità di cancerogeni chimici cui si è esposti sul luogo di lavoro possono essere alte rispetto a quelle presenti nel cibo, nell'aria o nell'acqua. A nostro parere, dato che l'aumento della velocità della divisione cellulare è un importante fattore mutageno e cancerogeno, l'estrapolazione dai risultati dei test sui tumori indotti esponendo gli animali da laboratorio ad alte dosi di cancerogeni a casi di esseri umani esposti a basse dosi degli stessi cancerogeni non può essere fatta senza considerare il meccanismo della carcinogenesi per ciascun agente chimico. Comunque, in passato, si sono verificati casi di esposizioni, sul luogo di lavoro, ad alte dosi di cancerogeni; in situazioni del genere, il grado di estrapolazione quantitativa necessario per estendere i risultati ottenuti sperimentalmente sui Roditori alla realtà delle esposizioni ad alte dosi sul luogo di lavoro può essere abbastanza basso. Dato che i casi di cancro di origine professionale sono limitati a piccoli gruppi esposti ad alte dosi di cancerogeni, è possibile controllarne o eliminarne i rischi, una volta che tali agenti siano stati identificati. Tuttavia, in contrasto con altri enti federali come la Environmental Protection Agency (EPA), l'Occupational Safety and Health Administration (OSHA) degli Stati Uniti regolamenta poche sostanze chimiche in quanto potenziali cancerogeni umani. Per i 75 agenti risultati cancerogeni per i Roditori per i quali l'OSHA ha stabilito limiti di esposizione consentita, sono stati recentemente classificati i potenziali rischi tramite un indice (PERP, Permitted Exposure/Rodent Potency) che confronta il tasso-dose di esposizione consentita con la dose cancerogena per i Roditori (v. Gold e altri, 1994). È risultato che per nove prodotti chimici le esposizioni permesse erano comprese entro un fattore 10 della dose cancerogena per i Roditori, e per 17 erano tra 10 e 100 volte più basse di tale dose. Questi valori sono alti in confronto con i rischi ipotetici regolamentati da altri enti federali. Per altre 120 sostanze cancerogene per i Roditori, l'OSHA non ha stabilito limiti di esposizione consentita; è pertanto necessario proseguire nella regolamentazione.
b) Esposizione al sole.
L'esposizione al sole è la causa principale dei tumori della pelle, in particolare del melanoma, e, specialmente nei casi in cui è causa di ustioni nei primi decenni di vita, sembra esserne il fattore dominante. Una corretta informazione e l'adozione di misure protettive costituiscono una efficace prevenzione per le persone di carnagione chiara.
c) Fattori iatrogeni.
Alcuni farmaci chemioterapici antineoplastici, in particolare agenti alchilanti, provocano formazioni maligne secondarie, più spesso leucemie, linfomi e sarcomi. Si pensa inoltre che taluni farmaci usati in passato, come la fenacetina e il dietilstilbestrolo, aumentino il rischio di contrarre un cancro. Anche potenti agenti immunosoppressori, come la ciclosporina, aumentano il rischio di contrarre diverse forme di cancro e, come già detto, la terapia estrogenica sostitutiva incrementa il rischio di contrarre il cancro dell'endometrio e quello della mammella. Gli stessi raggi X usati a fini diagnostici contribuiscono alla formazione di tumori maligni. Anche se si ritiene che questi effetti collaterali debbano esser presi in considerazione allorché si stabilisce una terapia, il contributo complessivo delle procedure terapeutiche e diagnostiche all'incidenza del cancro è modesto.
d) Inquinamento.
Gli inquinanti sintetici sono temuti da gran parte dell'opinione pubblica come principali cause di cancro, ma si tratta di un timore infondato. Anche se le più pessimistiche stime di rischio per gli inquinanti sintetici effettuate dall'Environmental Protection Agency fossero veritiere, la quota di casi di cancro prevenibile attraverso una regolamentazione sarebbe esigua (v. Gold e altri, 1992). Per di più, gli studi epidemiologici risultano particolarmente ardui per la difficoltà di accertare con precisione l'entità dell'esposizione ai diversi cancerogeni e per l'impossibilità di isolare gli effetti di fattori di confondimento, come il fumo, la dieta e la mobilità della popolazione. Dato che questo articolo si limita a trattare le cause del cancro, altri importanti argomenti relativi alla protezione ambientale e alla salute non verranno discussi.
1. Inquinamento dell'aria (v. Ames e altri, 1995). - Prenderemo in considerazione l'aria degli ambienti chiusi sia perché gli esseri umani passano il 90% del proprio tempo al chiuso, sia perché le concentrazioni di alcuni inquinanti tendono a essere più elevate che all'aperto. Probabilmente il più importante inquinante dell'aria con effetto cancerogeno è il radon, un gas radioattivo prodotto dal decadimento del radio presente in tracce nella crosta terrestre, che penetra nelle case principalmente attraverso l'aria che fuoriesce dal sottosuolo. In base a studi epidemiologici su minatori esposti alle alte concentrazioni di radon presenti nel sottosuolo, è stato stimato che negli Stati Uniti il radon provochi annualmente 15.000 casi di cancro del polmone, prevalentemente tra i fumatori, dato l'effetto sinergico del fumo. Studi epidemiologici sull'esposizione al radon nelle case non hanno dimostrato in modo convincente un rischio eccessivo. Si calcola che negli Stati Uniti i livelli medi annuali di radon riscontrati in 50-100 mila case (lo 0,1% circa) siano circa 20 volte superiori a quelli della media nazionale, e che i rispettivi abitanti ricevano dosi di radiazioni annuali che superano la quantità standard corrente cui sono esposti i minatori che lavorano nel sottosuolo. Le ricerche volte a identificare le case ad alta concentrazione di radon ne hanno indicata una maggiore frequenza in ben localizzate aree geografiche. Gli stessi abitanti delle case comprese nelle aree ad alta concentrazione di radon possono effettuarne una misurazione e ridurla a buon mercato con la tecnologia disponibile.
2. Inquinamento dell'acqua (v. IARC, 1991; v. Ames e altri, 1995). - Il rischio di cancro dovuto all'inquinamento dell'acqua è trascurabile. Tra i potenziali fattori di rischio presi in considerazione, i principali sono il radon (anche se di importanza modesta rispetto a quello presente nell'aria) e l'arseniato naturale, che è un noto cancerogeno umano; relativamente a quest'ultimo, sono necessarie ulteriori ricerche per chiarirne il meccanismo d'azione e le dosi da considerarsi cancerogene per gli esseri umani.
La clorazione dell'acqua, un importante intervento a favore della salute pubblica, produce come sottoprodotti molte sostanze chimiche contenenti cloro, alcune delle quali sono cancerogene per i Roditori; tuttavia, le prove di un'associazione tra il cancro umano e l'acqua clorata sono state considerate inadeguate. Un'associazione precedentemente ipotizzata con il cancro della vescica e con quello del colon non è stata confermata in un recente studio-intervista su casi-controllo, ma è stata osservata un'associazione con il cancro del retto.
6. Fattori ereditari
È chiaro che una certa percentuale di casi di cancro - specialmente nei bambini e in giovani adulti - dipende da fattori ereditari. I tassi relativi a tutte le forme di cancro aumentano esponenzialmente con l'età, a eccezione di una flessione nella curva del cancro infantile, che si pensa sia dovuto principalmente alla trasmissione ereditaria di un gene mutante che provoca il cancro. È probabile che la predisposizione a tutti i tipi di cancro sia influenzata da fattori ereditari, ma non è chiaro fino a che punto; tuttavia è lecito attendersi che la biologia molecolare possa presto chiarire questo aspetto. Quasi tutti i tumori più importanti sono imputabili a fattori diversi da quelli ereditari, come indicano le grandi differenze riscontrabili da paese a paese nei tassi di incidenza delle varie neoplasie, l'osservazione che tra gli immigrati le frequenze delle diverse forme tumorali sono prossime a quelle della popolazione ospite e il fatto che i tassi relativi a molti tumori hanno subito grandi mutamenti nel corso del tempo. Mentre non si hanno prove definitive dell'influenza dei fattori ereditari sul cancro del polmone, il rischio attribuibile a tali fattori nel caso del cancro della mammella sembra essere dell'ordine del 10%. Ciononostante, l'identificazione dei tumori ad alto rischio genetico potrà essere particolarmente importante, specie se si riuscirà a identificare dei fattori modificabili capaci di interagire con la predisposizione genetica e a mettere a punto metodi di screening sensibili, come, per esempio, la colonscopia.
7. Errori di valutazione; limiti di validità dei test sugli animali
L'idea che sia in atto un'epidemia di cancro umano causata da sostanze chimiche industriali sintetiche non è suffragata né da dati tossicologici, né da dati epidemiologici. Alcuni studi epidemiologici individuano una certa associazione tra cancro e inquinamento industriale, ma non tengono conto della dieta, che è un fattore potenzialmente importante di confondimento; inoltre, i livelli degli inquinanti, che in confronto con il sottofondo di sostanze chimiche naturali che risultano cancerogene per i Roditori sono bassi, raramente sembrano costituire un fattore causale (v. Ames e Gold, 1990; v. Gold e altri, 1992; v. Ames e altri, 1995).
In grandissima maggioranza le sostanze chimiche cui gli esseri umani sono esposti sono naturali e per ogni sostanza chimica esiste un determinato livello al quale risulta tossica. È quindi sbagliato il giudizio di Rachel Carson, secondo cui: ‟Per la prima volta nella storia del mondo, ogni essere umano è ora soggetto al contatto con sostanze chimiche pericolose, dal momento del concepimento alla morte" (v. Carson, 1962).
I test sui tumori animali sono condotti usualmente con sostanze chimiche sintetiche usando la massima dose tollerata (MDT) di ogni sostanza studiata, ma i risultati ottenuti sono stati erroneamente interpretati come una dimostrazione che basse dosi di sostanze chimiche sintetiche e di inquinanti industriali fossero rilevanti per il cancro umano. Alle alte dosi impiegate, ben una metà circa delle sostanze chimiche sottoposte a prova, sia sintetiche che naturali, risulta cancerogena per i ratti o per i topi (v. Gold e altri, 1992; v. Cunningham e altri, 1994; v. Ames e altri, 1996). Una spiegazione possibile per l'alta percentuale di risultati positivi è che il test alla MDT spesso può causare l'uccisione cronica di cellule e la loro conseguente sostituzione; ma in questo caso il rischio di cancro è legato ai dosaggi elevati.
Le sostanze chimiche ingerite dagli esseri umani sono in larghissima misura, sia per peso sia per numero, di origine naturale. Per esempio, quasi tutti i pesticidi presenti nei cibi (il 99,99%) costituiscono difese naturali di cui dispongono le piante contro gli Insetti e altri predatori. La metà dei pesticidi naturali provati (29 su 57) è cancerogena per i Roditori. Ridurre l'esposizione ai pochissimi (lo 0,01%) pesticidi sintetici, singole sostanze chimiche o loro miscele, non ridurrebbe apprezzabilmente i tassi di cancro. Al contrario, frutta e verdura, come abbiamo visto, esercitano un efficace ruolo di prevenzione del cancro; renderle più care riducendo l'uso dei pesticidi sintetici con ogni probabilità aumenterebbe l'incidenza del cancro. Individui con bassi redditi consumano meno frutta e verdura e spendono una quota maggiore del proprio reddito in alimenti.
Gli esseri umani ingeriscono anche numerosissime sostanze chimiche derivate dalla cottura dei cibi. Per esempio, nel caffè tostato sono state identificate più di mille sostanze chimiche; solo 26 sono state sottoposte a prova e di queste oltre la metà (19) è risultata cancerogena per i Roditori. Vi sono più cancerogeni naturali, in peso, in una singola tazza di caffè che residui di pesticidi sintetici potenzialmente cancerogeni nella dieta media americana di un anno. Ciò non significa necessariamente che il caffè sia pericoloso, ma che i test per il cancro condotti sugli animali e le valutazioni di rischio più pessimistiche portano a stabilire dei coefficienti di sicurezza esagerati che dovrebbero venir ridimensionati.
A causa della loro inusuale lipofilia e della loro tendenza a persistere a lungo nell'ambiente, si è manifestato un particolare interesse per un piccolo gruppo di sostanze chimiche sintetiche policlorurate, quali il DDT e i PCB (bifenili policlorurati). Non esistono riscontri epidemiologici convincenti, né sembra plausibile sotto il profilo tossicologico che i livelli normalmente riscontrati nell'ambiente contribuiscano significativamente all'insorgenza del cancro. La TCDD (tetraclorodibenzo-p-diossina), che è prodotta naturalmente per combustione in presenza di cloruri ed è un sottoprodotto industriale, è un cancerogeno insolitamente potente per i Roditori, ma è improbabile che il suo effetto cancerogeno sull'uomo sia rilevante, almeno ai livelli ai quali è esposta normalmente la popolazione.
La ragione per cui gli esseri umani possono ingerire col cibo l'enorme varietà di sostanze che sono cancerogeni naturali per i Roditori è che essi, come altri animali, sono estremamente ben protetti da molti sistemi enzimatici di difesa, la maggior parte dei quali sono inducibili (cioè quanto più un enzima è necessario, tanto più ne viene prodotto; v. Ames e altri, 1990). Gli enzimi protettivi sono efficaci contro sostanze chimiche sia sintetiche sia naturali, come certi reattivi chimici potenzialmente mutageni. Nei test a dosaggi elevati sui Roditori non è stata riscontrata una diversa capacità di indurre il cancro tra le sostanze di sintesi e quelle naturali (v. Gold e altri, 1992).
Sono stati classificati i possibili rischi derivanti da sostanze note come cancerogene per i Roditori usando un indice (HERP, Human Exposure/Rodent Potency) che mette in relazione l'esposizione umana con il potere cancerogeno per i Roditori di tali sostanze (v. Gold e altri, 1992). Tale classificazione non valuta i rischi in termini assoluti, cosa che la scienza non è ancora in grado di fare, ma confronta i possibili rischi derivanti dal contatto con sostanze chimiche sintetiche con i livelli di fondo di sostanze cancerogene per i Roditori presenti comunemente nella dieta. I livelli dei residui di pesticidi sintetici o degli inquinanti ambientali sono assai bassi in confronto ai livelli di fondo presenti in natura, anche se i rischi connessi a questi ultimi risultano assai poco evidenti da questo tipo di confronto, dato lo scarso numero di sostanze chimiche naturali sottoposte a test di cancerogenità nei Roditori. Nel caso degli agenti chimici sintetici, il fatto di aver estrapolato linearmente agli uomini, sottoposti a una moderata esposizione, i risultati ottenuti nei Roditori con la massima dose tollerata, ignorando gli alti livelli di fondo degli agenti naturali, ha portato a esagerare le stime di rischio di cancro e a uno squilibrio tra la valutazione del rischio e l'allocazione delle risorse.
8. Conclusione
I dati epidemiologici permettono di identificare parecchie ampie categorie di fattori cancerogeni la cui pericolosità è chiaramente dimostrata (v. Ames e altri, 1995). Dato che i rischi connessi a molti di questi fattori sono evitabili, è possibile ridurre i tassi di incidenza di vari tipi di cancro. Nella loro rassegna del 1981 sui rischi di cancro evitabili negli Stati Uniti, Doll e Peto (v., 1981) attribuirono il 30% delle morti per cancro al tabacco e circa il 35% a fattori alimentari, benché il valore del contributo della dieta variasse dal 10 al 70%. Altri fattori vennero giudicati molto meno determinanti. Da allora la percentuale dovuta al fumo sembra essere aumentata un po' (sembra più realistico attribuirgli una quota del 35%) - anche se fra gli Americani adulti il numero dei fumatori è leggermente diminuito - perché il rischio relativo dovuto al fumo è molto aumentato per quasi tutti i tipi di cancro, nonché per le malattie cardiovascolari (v. Peto e altri, 1994). Ciò è probabilmente dovuto al fatto che il rischio di morte per cancro tra i non fumatori è diminuito e che l'impatto, nel corso della vita, dell'abitudine di fumare sin dall'adolescenza si sta sperimentando soltanto ora. I dati disponibili sui rapporti fra dieta e cancro si sono moltiplicati dal 1981, e in genere suffragano la stima precedente, anche se sembra più plausibile attribuire ai fattori alimentari un intervallo leggermente più stretto, tra il 20 e il 40% (v. Willett, 1995). In generale, i nuovi dati hanno evidenziato un inadeguato consumo di fattori protettivi piuttosto che una eccessiva assunzione di fattori nocivi. La stima relativa alla dieta è stata leggermente ridimensionata, in larga misura perché le grandi differenze su scala internazionale per quel che riguarda la frequenza del cancro del colon sono probabilmente dovute a differenze nell'attività fisica oltre che nella dieta. La stima effettuata da Doll e Peto nel 1981, secondo cui il contributo della dieta al cancro della mammella è pari al 50%, è ancora plausibile, ma il rischio relativo può non essere, in pratica, evitabile, dal momento che i rapidi tassi di accrescimento sono il più importante fattore nutrizionale di base. La stima relativa alle bevande alcoliche può essere leggermente aumentata dal 3 ± 1% al 5 ± 1%, in quanto molti nuovi studi hanno comprovato una stretta relazione fra il consumo di alcool e i tumori della mammella e del colon. I dati successivi al 1981 non hanno fornito una base per modificare sostanzialmente le precedenti stime per altre cause.
Per valutare l'impatto sulla popolazione dell'adozione di importanti norme igieniche atte ad abbassare il rischio di contrarre un cancro, basta confrontare l'incidenza delle neoplasie e i tassi di mortalità nella popolazione generale degli Stati Uniti con quelli relativi agli Avventisti del Settimo Giorno, che generalmente non fumano, non bevono esageratamente, non mangiano molta carne e seguono una dieta ricca di frutta e verdura (v. Mills e altri, 1994). In questo gruppo si osservano tassi di mortalità per cancro del polmone, della vescica e del colon considerevolmente più bassi; la mortalità complessiva per cancro è circa la metà di quella della popolazione generale. Sebbene questo confronto abbia dei limiti, in quanto un miglior uso dei servizi medici può contribuire a ridurre la mortalità e, d'altra parte, un'imperfetta conformità alle raccomandazioni può far sottovalutare l'impatto dello stile di vita, i risultati suggeriscono decisamente che una notevole quota delle morti per cancro può essere evitata usando le conoscenze disponibili. I tassi di incidenza forniscono un quadro simile a quello offerto dai tassi di mortalità, benché le differenze siano un po' meno accentuate. Per il cancro della mammella il comportamento conforme alle norme igieniche degli Avventisti del Settimo Giorno non bastava a diminuire significativamente il rischio di contrarre un tumore.
La diminuzione dell'attività fisica e l'aumento del fumo, dell'obesità e dell'esposizione al sole hanno contribuito notevolmente ad aumentare l'incidenza di alcuni tumori nel mondo industriale moderno, mentre miglioramenti nelle misure igieniche hanno ridotto altre forme di cancro legate alle infezioni. Non esiste una buona ragione per credere che sostanze chimiche sintetiche siano alla base degli importanti cambiamenti nell'incidenza di alcune forme di cancro. Negli Stati Uniti e in altri paesi industrializzati la speranza di vita sta costantemente aumentando e aumenterà ancor più velocemente al venir meno dell'abitudine di fumare.
BIBLIOGRAFIA
Ames, B. N., Gold, L. S., Chemical carcinogenesis: too many rodent carcinogens, in ‟Proceedings of the National Academy of Sciences", 1990, LXXXVII, pp. 7772-7776.
Ames, B. N., Gold, L. S., Shigenaga, M. K., Cancer prevention, rodent high-dose cancer tests, and risk assessment, in ‟Risk analysis", 1996, XVI, 5, pp. 613-617.
Ames, B. N., Gold, L. S., Willett, W. C., The causes and prevention of cancer, in ‟Proceedings of the National Academy of Sciences", 1995, XCII, pp. 5258-5265.
Ames, B. N., Profet, M., Gold, L. S., Nature's chemicals and synthetic chemicals: comparative toxicology, in ‟Proceedings of the National Academy of Sciences", 1990, LXXXVII, pp. 7782-7786.
Ames, B. N., Shigenaga, M. K., Hagen, T. M., Oxidants, antioxidants, and the degenerative diseases of aging, in ‟Proceedings of the National Academy of Sciences", 1993, XC, pp. 7915-7922.
Block, G., Patterson, B., Subar, A., Fruit, vegetables and cancer prevention: a review of the epidemiologic evidence, in ‟Nutrition and cancer", 1992, XVIII, pp. 1-29.
Carson, R., Silent spring, Boston 1962 (tr. it.: Primavera silenziosa, Milano 19955).
Cunningham, M. L., Elwell, M. R., Matthews, H. B., Relationship of carcinogenicity and cellular proliferation induced by mutagenic noncarcinogens vs carcinogens, in ‟Fundamental and applied toxicology", 1994, XXIII, pp. 363-369.
Doll, R., Peto, R., The causes of cancer. Quantitative estimates of avoidable risks of cancer in the United States today, in ‟Journal of the National Cancer Institute", 1981, LXVI, pp. 1191-1308.
Gold, L. S., Garfinkel, G. B., Slone, T. H., Setting priorities among possible carcinogenic hazards in the workplace, in Chemical risk assessment and occupational health: current applications, limitations and future prospects (a cura di C. M. Smith, D. C. Christiani e K. T. Kelsey), Westport, Conn., 1994, pp. 91-103.
Gold, L. S., Slone, T. H., Stern, B. R., Manley, N. B., Ames, B. N., Rodent carcinogens: setting priorities, in ‟Science", 1992, CCLVIII, pp. 261-265.
Henderson, B. E., Ross, R. K., Pike, M. C., Toward the primary prevention of cancer, in ‟Science", 1991, CCLIV, pp. 1131-1138.
Henderson, B. E., Ross, R. K., Pike, M. C., Hormonal chemoprevention of cancer in women, in ‟Science", 1993, CCLIX, pp. 633-638.
IARC (International Agency for Research on Cancer), Chlorinated drinking-water; chlorination by-products, Lyon 1991.
IARC (International Agency for Research on Cancer), Schistosomes, liver flukes and Helicobacter pylori, Lyon 1994.
Miller, B. A., Ries, L. A. G., Hankey, B. F., Kosary, C. L., Harras, A., Devesa, S. S., Edwards, B. K., SEER cancer statistics review: 1973-1990, Bethesda, Md., 1993.
Mills, P. K., Beeson, W. L., Phillips, R. L., Fraser, G. E., Cancer incidence among California Seventh-day adventists, in ‟American journal of clinical nutrition", 1994, LIX, suppl., pp. 1136-1142.
Peto, R., Lopez, A. D., Boreham, J., Thun, M., Heath, C. Jr., Mortality from smoking in developed countries 1950-2000, Oxford 1994.
Vogelstein, B., Fearon, E. R., Kern, S. E., Hamilton, S. R., Preisinger, A. C., Nakamura, Y., White, R., Allelotype of colorectal carcinomas, in ‟Science", 1989, CCXLIV, pp. 207-211.
Willett, W., Diet, nutrition and avoidable cancer, in ‟Environmental health perspectives", 1995, CIII, suppl. 8, pp. 165-170.
Oncologia clinica di Georges Mathé e Paolo Pontiggia
SOMMARIO: 1. Premessa: a) il problema medico e sociale del cancro; b) aspetti biologici dei tumori. 2. L'oncologia clinica moderna: a) generalità; b) la diagnostica e l'inventario di un cancro o di una precancerosi; c) le risorse terapeutiche. 3. La strategia terapeutica: a) definizione e generalità; b) il caso della malattia percettibile; c) malattia impercettibile e cellule residue (o probabilmente residue) a un trattamento precedente; d) trattamenti preventivi. □ Bibliografia.
1. Premessa
a) Il problema medico e sociale del cancro.
L'oncologia clinica, identificabile nel complesso di strategie messe in opera dai medici sui portatori di tumori nel tentativo di guarirli o di alleviarne i sintomi, si presenta alle soglie del 2000 gravata da un duplice problema: da un lato, da un complesso di inferiorità in rapporto a certe specializzazioni della patologia, perché l'incontestabile progresso che ha registrato in cinquant'anni di sforzi intensivi e molto costosi (ha, tra l'altro, consentito di curare bambini e giovani adulti colpiti da tumore) appare - se valutato in assoluto come guadagno di quantità e anche di qualità della vita (v. fig. 1) - molto più modesto di quello raggiunto da altre discipline, quali per esempio l'ortopedia e la cardiologia; dall'altro lato, da un complesso di superiorità, poiché è la disciplina biomedica che ha acquisito le maggiori conoscenze biologiche e ha messo a punto tecniche e metodi nuovi, anche se in gran parte ancora inesplorati malgrado che le incoraggianti premesse avessero fatto assumere impegni che solo parzialmente sono stati mantenuti (v. Mathé, 1988; v. Andreiu, 1995)
Ma quest'ultimo aspetto delle nostre conoscenze, troppo sfruttato dai media e non abbastanza dai terapeuti, non è privo di effetti negativi sui responsabili sanitari, i quali non esitano a ironizzare sul fatto che i risultati vengono più spesso promessi che ottenuti e, giudicando soltanto sulla base della situazione odierna, ritengono ormai persa la ‛guerra' che il governo americano aveva dichiarato al cancro nel 1971 (v. Cohen e Diamond, 1986; v. Murr e Hager, 1995).
In effetti, quando si considera il bilancio di mezzo secolo di sforzi scientifici e finanziari (v. figg. 2 e 3), in prima approssimazione non si può che restare delusi: l'aumento della mortalità globale per cancro tende a elevarsi ulteriormente dopo il 1971, anno del Cancer act; non si registra alcun progresso terapeutico nei confronti del cancro del polmone, la più frequente delle neoplasie, e discutibili sembrano i progressi acquisiti nella terapia del cancro della mammella e di quello della prostata, rispettivamente secondo e terzo in termini d'incidenza; soprattutto la mortalità dovuta a tali tumori continua a elevarsi, o al massimo rimane stazionaria (v. American Cancer Society, 1989; v. Mathé, 1992).
Di fronte a questi dati, il medico oncologo deve prospettare ai responsabili ed esperti della politica sanitaria i termini di un importante problema biologico e medico-sociale: l'esistenza degli esseri viventi, nella scala sia individuale sia cellulare, si svolge secondo due ‛programmi', quello della vita e quello della morte; i tumori - la cui incidenza significativa inizia a 40 anni (v. fig. 4) e si eleva esponenzialmente dopo questa età - rappresentano manifestamente una delle espressioni del programma di morte degli organismi. Negli Stati Uniti, la constatazione che tra il 1950 e il 1980 la mortalità per cancro si è elevata (v. fig. 1), non ha comunque determinato l'inizio di una ricerca oncologica dinamica: il Cancer act presidenziale, peraltro, viene emanato solo nel 1971 (v. Magnan, 1990; v. Studzinski, 1995).
b) Aspetti biologici dei tumori.
L'aspetto biologico del problema pone quindi i tumori in rapporto di dipendenza con l'invecchiamento, che ne facilita l'insorgenza, non fosse altro che in conseguenza della fisiologica riduzione delle resistenze immunitarie (v. fig. 5) e dell'accumularsi di mutazioni fisiche, chimiche e biologiche (specialmente quelle indotte da virus) che subiscono gli organismi, di cui la senescenza è una delle espressioni (v. Bruley-Rosset e altri, 1980).
Tuttavia, le mutazioni che rappresentano fattori di rischio per lo sviluppo di tumori nell'uomo sono evitabili in misura elevata (pari all'80%), in quanto dipendono da errori comportamentali, volontari e coscienti (come nel caso del tabagismo o dell'alcolismo), o da negligenza, perfino da ignoranza dell'igiene elementare (in particolare alimentare e sessuale; v. fig. 6). Vale la pena di ricordare che anche l'igiene psicologica sembra proteggere indirettamente contro certi rischi di sviluppo di tumori.
I tumori la cui ereditarietà, legata a una mutazione monogenica, obbedisce ai comportamenti mendeliani classici sono rari; un esempio è quello del retinoblastoma, di cui è responsabile l'inattivazione di un gene, recentemente caratterizzato e localizzato, detto oncosoppressore o antioncogene (v. neoplasie: Oncologia sperimentale, vol. XI): la mutazione di tale gene priva le cellule del loro potere regolatore sulla proliferazione. La maggior parte dei tumori per i quali gli epidemiologi hanno messo in evidenza una tendenza ereditaria non obbedisce invece alle leggi della trasmissione secondo gli schemi mendeliani, in quanto il fattore genetico potrebbe non essere il solo fattore causale, ma cooperare con altri fattori cancerogeni, oppure il tumore potrebbe non risultare prodotto dalla lesione di un solo gene, ma essere di ordine poligenico (v. Knudson, 1971; v. Mathé, 1983).
Sono stati scoperti e catalogati, a partire dagli anni ottanta, più di 20 geni, detti protoncogeni, che possono essere attivati a oncogeni da meccanismi vari (fratture, traslocazioni, delezioni cromosomiche; intervento di geni regolatori; mutazioni puntiformi di alcune sequenze) e contribuire così alla trasformazione cellulare e alla cancerogenesi. Peraltro, si pensa che generalmente per trasformare una cellula normale in una neoplastica debbano cooperare due o più oncogeni, i cui interventi raramente sono simultanei: la trasformazione della cellula o la sua sopravvivenza sottostanno alle leggi della probabilità e questo spiega come il tempo necessario allo sviluppo di un cancro possa misurarsi in anni (v. Cooper, 1990; v. Marshall, 1991; v. Cowell, 1995).
I protoncogeni, comuni alle diverse specie animali, come abbiamo detto possono essere attivati da mutazioni geniche o da alterazioni cromosomiche indotte dalla maggior parte dei fattori cancerogeni - Virus, sostanze chimiche, agenti fisici - e soprattutto dalle radiazioni, sia ionizzanti che ultraviolette. Per esempio, il tabagismo può mutare l'antioncogene p53 (v. neoplasie: Oncologia sperimentale, vol. XI). Uno stesso oncogene può essere individuato in tumori indotti da virus differenti o da altri fattori cancerogeni e può quindi intervenire nella genesi di diversi tipi di tumore. Inoltre, la necessità che molti oncogeni vi cooperino spiega perché lo sviluppo di un tumore avvenga solitamente in diverse tappe (v. Sandberg e Turx Carel, 1987; v. Mitelman, 19882; v. Brennan e altri, 1995).
Molti oncogeni e protoncogeni codificano per la sintesi di proteine dette fattori di crescita o di differenziazione che, secrete dalla cellula trasformata, esercitano azioni a carattere decentralizzato e localizzato. Contrariamente agli ormoni, prodotti nelle cellule delle ghiandole endocrine e immessi nel sangue (meccanismo endocrino), i fattori di crescita possono essere prodotti da diversi tipi di cellule, anche se non da tutti, e sono destinati alle cellule loro vicine (meccanismo paracrino), e in alcuni casi perfino alle cellule stesse (meccanismo autocrino; v. fig. 7). Le concentrazioni che possono essere raggiunte da alcuni fattori di regolazione cellulare permettono di misurarli e di individuare così possibili anomalie di funzionamento: almeno con i mezzi di routine attualmente a disposizione, tale è il caso delle cicline, i cui geni possono esprimersi in eccesso, o dei fattori che controllano le cellule endoteliali (v. Aaronson, 1991; v. Motokura e altri, 1991; v. Brooks e altri, 1994).
Si comprende dunque come una neoplasia possa manifestarsi come tale solo dopo una lunga fase di displasia precancerosa benigna, la cui individuazione potrebbe permettere un trattamento preventivo del tumore maligno; quale che sia la durata del processo di cancerizzazione, un tumore rappresenta la discendenza di una sola cellula, e per tale motivo viene detto monoclonale (v. Ackerman e Rosai, 1971; v. Nowell, 1976). L'influenza di vari agenti, variabili a seconda del tipo di tessuto al quale appartiene la cellula progenitrice, stimola la divisione di quest'ultima e delle cellule sue discendenti, determinando in tal modo l'instabilità dell'intera popolazione cellulare con conseguenti alterazioni cromosomiche (v. Nicolson, 1987). La cellula della ghiandola mammaria, per esempio, può conservare certe strutture, i recettori estrogenici, proprie della sua stirpe e venire cronicamente stimolata da questi ormoni; ma se è troppo indifferenziata, e quindi priva di recettori specifici, esprime anche altri recettori che vengono stimolati dai fattori di crescita talvolta prodotti dalla stessa cellula (autocrinia). È una tale condizione d'autonomia massimale a dirigere la tendenza allo sviluppo del tumore: un cancro mammario costituito da cellule provviste di recettori estrogenici ha un andamento cronico, mentre è acuto quello di un cancro costituito da cellule prive dei recettori estrogenici, ma provviste di recettori per i fattori di crescita (v. Gerdes e altri, 1986).
La proliferazione clonale, tuttavia, trova nell'organismo non soltanto fattori che la favoriscono, ma anche reazioni contrarie - le reazioni immunitarie - le quali mettono in atto quello che viene considerato un meccanismo di ‛resistenza' e/o di ‛difesa' dell'organismo (v. fig. 8). Il ruolo di queste reazioni è stato per lungo tempo messo in dubbio per il fatto che non erano stati individuati antigeni specifici in tutti i tumori, sebbene antigeni particolari fossero stati evidenziati sulle cellule di alcuni tumori di origine virale. L'aumento considerevole, nei soggetti terapeuticamente immunodepressi per trapianto e in quelli colpiti da AIDS, della frequenza di linfomi indotti dal virus di Epstein-Barr, dei tumori del tratto vulva-vagina-collo dell'utero e dell'ano (negli omosessuali) - che sono legati ai Papillomavirus 16 e 18 - e infine del tumore di Kaposi, il cui agente eziologico potrebbe essere l'Herpesvirus 8, pone in evidenza l'importanza del controllo immunologico sullo sviluppo delle neoplasie. Le endorfine, infine, sembrano capaci di potenziare l'immunità, che influenza anche il sistema neurovegetativo essendone a sua volta influenzata (v. Des Jarlais e altri, 1984; v. Byers e Baldwin, 1987; v. Daling e altri, 1987; v. Niedobitek e Young, 1994).
I diversi fattori della cancerogenesi si potenziano tra di loro, non solo nei casi in cui vi sia un'interazione prevedibile - alcool e tabacco, nel caso di tumori delle vie respiratorie e in quello delle vie digestive superiori - ma anche in casi meno prevedibili (ad esempio, tabacco e Papillomavirus per il cancro del collo uterino; v. Mathé, 1987).
La possibilità che una cellula normale, tra i 60-70 mila miliardi di cellule che conta un organismo adulto, si trasformi in cellula tumorale capace di produrre una neoplasia, è bassa, poiché essa deve far parte di cellule che hanno tra le loro funzioni specifiche il dividersi ed essere al contempo ‛vittima' di molte lesioni genetiche. Certo, il numero di cellule che si dividono o possono dividersi normalmente è elevato e i fattori che favoriscono queste divisioni sono numerosi; inoltre, la durata della vita di un individuo dà alle cellule il tempo di subire molte mutazioni. In conclusione, il rischio di trasformazione nella popolazione delle cellule che devono dividersi è presente, ma non è molto elevato. Il rischio che una cellula mutata, e il clone da essa derivato, sopravviva è ugualmente molto limitato, visto che deve affrontare diverse difficoltà, tra le quali l'eventuale riconoscimento dei suoi antigeni da parte del sistema immunitario che può distruggere le cellule che ne sono portatrici.
2. L'oncologia clinica moderna
a) Generalità.
La nascita e lo sviluppo dei tumori sono condizionati dalla catena di fenomeni che portano alla trasformazione delle cellule capaci di dividersi e dalla progressione della divisione delle cellule trasformate e delle cellule loro discendenti; tali eventi subiscono inoltre l'influenza di condizioni genetiche, tessutali (particolarmente secondo il sesso e gli organi), comportamentali, geosociali e temporali (soprattutto riguardo all'età). L'oncologo clinico non può quindi pensare di esercitare il suo lavoro con la stessa tranquillità intellettuale di chi si occupa di una disciplina ‛stabile', ma deve adattarsi e adattare le sue cognizioni allo sviluppo di diversi parametri.
L'evoluzione delle conoscenze nel campo della cancerogenesi, della prognosi e della terapia dei tumori da un lato ha consentito l'individuazione dei rischi oncogeni e la messa in opera di misure preventive quando possibile, e dall'altro offre la possibilità di diagnosticare le neoplasie in uno stadio precedente la loro manifestazione clinica e, soprattutto, di instaurare trattamenti precoci.
Certi esperti e/o responsabili della sanità pubblica che discutono sul ‛rendimento' dell'individuazione di un tumore, dichiarano non redditizie la diagnosi precoce del cancro alla prostata e la mammografia eseguita prima dei 50 anni di età: in realtà, tale valutazione dovrebbe riguardare soltanto l'individuazione sistematica perseguita da alcune istituzioni e organizzazioni sanitarie, il cui rendimento, dopo un certo periodo che la ricerca è applicata a una popolazione, mostra fatalmente una certa riduzione (v. Melvin Howe, 1986; v. IARC, 1992; v. McConnell, 1995). È invece vero che nella diagnosi delle lesioni precoci e nell'individuazione dei rischi dovrebbero verificarsi due condizioni: 1) sarebbe opportuno che le istituzioni che si dedicano all'individuazione dei tumori in fase precoce - effettuata in base a considerazioni di tipo statistico (per esempio, sesso, età, tipo di lavoro) - integrassero nell'esercizio dei controlli il praticante generico, spesso inesperto, e specialmente il medico di base, mettendolo in grado di offrire le cure necessarie ai suoi pazienti di interesse oncologico; 2) la diagnostica precoce dovrebbe riguardare non solo tumori già manifesti, ma anche le displasie e le lesioni precancerose e, soprattutto, dovrebbe individuare i rischi (v. tabb. I e II).
In un prossimo futuro si imporrà anche l'individuazione dei rischi biologici, cioè di determinate infezioni virali e di particolari condizioni genetiche, somatiche o germinali, predittive dello sviluppo di un tumore (v. tab. III). Conviene tuttavia precisare che sul piano clinico, benché ai fini pratici e d'informazione per il medico di base sia possibile operare una distinzione tra la diagnosi dei tumori manifesti e quella di lesioni precancerose, in un caso o nell'altro la condotta terapeutica non dovrebbe variare di molto.
b) La diagnostica e l'inventario di un cancro o di una precancerosi.
A partire dal 1950, le possibilità diagnostiche dell'oncologia clinica si sono estremamente arricchite grazie ai progressi conseguiti dalle scienze di base: ingegneria, biologia, medicina, biochimica, fisica, genetica e, in particolare, immunologia e biologia molecolare.
Nonostante il considerevole avanzamento delle tecniche diagnostiche, in particolare della radiologia (la cosiddetta stadiazione dei tumori già manifesti o individuati in fase precoce), resta basilare e assolutamente indispensabile il corretto esame clinico del malato. Sono sempre più numerosi gli oncologi che tendono a sostituire il colloquio che deve segnare l'inizio del rapporto tra medico e paziente - l'intervista, cioè, in grado di fornire notizie attendibili su anamnesi e sintomatologia in atto - con un questionario al quale i soggetti sono invitati a rispondere con dei ‛sì' o dei ‛no'. Peraltro, i questionari sistematici, che permettono di evitare le omissioni e facilitano il confronto degli stati pre- e post-terapeutici, possono essere soprattutto preziosi per gli esami di orientamento o individuazione.
Infine, va da sé che l'oncologo clinico non dovrebbe trascurare le malattie non tumorali, dovrebbe cioè evitare di applicarsi soltanto a ricercare l'esistenza di un cancro senza fare il punto sulle altre principali affezioni e sugli altri rischi che minacciano il soggetto in esame, non dimenticando che l'età a rischio di cancro è anche, per esempio, a rischio di infarto del miocardio e che per di più quest'ultima malattia condivide con i tumori della mammella, della prostata e del colon gli stessi fattori di rischio di ordine nutrizionale (dieta ipercalorica, eccessivo consumo di grassi animali).
L'esame clinico rimane insostituibile; purtroppo molti medici credono, erroneamente, di poter tralasciare esami poco piacevoli, quali l'esplorazione rettale o l'esame completo della vagina e del collo dell'utero, particolarmente con lo speculum. Anzi, l'esame ginecologico deve essere completato (specialmente in considerazione dell'attuale condizione epidemiologica) con l'allestimento di strisci che consentano di studiare le cellule desquamate a diversi livelli, per la ricerca sia di anomalie ‛displasiche' o ‛neoplastiche' (cancerose) sia del Papillomavirus attraverso la PCR (Polymerase Chain Reaction o amplificazione genica; v. biotecnologie, vol. X; v. genetica, vol. X). L'oncologo ha poi la possibilità di disporre di varie metodiche di indagine, quali ad esempio: l'ecografia, che consente, tra gli altri vantaggi, di guidare le punture-biopsie, così preziose per completare l'esame clinico, e di individuare la maggior parte delle anomalie della tiroide, pur senza evidenziarne l'eventuale carattere secretivo; la tomodensitometria, che ha permesso di esplorare organi un tempo inaccessibili come il pancreas; la scintigrafia ai polifosfati marcati con tecnezio (99mTc), che rappresenta il miglior esame dell'intero scheletro per rivelare la più piccola localizzazione tumorale. La puntura-biopsia mirata in ecografia o in endoscopia e il successivo esame citologico della ‛carota' ritirata o del materiale ottenuto per puntura ad ago fine possono completare l'esame istologico e perfino, in alcuni casi, sostituirlo (v. Gompel, 1978). È così possibile, attraverso la dimostrazione delle anomalie cellulari fornita dallo studio citologico, determinare tipi e gradi di neoplasie non definibili dalla sola struttura tessutale, messa in evidenza dall'esame istologico classico. Ben noto è infatti il ruolo della citologia nell'individuazione delle lesioni displasiche e/o tumorali, non solamente delle vie genitali femminili, ma anche degli altri organi e tessuti, e quindi la sua importanza in termini di sopravvivenza dei soggetti esaminati.
La definizione del grado di malignità è rafforzata dalla misura della ploidia o ricchezza in DNA delle cellule, la quale può essere effettuata mediante la citometria a flusso. La proliferazione cellulare può essere valutata con la ricerca di determinati marcatori cellulari e di alcuni fattori di crescita, come la proteina KI67, assente nelle cellule che non si dividono. Questi test permettono di determinare le fasi del ciclo cellulare, dunque la loro lunghezza, e di conseguenza la velocità della crescita tumorale.
La ricerca dei recettori cellulari di ormoni è preziosa nel caso di tumori ormonodipendenti a sviluppo cronico, mentre la ricerca dei recettori cellulari per i fattori di crescita lo è per quelli a sviluppo acuto, solitamente svincolati dall'influenza ormonale. Ovviamente, gli esami clinici e paraclinici devono mirare anche a individuare lesioni pretumorali.
L'inventario topografico e volumetrico di una neoplasia, infine, non è solamente basato sull'ispezione, sulle immagini radiologiche, sonografiche, endoscopiche, ecc., ma deve essere completato dalla ricerca e dal dosaggio delle sostanze chimiche che chiamiamo marcatori sanguigni, i quali possono essere specifici di certi tumori, o aspecifici, cioè comuni alla maggior parte delle neoplasie. I marcatori costituiscono un test prezioso per la diagnosi di molti tumori solidi, sebbene l'aumento anormale di un marcatore non dipenda solo da un fattore volumetrico (cioè dalle dimensioni raggiunte dal tumore), ma anche dalla capacità delle cellule che lo costituiscono di rilasciare tali sostanze (v. tab. IV; v. Bates e Longo, 1987; v. Jacobs e Haskell, 1991).

c) Le risorse terapeutiche.
Dal punto di vista clinico, si possono distinguere due fasi nello sviluppo di un tumore: quella nella quale la neoplasia è ancora impercettibile - essendo costituita da un numero di cellule che non raggiunge valori tali da renderla visibile o palpabile (1 g di tessuto conta circa 1 miliardo di cellule) o dimostrabile all'esame radiologico - e quella nella quale è percettibile. È intuitivo come questa nozione di percettibilità sia essenziale per la possibilità non solo d'individuare più facilmente un tumore, ma anche per valutare gli effetti a breve termine delle terapie messe in atto, frequentemente espressi in termini di ‛risposta', di ‛remissione incompleta' oppure di ‛remissione completa': è bene infatti tener presente che aver ottenuto il primo tipo di remissione significa che, dopo il trattamento di un tumore del peso ad esempio di 10 g e quindi costituito da 1010 cellule, residuano meno del 50% degli elementi neoplastici, mentre avere ottenuto una remissione (apparentemente) completa può significare che il tumore residuo al trattamento pesa 1 g (109 cellule) e non è percettibile con gli strumenti oggi disponibili. Per mettere in evidenza masse neoplastiche residue di dimensioni microscopiche, sono state sviluppate metodiche di indagine basate sull'impiego di anticorpi diretti contro antigeni associati al tumore o di sostanze chimiche captate specialmente dai macrofagi infiltranti i tumori, marcati da isotopi radioattivi rilevabili con i contatori di radioattività (v. Le Pape e altri, 1987).
Si capisce pertanto che mentre i risultati del trattamento di un tumore impercettibile debbono essere valutati indirettamente sulla base della durata della remissione o addirittura della sopravvivenza, i risultati ottenuti su un tumore percettibile sono invece misurabili direttamente dal livello di regressione del suo volume o dalla scomparsa del carattere di percettibilità. La validità dei saggi terapeutici ai quali ricorre la cancerologia moderna per la valutazione di questi parametri merita di essere discussa: la durata delle remissioni o delle sopravvivenze dopo trattamento non può, in effetti, essere valutata che confrontandola con quelle che sarebbero state osservate in assenza di trattamento.
I mezzi terapeutici di cui l'oncologia dispone per il trattamento di una neoplasia percettibile sono principalmente di due tipi: quelli ad azione locale, che vengono applicati a una massa tumorale, e quelli ad azione generale, il più sovente sistemici, che mirano ad attaccare tutte le cellule neoplastiche disseminate nell'organismo. Tra i primi s'inscrivono la chirurgia, che tenta di eradicare i tumori, la radioterapia con radiazioni di tipo ionizzante, che blocca la divisione delle cellule nei tumori localizzati, e la somministrazione di farmaci per via arteriosa loco-regionale o per infiltrazione diretta nella massa in trattamento.
1. La chirurgia. - I canoni classici della chirurgia oncologica prescrivono l'ablazione degli organi o dei distretti sedi di tumori: ad esempio, la mastectomia nei casi di cancro della mammella, o una larga exeresi (a 5 cm dai limiti del tumore) nel caso di melanoma cutaneo, con asportazione dei linfonodi satelliti.
Alcuni test terapeutici comparativi hanno dimostrato che spesso, nel momento dell'intervento chirurgico, le cellule che daranno origine alle metastasi sono già partite dal tumore e arrivate ai siti di colonizzazione: quindi, un intervento chirurgicamente limitato può non essere inferiore, in termini di risultati, a interventi mutilanti, tanto più che, dopo un'operazione conservativa, eventuali cellule residue in un organo possono essere sterilizzate da radioterapia complementare (v. Veronesi e altri, 1981; v. Fischer e altri, 1988). Così è stato documentato che nel caso del cancro della mammella i risultati della tumorectomia completata da radioterapia o della quadrantectomia con dissezione della regione linfonodale ascellare, quando l'invasione è ancora limitata, sono sovrapponibili a quelli della mastectomia radicale. Un particolare approccio chirurgico, in questi casi, è rappresentato dalla biquadrantectomia, più favorevole alla ricostruzione (riparazione cosmetica) rispetto alla monoquadrantectomia (v. fig. 9; v. Garofalo e altri, 1992).
Nel trattamento dei sarcomi dei tessuti molli, i risultati della chirurgia detta conservativa, seguita da radioterapia, sono stati valutati statisticamente equivalenti a quelli della classica exeresi massimale (v. Lindberg e altri, 1981).
Poiché anche nei casi di melanoma al primo stadio l'ablazione sistematica dei linfonodi satelliti non si è rivelata in grado di influire favorevolmente sulla prognosi, ma si impone in caso d'invasione accertata o sospetta dei linfonodi stessi, questa eventualità deve essere attentamente ricercata con l'aiuto di una linfografia, una tomodensimetria scanner e/o un'ecografia.
Ancora non è possibile valutare con certezza i risultati della chirurgia laparoscopica, attualmente in sperimentazione; tuttavia, dopo un trattamento chirurgico di cui si sospetti l'incompletezza si pratica l'intervento detto second look (di controllo secondario), seguito da una chemioterapia per ridurre il rischio di un tumore residuo. Tale intervento laparoscopico è importante anche per valutare, ad esempio, i risultati del trattamento chemioterapico di un cancro all'ovaio, oppure può essere prezioso in un paziente in cui dopo un'operazione radicale di un determinato tumore persistano alti valori di più marcatori tumorali sanguigni (v. Piver e altri, 1978; v. Attiyeh e Stearns, 1981).
L'intervento per exeresi delle metastasi, specialmente del fegato e del polmone, è efficace soprattutto nei casi di metastasi unica comparsa dopo l'escissione del cancro primitivo, e talvolta può anche indurre la guarigione.
2. La radioterapia. - La radioterapia è una forma localizzata di trattamento dei tumori che, impiegata in passato in maniera estensiva, viene oggi utilizzata, in modo più corretto, solo in base a indicazioni precise. L'efficacia terapeutica di un trattamento radiante dipende dalla sensibilità specifica del tumore alle radiazioni ionizzanti, che deve essere necessariamente superiore a quella dei tessuti sani circostanti: classicamente considerati radiosensibili sono per esempio i linfomi. Quando la sensibilità specifica di un tessuto neoplastico è non molto superiore a quella dei tessuti circostanti normali, gli effetti nocivi su questi ultimi possono essere minimizzati da particolari conformazioni degli schemi di irradiazione che consentono di concentrare le radiazioni sul tumore; gli artifizi tecnici oggi disponibili consentono inoltre di trattare anche tumori scarsamente sensibili alle radiazioni. Un tumore, invece, la cui radiosensibilità è pari o inferiore a quella delle cellule normali, deve essere considerato radioresistente e perciò intrattabile.
Nella pianificazione di un trattamento, si deve inoltre tenere conto del fatto che i danni cellulari da radiazioni sono progressivi e spesso irreversibili: sono questi i motivi principali della relativa inefficacia della radioterapia in alcuni tumori molto diffusi, quali ad esempio neoplasie polmonari o tumori cerebrali primitivi o metastatici.
Nel trattamento dei tumori sensibili, la radioterapia ha il vantaggio, rispetto alla terapia chirurgica, di non essere mutilante e di indurre un'analoga percentuale di regressioni. Le indicazioni preferenziali per la radioterapia, sola o combinata con terapia chirurgica e/o con chemioterapia, sono numerose: il linfoma di Hodgkin al I e II stadio, alcuni linfomi sicuramente localizzati, i tumori delle corde vocali, il primo stadio del carcinoma della cervice uterina, alcuni tumori cutanei e le disseminazioni linfonodali del seminoma costituiscono alcuni esempi. L'uso della radioterapia è invece controindicato nel trattamento del carcinoma ovarico e praticamente inefficace in quello del tumore del polmone. Comunque, i risultati ottenuti dalla radioterapia dipendono in larga misura dai mezzi tecnici a disposizione delle varie istituzioni, oltre che naturalmente dalla preparazione specifica degli operatori (v. Deeley, 1976).
3. La chemioterapia. - La terapia chirurgica elimina mediante escissione le cellule tumorali; la radioterapia le danneggia attraverso lesioni solo parzialmente reversibili del loro materiale genetico che viene alterato tanto da non permetterne più la divisione; la chemioterapia, invece, agisce con meccanismi che variano a seconda del tipo di molecola utilizzata e del bersaglio molecolare, cercando di ‛avvelenare' sia il patrimonio cromosomico che altri organuli cellulari. La fig. 10 schematizza l'azione sui processi metabolici cellulari di alcuni dei chemioterapici di più largo impiego in clinica.
Nell'organismo, gli agenti cosiddetti citotossici, dopo la loro conversione molecolare (metabolismo) e durante la loro degradazione (catabolismo), esplicano la loro efficacia e la loro tossicità, colpendo siti attivi a livello sia delle cellule tumorali che delle cellule sane. Al pari di quello radioterapico, anche il trattamento chemioterapico si basa sul riscontro di una frequente maggiore sensibilità alle sostanze citotossiche delle cellule tumorali rispetto alle cellule normali.
La chemioterapia ha guarito, o contribuito a guarire, alcuni tipi di tumore in bambini e in adolescenti (osteosarcoma) e in giovani adulti (cancro del testicolo); ha inoltre indotto la guarigione di adulti colpiti da coriocarcinoma o da linfoma di Hodgkin (v. Clarysse e altri, 1976; v. Prosnitz e altri, 1988).
Si distinguono chemioterapie utilizzate come primo trattamento, detto induttore della regressione massimale, e chemioterapie praticate come trattamento adiuvante dopo intervento chirurgico. Vi è poi una chemioterapia praticata prima della chirurgia, detta neoadiuvante: specialmente in alcuni casi di cancri acuti della mammella, di solito a decorso rapidamente mortale, questa particolare sequenza terapeutica è in grado di indurre la guarigione (v. Rouëssé e altri, 1986). In realtà, poiché la distruzione cellulare provocata dai chemioterapici segue una cinetica di primo ordine, vale a dire il numero delle cellule distrutte corrisponde a una percentuale costante del numero complessivo delle cellule neoplastiche, il trattamento chemioterapico non è in grado di uccidere l'intera popolazione di tali cellule (v. fig. 11). Occorre poi tenere presente che una parte delle cellule tumorali può sviluppare una resistenza a determinati agenti antineoplastici. Questo fenomeno, che si è cercato di contrastare con l'impiego di combinazioni di farmaci, coinvolge tutte le famiglie degli agenti antitumorali, a eccezione di quelli che inibiscono le sintesi e di quelli che danneggiano gli acidi nucleici secondo un processo di sostituzione detto ‛alchilazione'. La ‛resistenza multipla comune' o MDR (Multidrug Resistance) sembra dipendere in larga misura dalla glicoproteina di membrana P-170, la cui sintesi è codificata da un particolare gene, denominato MDRI.
La corretta esecuzione della chemioterapia nel trattamento dei tumori, messa a punto in base alla regola della cinetica di primo ordine, consta generalmente di cicli ripetitivi, separati da intervalli destinati a lasciare ai precursori delle cellule sanguigne normali e alle loro cellule madri il tempo di moltiplicarsi per correggere l'eventuale deficit nel sangue periferico degli elementi normali differenziati: questa tattica è evidentemente efficace solo nel caso in cui le cellule tumorali siano più sensibili alla chemioterapia di quelle normali (v. Armitage e Antman, 19952). Che fare quando, dopo 6 cicli di efficacia apparente, alla fine si riscontra che il tumore, regredito solo in parte, riprende la sua crescita? Per far fronte alla resistenza, causa di questo insuccesso, o per prevenirla, sono attualmente in competizione due approcci: l'aumento delle dosi dei farmaci o le combinazioni a rotazione. Per quanto riguarda il primo, un grande entusiasmo suscitano attualmente le chemioterapie dette intensive, che noi stessi abbiamo introdotto nel 1966 in pazienti ai quali veniva poi reinfuso il midollo osseo prelevato prima dell'inizio della chemioterapia. Oggi, in luogo del midollo si utilizzano le cellule staminali isolate dal sangue periferico, che possono anche essere depurate dalle cellule tumorali che occasionalmente ne contaminino la preparazione. L'aggiunta dei fattori di crescita (CSF, Colony Stimulating Factor, fattore stimolante la formazione delle colonie derivate, per divisione, dalle cellule progenitrici) che stimolano le cellule progenitrici comuni dei granulociti e dei monociti (GMCSF), o quelle dei granulociti (GCSF), permette di evitare le complicazioni delle citopenie sanguigne di durata troppo lunga (v. McCarthy e Goldman, 1984; v. Aurer e altri, 1990; v. sangue: Trapianto del midollo, vol. XI). I risultati variano a seconda della sensibilità alla chemioterapia dei differenti tessuti neoplastici: sono sembrati notevoli per certi linfomi non Hodgkin, classificati ad ‛alto rischio', e per alcune forme severe di linfoma di Hodgkin e di leucemia mieloide e linfoide acuta. Nei casi di tumori solidi, i risultati sono troppo variabili per essere valutati positivamente; per esempio, nel trattamento del cancro della mammella meritano una seria valutazione i rischi della tossicità, a volte letale, rispetto all'eventuale beneficio; in quello dei tumori dell'ovaio e del polmone le risposte sembrano molto modeste.
Le cause di questi dati relativamente deludenti vanno essenzialmente ricercate nell'inesattezza della cosiddetta regola di Skipper, secondo la quale ogni ciclo di chemioterapia distrugge la stessa percentuale di cellule neoplastiche. Più vicine alla realtà sono invece le cosiddette regole di Norton (v. fig. 12) e di Stout, che prevedono, rispettivamente, una riduzione della frazione di cellule neoplastiche distrutte a ogni ciclo di chemioterapia e la necessità di un aumento logaritmico della dose del chemioterapico a ogni ciclo per mantenere costante la quota di elementi tumorali distrutti; purtroppo, i margini di sicurezza nella terapia con i citostatici sono molto ristretti per la facile comparsa di fenomeni di tossicità non controllabili.
Sembra invece senz'altro preferibile praticare, prima della chemioterapia intensiva, il trattamento con combinazioni di tre farmaci, somministrati a rotazione in dosi massimali, meglio tollerate perché i fenomeni tossici secondari sono spesso differenti a seconda dei farmaci impiegati: si possono così evitare sovrapposizioni di tossicità (v. Peters e altri, 1988; v. Extra e altri, 1989; v. Lazarus, 1993).
4. L'immunoterapia. - Negli anni sessanta abbiamo iniziato il trattamento di malati neoplastici con questa metodica, che può essere classificata schematicamente in tre forme: immunoterapia passiva, immunoterapia adottiva e immunoterapia attiva.
L'immunoterapia passiva consiste nella somministrazione al paziente portatore di tumore di anticorpi contro le cellule di quel tumore prodotti da un organismo della stessa specie, o di specie diversa. Il deludente risultato di un nostro pionieristico esperimento con questa metodica si spiega con il fatto che gli anticorpi, appartenenti alla fase umorale dell'immunità, non esercitano un ruolo notevole nelle resistenze e difese antitumorali (v. Dillman, 19912). La sostanziale inefficacia dell'immunoterapia passiva è stata poi confermata dalla più vasta esperienza maturata con l'impiego degli anticorpi monoclonali.
L'immunoterapia adottiva è basata sul trapianto allogenico, in un paziente portatore di tumore precedentemente immunodepresso con trattamento radiante e/o farmacologico, di midollo osseo i cui linfociti T (timo-dipendenti) o NK (Natural Killer) reagiscono contro le cellule tumorali dell'ospite (v. immunologia clinica e immunopatologia, vol. VIII). Per primi abbiamo osservato questa reazione immunoterapica che si manifesta nel corso delle leucemie sperimentali, da cui il nome di ‛reazione del trapianto contro la leucemia' (GvL, Graft versus Leukemia). Tuttavia, la reazione dei linfociti T trapiantati non si esercita esclusivamente nei confronti delle cellule tumorali, ma, fatalmente, anche contro le cellule normali, inducendo quello che noi abbiamo chiamato la malattia secondaria all'innesto di midollo osseo non compatibile, o ‛reazione del trapianto contro l'ospite' (GvH, Graft versus Host; v. sangue: Trapianto del midollo, vol. XI; v. Mathé e altri, Essai..., e Transfusions..., 1959).
Questo tipo di immunoterapia si è sviluppato in base alle seguenti osservazioni, da noi descritte nell'uomo: 1) nel 1959 abbiamo verificato la possibilità di innestare transitoriamente midollo osseo incompatibile in pazienti irradiati accidentalmente a dosi non sufficientemente elevate e regolarmente distribuite; 2) sempre nel 1959, abbiamo notato l'insorgenza di GvH nei primi soggetti leucemici nei quali avevamo tentato trapianti di midollo osseo incompatibile dopo irradiazioni totali a dosi troppo deboli per permettere un innesto definitivo; 3) infine, nel 1963, in un paziente in remissione, è stato possibile l'innesto definitivo, con la reazione contro l'ospite e contro la leucemia. Due anni più tardi, nel 1965, Thomas ha confermato la possibilità dell'attecchimento definitivo di midollo osseo incompatibile, e molte équipes hanno confermato la reazione del trapianto contro la leucemia (v. Thomas e altri, 1975). Noi stessi abbiamo poi osservato, dopo semplici trasfusioni di linfociti non compatibili in soggetti immunodepressi, reazioni contro l'ospite e contro la leucemia simili a quelle indotte da trapianto di midollo osseo.
S. A. Rosenberg ha proposto una forma di immunoterapia anch'essa detta ‛adottiva', peraltro impropriamente perché in realtà gli organismi non possono ‛adottare' le proprie cellule: si tratta, infatti, di una forma d'immunoterapia attiva cellulare basata sulla reiniezione, in soggetti portatori di tumore, di linfociti autologhi attivati in vitro (LAK, Lymphokine Activated Killers, e TIL, Tumor Infiltrating Lymphocytes; v. Rosenberg, 1984; v. Rosenberg e altri, 1986).
L'immunoterapia attiva, che abbiamo potuto sperimentare valutandone la capacità di ‛stimolazione' dell'immunità negli animali da laboratorio, ha confermato la sua efficacia clinica anche nell'uomo; essa è stata impiegata come trattamento antitumorale in fase di terapia (dopo la stabilizzazione del tumore) o in fase di prevenzione (anche di ricadute). Tale terapia può essere specifica o non specifica: la prima è basata sull'impiego di materiale vettore di antigeni associati al tumore, la seconda comporta la somministrazione dei cosiddetti modulatori di immunità.
Per i nostri primi lavori ed esperimenti, tra gli immunomodulatori esistenti sotto il nome di ‛coadiuvanti' noi abbiamo scelto il BCG (Bacillus Calmette-Guérin), la cui azione primaria si esplica essenzialmente sui macrofagi. Successive osservazioni hanno consentito di dimostrare che, impiegati nella forma locale di immunoterapia attiva non specifica, interferone α e BCG inducono lo stesso numero di regressioni, ma soltanto quest'ultimo può determinare vere guarigioni (v. Mathé, 1968; v. Kalble e altri, 1994).
Come abbiamo recentemente osservato, l'ipertermia applicata direttamente sui tumori o in una cavità (particolarmente il peritoneo), oltre a determinare una parziale regressione tumorale per la spiccata termosensibilità delle cellule neoplastiche, è anche in grado d'indurre un'intensa esocitosi lisosomiale, visibile al microscopio elettronico (v. fig. 13), fungendo così da modulatore di immunità (v. Pontiggia e Mathé, 1994).
L'interferone α, che agisce essenzialmente sui macrofagi e sulle cellule NK, induce regressioni anche quando viene somministrato per via sistemica nel trattamento della tricoleucemia, di alcuni linfomi di basso grado, dei melanomi e dei tumori renali (v. Fossa e altri, 1991; v. Kedar e Segal, 1995).
L'interleuchina 2 agisce sui linfociti T killer e NK, e, specialmente in associazione con l'interferone α, è impiegata nell'immunoterapia di melanomi e tumori renali. La percentuale dei risultati è stata espressa solo in termini di ‛risposta'; tuttavia, i tassi di risposta favorevole, molto variabili, sono per lo più ‛bassi': va però osservato che generalmente gli esperimenti sono stati condotti su tumori in avanzata fase di evoluzione e di volume considerevole.
In base alle nostre constatazioni sperimentali, pubblicate al momento dell'introduzione dell'immunoterapia attiva nel trattamento dei tumori, tale terapia, praticata in vivo per via sistemica, è in grado di guarire solo soggetti portatori di un piccolissimo numero di cellule tumorali (meno di 105 nel topo) e deve dunque essere considerata un trattamento indicato nella malattia tumorale impercettibile e nella prevenzione di ricadute. A questo punto s'impongono però due osservazioni: 1) poiché la maggior parte dei test negativi si riferisce in realtà a trattamenti combinati chemio-immunoterapici comprendenti anche l'impiego di potenti agenti immunosoppressori, va notato che l'efficacia clinica dell'immunoterapia è annullata dalla combinazione con tali agenti; 2) l'efficacia dell'immunoterapia si manifesta in soggetti con determinate caratteristiche genetiche, come dimostra la differenza della ripartizione dei fenotipi HLA1 tra i malati apparentemente guariti, o comunque con lunga sopravvivenza, dopo chemioterapia seguita da immunoterapia attiva, o dopo sola chemioterapia; Ochiai (v., 1985) ha fatto la stessa constatazione per quel che riguarda i fenotipi HLA2 in pazienti trattati per cancro dello stomaco.
5. La terapia ormonale e antiormonale. - Le terapie con sostanze di tipo ormonale (o antiormonale) sono basate sul presupposto del ruolo promotore, se non induttore, della cancerogenesi svolto da alcuni ormoni, particolarmente quelli sessuali, su determinati organi o tessuti, soprattutto quelli detti sessuali secondari. È questo il caso per esempio del tumore della prostata nell'uomo, le cui forme differenziate sono promosse sia dal testosterone sia dal diidrotestosterone: gli analoghi dell'LHRH (Luteinizing Hormone Releasing Hormone), che inibiscono la produzione testicolare di ormone, e gli antiandrogeni, che bloccano i recettori a livello cellulare, riducono la proliferazione tumorale anche per anni; tale effetto è documentato dalla diminuzione dei livelli dell'antigene specifico prostatico (PSA, Prostatic Specific Antigen) e di fosfatasi acida prostatica.
Anche nel trattamento del cancro della mammella, che esprime recettori per gli estrogeni, hanno dato prova certa di efficacia sia i bloccanti di tali recettori (particolarmente il tamoxifene), sia gli analoghi dell'LHRH. In stretta interazione con gli ormoni agiscono nella modulazione della crescita neoplastica anche i fattori di crescita (v. Heldin e Westermark, 1984; v. Harris e altri, 1989).
Finora hanno dimostrato attività terapeutiche di tipo antiormonale anche gli inibitori dell'aromatasi e, tra i fattori di crescita, gli analoghi della somatostatina, alcuni dei quali sembrano essere più attivi su determinati cicli cellulari, altri sulle cellule mammarie, altri ancora su quelle pancreatiche. In realtà, la terapia delle neoplasie con ormoni, antiormoni e fattori di crescita non è stata oggetto di studi particolarmente numerosi: così, ad esempio, per il trattamento del cancro alla prostata Schally consiglia la combinazione di un analogo della somatostatina e di un analogo dell'LHRH, mentre per quello del carcinoma mammario noi ci avvaliamo dell'uso combinato di tamoxifene, di un antiaromatasi e di un analogo dell'LHRH (v. Schally e altri, 1980; v. Nicholson e altri, 1994).
6. La terapia genica. - Le strette connessioni tra insorgenza (e sviluppo) del cancro e alterazioni genetiche sono ampiamente documentate. L'attuale disponibilità di tecniche sofisticate che consentono d'indurre modificazioni genetiche controllate ha fatto ipotizzare la possibilità di incidere sulla crescita dei tumori con manipolazioni genetiche.
I tentativi effettuati sono stati finora deludenti, anche se la speranza di poter in futuro ottenere risultati concreti è basata su ipotesi fondate. Le modalità di intervento ipotizzate e già in fase di sperimentazione riguardano da una parte la possibilità di provocare una modificazione della configurazione genetica di una cellula tumorale, oppure di indurre o di inibire la produzione di enzimi o proteine particolari (come ad esempio la glicoproteina P-170, probabilmente responsabile della resistenza multipla ai farmaci antiblastici); dall'altra la possibilità, ancora più interessante, di aumentare le potenzialità della risposta immunitaria antineoplastica di linfociti e macrofagi, così da rendere l'organismo capace di reagire alle neoplasie. La scarsità dei risultati finora ottenuti non significa che orizzonti oggi non prevedibili non possano in futuro aprirsi per questa nuova branca dell'oncologia sperimentale e (forse) clinica.
3. La strategia terapeutica
a) Definizione e generalità.
La strategia terapeutica si definisce come una programmazione d'interventi e manovre per l'attuazione dei trattamenti, razionalmente combinati in vista di ottenere la guarigione. Due nozioni ci sembrano basilari per un moderno concetto di strategia terapeutica: quella della malattia residua, che può essere, come di frequente avviene, la causa di una ricaduta dopo il primo trattamento; e quella dell'esistenza, nel caso di un tumore localizzato, di forme a sviluppo cronico e forme a sviluppo acuto. Per semplici che siano, queste nozioni sono recenti, e in ogni caso lo è la loro utilizzazione nella pratica clinica, tanto che non sono ancora applicate da tutti i medici, chirurghi e radioterapisti che operano nell'oncologia. Molte pazienti affette da cancro acuto della mammella subiscono ancora, come primo trattamento, una exeresi chirurgica, per il solo fatto di essere state indirizzate a un chirurgo, quando le loro cellule tumorali sono già, a causa dello sviluppo acuto della malattia, disseminate in tutto l'organismo e il solo trattamento che offra speranza di guarigione è la combinazione sequenziale dapprima della chemioterapia, in seconda istanza della chirurgia (coadiuvante), ed eventualmente della terapia supplementare con antagonisti di ormoni o di fattori di crescita quali gli analoghi della somatostatina.
Vi sono casi di pazienti affetti da linfoma diagnosticato a livello di un distretto linfonodale superficiale, che hanno subito il trattamento radiante come prima se non unica terapia solo perché erano stati inviati a un radioterapista; in tali pazienti l'attento inventario sistematico che consente di mettere in evidenza la presenza di lesioni profonde avrebbe indicato come primo trattamento la chemioterapia.
Da queste brevi considerazioni risulta evidente la necessità di organizzare l'oncologia clinica come specialità multidisciplinare, così che le decisioni possano essere prese da un comitato di medici comprendente l'internista oncologo, il chirurgo e il radioterapista.
b) Il caso della malattia percettibile.
Un tumore localizzato, del quale, alla stadiazione, non risultino evidenti disseminazioni, particolarmente ai linfonodi satelliti (stadio I), il cui sviluppo ha tutte le probabilità di essere cronico (basso grado), rientra nell'ambito della sola chirurgia. Un'incertezza riguardante o uno solo di questi due parametri clinici, o la validità dell'intervento chirurgico (exeresi limitata al tumore ma insufficiente ad arginare la dispersione di cellule maligne, possibili fughe cellulari provocate da incaute manovre chirurgiche), pone l'indicazione per una radioterapia complementare. Questo può verificarsi, ad esempio, nel caso di una tumorectomia o di una quadrantectomia eseguita per ragioni estetiche su una malata con un cancro della mammella di piccole dimensioni.
Conviene qui considerare uno dei difetti della radioterapia, senza dubbio dipendente dal carattere continuo della sua applicazione: infatti, praticata secondo il metodo classico, anche localmente, su una parte sia pure relativamente limitata del corpo e non comprendente il timo né un volume linfonodale notevole, essa induce una linfopenia notevole e irreversibile; la radioterapia non induce linfopenia, invece, se praticata in maniera intermittente e in combinazione con una chemioterapia discontinua, secondo uno schema in cui ogni ciclo è costituito da un trattamento chemioterapico che precede un trattamento radioterapico, e mantenendo i cicli successivi separati da intervalli abbastanza lunghi per lasciare all'organismo il tempo necessario al rinnovamento delle cellule del sangue periferico. La linfopenia, che complica il trattamento radioterapico continuo, è in particolare temibile nei pazienti portatori di tumori che già spontaneamente si accompagnano a immunodeficienza, quali il sarcoma di Kaposi, frequente complicanza dell'AIDS, e nei soggetti sottoposti a trapianto e affetti da neoplasia.
Nel caso di alcuni tumori particolarmente radiosensibili, come ad esempio il basalioma cutaneo, è sufficiente anche il solo trattamento radioterapico, il quale risulta altrettanto efficace, in termini di guarigione, che quello chirurgico.
Vi sono, infine, casi in cui la radioterapia può essere praticata sistematicamente in prima istanza per rendere ottimale l'effetto della chirurgia: tale possibilità, si ricorda, deve essere esclusa per il trattamento di cancri acuti, come quelli della mammella, nei quali l'immunodepressione provocata dall'irradiazione favorirebbe la disseminazione di cellule neoplastiche. La radioterapia come prima scelta, seguita poi da chemioterapia, si pratica nel trattamento dei linfomi localizzati, mentre la sequenza si inverte nel caso dei linfomi disseminati: i tassi di guarigione sono apprezzabili per i tipi di basso e medio grado, mentre sono molto scarsi per le forme di alto grado, nelle quali solo le radio-chemioterapie intensive seguite da un trapianto autologo di cellule emopoietiche staminali sono in grado di indurre guarigioni.
È statisticamente dimostrato che la radioterapia non solo non migliora, ma in molti casi addirittura compromette il risultato di un trattamento chirurgico, o chirurgico e chemioterapico, di un cancro all'ovaio; tale effetto può forse essere posto in relazione all'immunodepressione determinata dalle radiazioni. Queste restrizioni di indicazioni della radioterapia, imposte dalla conseguente immunodepressione (che è tuttavia possibile correggere con BCG, stimolatore dell'ematopoiesi, o con i fattori di crescita - quali il GMCSF e il GCSF, per quanto riguarda monociti e granulociti - e con l'impiego dell'antienzima bestatina, che è in grado di proteggere le piastrine), spiegano il ruolo assunto come primo trattamento dalla chemioterapia, la cui azione è completata dalla chirurgia nel caso di tumori acuti, particolarmente della mammella.
c) Malattia impercettibile e cellule residue (o probabilmente residue) a un trattamento precedente.
In situazioni particolari sono proponibili trattamenti coadiuvanti diversi: dalla chirurgia detta di second look, complementare alla chemioterapia di un tumore ovarico inizialmente inoperabile, alla chemioterapia di consolidamento dopo chemioterapia primaria - eventualmente completata da immunoterapia nei casi di neoplasia disseminata, come in alcune leucemie - alla chemioterapia e/o immunoterapia e/o ormonoterapia, o impiego degli antagonisti dei fattori di crescita, nel caso di tumori solidi come quelli della mammella o del colon.
1. Chirurgia e radioterapia quali trattamenti coadiuvanti locali. - Seguendo l'ordine degli esempi citati, nel caso di tumori come il cancro dell'ovaio non operabile radicalmente perché troppo esteso, se la chemioterapia determina la regressione completa un second look chirurgico consente l'asportazione di qualche linfonodo ancora interessato da metastasi: è appunto questo che si definisce intervento coadiuvante. Questa strategia terapeutica, che fa del cancro dell'ovaio il secondo tumore dell'adulto (dopo quello del testicolo) curabile con mezzi medici, ha consentito notevoli risultati. Le pazienti inseribili in questo schema di trattamento devono essere seguite per una dozzina di anni, particolarmente attraverso il monitoraggio del marcatore specifico CA125, di cui sia stato riscontrato un tasso elevato nel periodo iniziale.
Al contrario, le indicazioni della radioterapia coadiuvante sono più sfumate in ragione del suo effetto immunodepressivo: così, se la radioterapia ha un ruolo indiscutibile nel completare il trattamento chirurgico primario deliberatamente incompleto (per ragioni estetiche) di un cancro della mammella, come la tumorectomia o la quadrantectomia, essa è invece controindicata quale terapia coadiuvante locale di un trattamento localmente completo di un tumore, perché in questo caso il rischio di ricadute non può presentarsi che a distanza di anni.
2. Immunoterapia adottiva a completamento di radio-chemioterapia intensiva, o radio-chemioterapia intensiva seguita dal ripristino dell'ematopoiesi autologa. - Le leucemie acute rappresentano un modello di malattie tumorali disseminate fin dall'inizio e a rapido sviluppo: verso il 1960, dopo l'induzione delle prime remissioni apparentemente complete e durature, si constatò rapidamente che tutte erano seguite da ricadute, e si comprese che il trattamento della malattia residua, rappresentata dalla sopravvivenza di cellule resistenti, avrebbe dovuto essere effettuato secondo schemi differenti da quelli utilizzati per indurre la remissione. Furono allora proposti, e applicati in tempi successivi, differenti metodi: 1) l'immunoterapia adottiva, già descritta (v. cap. 2, § c), che comportava il pericolo della reazione del trapianto contro l'ospite (GvH); 2) l'irradiazione totale del corpo e la chemioterapia massimale seguita da trapianto di midollo osseo compatibile (dunque senza GvL), oppure chemioterapie d'intensità massimale seguite dalla restaurazione della funzionalità emopoietica grazie alla reinfusione di midollo osseo autologo. I risultati, come dimostrano oggi le numerose statistiche disponibili, sono ancora più interessanti di quanto le basi sperimentali lasciassero prevedere. Nel trattamento dei tumori solidi, la chemioterapia intensiva seguita da innesto di midollo osseo autologo consente di conseguire risultati di un certo interesse, malgrado manchi ancora una valutazione attenta del rapporto costo-beneficio e, soprattutto, dei risultati a lungo termine.
3. Immunoterapie attive in vivo, chemioterapie a dosi convenzionali e ormonoterapie quali trattamenti coadiuvanti sistemici. - Attualmente, onde evitare il pericolo di una spesso letale GvH associata alla GvL, si tenta di ottenere la distruzione ‛dell'ultima cellula tumorale' con un'altra forma d'immunoterapia, la forma attiva.
L'immunoterapia attiva diretta, costituita da un'attivazione in vivo dei linfociti (essenzialmente T) e dei macrofagi, ha avuto inizio nel 1966 come trattamento supplementare della chemioterapia di consolidamento dopo chemioterapia primaria inducente la remissione. Essa prevede essenzialmente l'impiego di BCG come immunomodulatore e la somministrazione di cellule tumorali sterilizzate mediante irradiazione in vitro, utilizzate come ipotetico vaccino specifico.
Le statistiche più recenti mostrano un aumento dei livelli di sopravvivenza dei bambini trattati per leucemie acute linfoidi, probabilmente in funzione del perfezionamento degli schemi di chemioterapia d'attacco; tuttavia, se si confrontano i risultati della chemioterapia seguita dall'immunoterapia con quelli della sola chemioterapia, si constata che la percentuale dei soggetti viventi in remissione è generalmente più elevata tra i pazienti sottoposti al primo dei due metodi. I risultati dei National Institutes of Health negli Stati Uniti confermano l'inferiorità della sola chemioterapia in confronto alla sequenza chemioterapia-immunoterapia attiva.
Per quanto riguarda la modalità di esecuzione della combinazione della chemioterapia e dell'immunoterapia, se debba essere sequenziale, simultanea o alternata, esistono dati apparentemente non univoci: alcuni risultati sperimentali hanno mostrato che il trattamento con chemioterapia alternata all'immunoterapia può avere esito sfavorevole se gli agenti chemioterapici impiegati sono anche immunosoppressori, come si è verificato nel caso del melanoma; per contro, Ochiai (v., 1985) aveva ottenuto, con una combinazione simultanea di chemioterapia e di immunoterapia coadiuvante, un notevole beneficio in alcuni casi di cancro gastrico. Nei pazienti operati per cancro del colon-retto, mentre il chemioterapico fluorouracile, somministrato solo come terapia adiuvante, può addirittura indurre una significativa riduzione della sopravvivenza a 5 anni dall'intervento e l'immunomodulatore levamisolo, usato da solo, determina soltanto un modesto beneficio, la somministrazione combinata di levamisolo e fluorouracile è in grado di indurre un beneficio considerevole e altamente significativo.
I casi considerati provano dunque che l'immunoterapia attiva è efficace, in molti casi più efficace della chemioterapia, e che ancor più efficace può essere la loro combinazione, purché si abbia cura di impiegare citostatici non particolarmente immunodeprimenti.
Nel trattamento del cancro della mammella si è dimostrata utile la combinazione di immunoterapia, chemioterapia e ormonoterapia: l'esperienza clinica mostra infatti che l'immunoterapia agisce sia prima che dopo la menopausa, ma solamente nei casi in cui i linfonodi colpiti sono poco numerosi, vale a dire che è efficace solo sulle malattie tumorali con poche cellule residue; la chemioterapia è efficace qualunque sia il numero dei linfonodi invasi, ma solamente prima della menopausa; l'ormonoterapia con antiestrogeni (ad esempio, il tamoxifene) agisce soprattutto dopo la menopausa. Il trattamento coordinato e stratificato con immunoterapici, antiormoni e citostatici sembra essere più attivo di ciascuno dei singoli provvedimenti.
Altri modulatori dell'immunità sono in corso di valutazione clinica preliminare: la bestatina, ad esempio, è attiva nei casi di leucemia acuta mieloblastica; sia la bestatina che la tuftsina, applicate sistematicamente dopo l'età di 16 mesi nel topo (età corrispondente ai 40 anni dell'uomo), ripristinano le funzioni immunitarie e riducono a un tasso molto basso la frequenza dei tumori spontanei in confronto ai controlli non trattati (v. Bruley-Rosset e altri, 1979).
d) Trattamenti preventivi.
Dal lato clinico-sperimentale, si deve introdurre la nozione di trattamenti preventivi di condizioni a rischio di tumore, legate a immunodepressione dovuta all'età e a particolari condizioni genetiche, per le quali potrà essere ragionevolmente indicato un tentativo di impiego di bestatina o tuftsina. Analogamente, un trattamento preventivo è praticabile nelle displasie, per esempio nei casi di condilomi del collo uterino associati a Papillomavirus, nei quali abbiamo osservato l'effetto positivo degli analoghi dell'LHRH.
Va da sé che devono essere considerati come essenziali i trattamenti antivirali nel caso di infezioni di virus epatitici B e C, che provocano il cancro al fegato in percentuale relativamente elevata. Il fatto che il trattamento con interferone e AZT di linfomi associati al virus leucemogeno HTLV1 abbia fatto regredire lesioni tumorali, indica che la terapia antivirale può assumere valore preventivo nei portatori di Virus che ancora non presentino tumori (v. neoplasie: Oncologia sperimentale, vol. XI).
Le indicazioni terapeutiche sono più delicate nel caso della prevenzione dei tumori della mammella e nel trattamento delle displasie mammarie: gli antiestrogeni sono indicati, e possono dare risultati notevoli, nel caso che le cellule di una displasia siano portatrici di recettori per gli estrogeni, mentre per le cellule che presentano recettori per i fattori di crescita vi è un'indicazione al trattamento con gli analoghi della somatostatina.
Noi abbiamo sottoposto a trattamento con un differenziatore, come l'acido cis-retinoico, un gruppo di fumatori che presentavano metaplasie e displasie bronchiali di grado elevato, diagnosticate mediante biopsia eseguita in broncoscopia: queste lesioni precancerose sono regredite e non sono riapparse, ma solo nei soggetti che hanno smesso definitivamente di fumare.
È evidente che prima di considerare la prevenzione dei tumori attraverso le cosiddette manipolazioni genetiche, dagli ipotetici effetti benefici ma dall'innocuità incerta, sarà più ragionevole applicare le misure accessibili e prive di rischio qui sopra elencate, derivate dall'esperienza dell'oncologia clinica, seguendo gli stessi criteri usati per il trattamento dei tumori documentati.
BIBLIOGRAFIA
Aaronson, S. A., Growth factors and cancers, in ‟Science", 1991, CCLIV, pp. 1146-1153.
Ackerman, L. V., Rosai, J., The pathology of tumors. 1. Introduction, precancerous lesions, benign lesions that resemble cancers, in ‟Ca. A cancer journal for clinicians", 1971, XXI, 3, pp. 163-173.
American Cancer Society, Cancer: facts and figures. 1989, New York 1989.
Andreiu, J. M. (a cura di), Biologie des cancers, Paris 1995.
Armitage, J. O., Antman, K. H. (a cura di), High dose cancer therapy: pharmacology, hematopoietins, stem cells, Baltimore, Md., 19952.
Attiyeh, F. F., Stearns, M. W. Jr., Second-look laparotomy based on CEA elevations in colorectal cancer, in ‟Cancer", 1981, XLVII, pp. 2119-2125.
Aurer, I., Ribas, A., Gale, R. P., What is the role of recombinant colony stimulating factors in bone marrow transplantation?, in ‟Bone marrow transplantation" 1990, VI, pp. 79-87.
Bates, S. E., Longo, D. L., Use of serum tumor markers in cancer diagnostic and management, in ‟Seminars in oncology", 1987, XIV, pp. 102-138.
Brennan, J. A. e altri, Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck, in ‟New England journal of medicine", 1995, CCCXXXII, pp. 712-717.
Brooks, P. C., Clark, R. A. F., Cheresh, D. A., Requirement of vascular integrin αvb3 for angiogenesis, in ‟Science", 1994, CCLXIV, pp. 569-571.
Bruley-Rosset, M., Florentin, I., Mathé, G. e altri, Restoration of impaired immune functions of aged animals by chronic bestatin treatment, in ‟Immunology", 1979, XXXVIII, pp. 75-83.
Bruley-Rosset, M., Florentin, I., Mathé, G. e altri, Age related changes of the immune response and immunorestoration by stimulating agents, in The immune system: functions and therapy of dysfunction (a cura di G. Doria e A. Eshkol), New York 1980, pp. 171-187.
Byers, V. S., Baldwin, R. W. (a cura di), Immunology of malignant diseases, Lancaster 1987.
Clarysse, A., Kenis, Y., Mathé, G., Cancer chemotherapy: its role in the treatment strategy of hematologic malignancies and solid tumors, Berlin-New York 1976.
Cohen, M. M., Diamond, J. M., Are we losing the war against cancer?, in ‟Nature", 1986, CCCXXIII, pp. 488-489.
Cooper, G. M., Oncogenes, Boston 1990.
Cowell, J. K. (a cura di), Molecular genetics, Oxford 1995.
Daling, J. R., Weiss, N. S., Hislop, T. G. e collaboratori, Sexual practices, sexually transmitted diseases and the incidence of anal cancer, in ‟New England journal of medicine", 1987, CCCXVII, pp. 973-977.
Deeley, T. J., Principles of radiation therapy, London 1976.
Des Jarlais, D. C., Marmor, M., Thomas, P. e collaboratori, Kaposi's sarcoma among four different AIDS risk groups (Letter to the Editor), ‟New England journal of medicine", 1984, CCCX, p. 1119.
Dillman, R. O., Antibody therapy, in Principles of cancer biotherapy (a cura di R. K. Oldham), New York 19912, pp. 396-432.
Extra, J. M., Dieras, V., Espié, M. e altri, Intensification thérapeutique avec autogreffe médullaire au cours des adénocarcinomes ovariens et étude de phase I-II- de 21 sujets, in ‟Cahiers Cancer", 1989, I, pp. 81-84.
Fischer, B., Redmond, C., Posson, R. e altri, Eight-year results of randomized trial comparating total mastectomy and lumpectomy with or without irradiation in the treatment of breast cancer, in ‟New England journal of medicine", 1988, CCCXX, pp. 825-828.
Fossa, S. D., Lien, H. H., Lindegaard, M., Effect of recombinant interferon alpha on bone metastases of renal-cell carcinoma, in ‟New England journal of medicine", 1991, CCCXXIV, pp. 633-634.
Garofalo, R., Borioni, R., Garofalo, R. L., Lavanga, G., Mathé, G., Radical tumor excision and cosmetic balance in the surgical treatment of breast carcinoma: biquadrantectomy, in ‟Biomedicine and pharmacotherapy", 1992, XLVI, pp. 401-404.
Gerdes, J., Lelle, R. J., Pickartz, H., Heidenreich, W., Schwarting, R., Kurtsiefer, L., Stauch, G., Stein, H., Growth fractions in breast cancers determined in situ with monoclonal antibody ki-67, in ‟Journal of clinical pathology", 1986, XXXIX, pp. 977-980.
Gompel, C., Atlas of diagnostic cytology, New York 1978.
Harris, A. L., Nicholson, S., Sainsbury, J. R. e altri, Epidermal growth factor receptors in breast cancer: association with early relapse and death, poor response to hormones and interaction with neu, in ‟Journal of steroid biochemistry", 1989, XXXIV, pp. 123-131.
Heldin, C. H., Westermark, B., Growth factors. Mechanism of action and relation to oncogens, in ‟Cell", 1984, XXXVII, pp. 9-20.
IARC (International Agency for Research on Cancer), Cancer incidence in five continents, vol. IV (a cura di J. Waterhouse, K. Shanmuguratham e C. Muir), Lyon 1992.
Jacobs, E. L., Haskell, C. M., Clinical use of tumors markers in oncology, in ‟Current problems in cancer", 1991, XV, pp. 299-360.
Kalble, T., Beer, M., Mendoza, E., Ikinger, U., Link, M., Reichert, H. E., Frangenheim, T., Klein, E., Fabricius, P. G., BCG vs interferon alpha for prevention of recurrence of superficial bladder cancer. A prospective randomized study, in ‟Der Urologe. Ausgabe A", 1994, XXXIII, pp. 133-137.
Kedar, I., Segal, A., Interferon in multiple myeloma: does it pay?, in ‟Israel journal of medical sciences", 1995, XXXI, pp. 635-637.
Knudson, A. G. Jr., Mutation and cancer: statistical study of retinoblastoma, in ‟Proceedings of the National Academy of Sciences", 1971, LXVIII, pp. 820-823.
Lazarus, H. M., Autologous bone marrow transplantation for the treatment of lung cancer, in ‟Seminars in oncology", 1993, XX, pp. 72-79.
Le Pape, A., Barot-Ciorbaru, R., Musset, M., Baulieu, J. L., Jubalt, C., Lemarié, E., Pourcelot, L., Besnard, J. C., Nicolau, C., Mathé, G., An original method for submacroscopic metastases visualization in cancer residual minimal disease, in ‟Biomedicine and pharmacotherapy", 1987, XLI, pp. 392-398.
Lindberg, R. D., Martin, R. G., Romsdahl, M. M. e altri, Conservative surgery and postoperative radiotherapy in 300 adults with soft-tissue sarcomas, in ‟Cancer", 1981, XLVII, pp. 2391-2397.
McCarthy, D. M., Goldman, J. M., Transfusion of circulating stem cells, in ‟Critical reviews in clinical laboratory sciences", 1984, I, pp. 20-24.
McConnell, J., Screening tests: are they worth doing?, in ‟The lancet", 1995, CCCXLV, p. 1629.
Magnan, C., Et Newton croqua la pomme, Paris 1990.
Marshall, C. J., Tumor suppressor genes, in ‟Cell", 1991, LXIV, pp. 313-326.
Mathé, G., L'immunothérapie active de la leucémie L1210 appliquée après la greffe tumorale, in ‟Revue française d'études cliniques et biologiques", 1968, XIII, pp. 454-459.
Mathé, G., Génétique comparée des cancers héréditaires et des cancers non héréditaires, in ‟Biomedicine and pharmacotherapy", 1983, XXXVII, pp. 309-312.
Mathé, G., Alcohol, a drug as a remedy and/or as a dope? An individual and/or a social problem, in ‟Biomedicine and pharmacotherapy", 1987, XLI, pp. 61-62.
Mathé, G., Cancer ‟cures" in children, adolescents and young adults vs general cancer cure failure after 50 years of age, in ‟Biomedicine and pharmacotherapy", 1988, XLII, pp. 227-228.
Mathé, G., Is the ‟war against cancer" lost?, in ‟Biomedicine and pharmacotherapy", 1992, XLVI, pp. 433-438.
Mathé, G., Bernard, J., Schwarzenberg, L., Larrieu, M. J., Lalanne, C., Dutreix, A., Denoix, P., Schwarzmann, V., Ceoara, B., Essai de traitement de sujets atteints de leucémie aiguë par irradiation suivie de transfusion de moelle osseuse homologue, in ‟Revue française d'études cliniques et biologiques", 1959, IV, pp. 675-704.
Mathé, G. e altri, Transfusions et greffes de moelle osseuse homologue chez des humains irradiés à haute dose accidentellement, in ‟Revue française d'études cliniques et biologiques", 1959, IV, pp. 226-238.
Melvin Howe, G., Global geocancerology, Edinburgh 1986.
Mitelman, F., Catalog of chromosome aberrations in cancer, New York 19882.
Motokura, T. e altri, A novel cyclin encoded by a bcl1-linked candidate oncogene, in ‟Nature", 1991, CCCL, pp. 512-515.
Murr, A., Hager, M., Promises, promises, in ‟Newsweek", 16 ottobre 1995, pp. 42-44.
Nicholson, R. I., McClelland, R. A., Gee, J. M., Manning, D. L., Cannon, P., Robertson, J. F., Ellis, I. O., Blamey, R. W., Epidermal growth factor receptor expression in breast cancer; association with response to endocrine therapy, in ‟Breast cancer research treatment", 1994, XXIX, pp. 117-125.
Nicolson, G. L., Tumor cell instability, diversification and progression to the metastatic phenotype: from oncogene to oncofetal expression, in ‟Cancer research", 1987, XLVII, pp. 1473-1487.
Niedobitek, G., Young, L. S., Epstein-Barr virus persistence and virus-associated tumours, in ‟The lancet", 1994, CCCXLIII, pp. 333-335.
Nowell, P. C., The clonal evolution of tumor cell populations, in ‟Science", 1976, CXCIV, pp. 23-28.
Ochiai, T., Gastric tumors: immunotherapy, in Therapeutical trials in oncology: methods, ethics, results (a cura di G. Mathé e altri), Geneva 1985, pp. 249-254.
Peters, W. P., Shpall, E. J., Jones, R. B. e altri, High-dose combination alkylating agents with bone marrow support as initial treatment for metastatic breast cancer, in ‟Journal of clinical oncology", 1988, I, pp. 1368-1376.
Piver, M. S., Barlox, J. J., Lele, S. B., Incidence of subclinical metastasis in stage I and II ovarian carcinoma, in ‟Obstetrics and gynecology", 1978, LII, pp. 100-104.
Pontiggia, P., Mathé, G., A new mode of cancer cell death induced by hyperthermia and non specific (macrophagic) cancer immunotherapy: lysosomal exocytosis, in ‟Biomedicine and pharmacotherapy", 1994, XLVIII, p. 331.
Prosnitz, L. R., Farber, L. R., Kapp, D. S., Combined modality therapy for advanced Hodgkin's disease: 15 year follow-up data, in ‟Journal of clinical oncology", 1988, VI, pp. 603-612.
Rosenberg, S. A., Adoptive immunotherapy of cancer: accomplishments and prospects, in ‟Cancer treatment reports", 1984, LXVIII, pp. 233-255.
Rosenberg, S. A., Aebersold, P., Cornetta, K., Kasid, A., Morgan, R. A., Moen, R., Karson, E. M. e altri, Gene transfer into humans. Immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction, in ‟The New England journal of medicine", 1990, CCCXXIII, pp. 570-578.
Rosenberg, S. A., Spiess, P., Lafrenière, R., A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes, in ‟Science", 1986, CCXXXIII, pp. 1318-1321.
Rouëssé, J., Friedman, S., Sarrazin, D. e altri, Primary chemotherapy in the treatment of inflammatory breast carcinoma. A study of 230 cases, in ‟Journal of clinical oncology", 1986, IV, pp. 1765-1771.
Sandberg, A. A., Turx Carel, C., The cytogenetics of solid tumors. Relations with diagnosis classification and pathology, in ‟Cancer", 1987, LIX, pp. 387-389.
Schally, A. V., Coy, D. H., Arimura, A., LH-RH agonists and antagonists, in ‟International journal of gynaecology and obstetrics", 1980, XVIII, pp. 318-324.
Srkalovic, G., Cai, R. Z., Schally, A. V., Evaluation of receptors for somatostatin in various tumors using different analogs, in ‟Journal of clinical endocrinology and metabolism", 1990, LXX, pp. 661-667.
Studzinski, G. P., Cell growth and apoptosis, Oxford 1995.
Thomas, E. D., Storb, R., Clift, R. A. e altri, Bone marrow transplantation, in ‟New England journal of medicine", 1975, CCXCII, pp. 832-843.
Veronesi, U., Saccozzi, R., Del Vecchio, M. e altri, Comparing radical mastectomy with quadrantectomy, axillary dissection and radiotherapy in patients with small cancers of the breast, in ‟New England journal of medicine", 1981, CCCV, pp. 6-11.
Oncologia sperimentale di Giancarlo Vecchio
SOMMARIO: 1. Basi biologiche dell'oncologia: a) le colture cellulari; b) la trasformazione neoplastica. 2. Cancerogenesi chimica: a) premessa; b) metabolismo dei cancerogeni chimici e loro interazione con il DNA; c) la cancerogenesi come processo multifasico; d) meccanismo d'azione degli agenti cocancerogeni. 3. Cancerogenesi virale: a) premessa; b) virus oncogeni a DNA; c) virus oncogeni a RNA (Retrovirus). □ 4. Oncogeni: a) introduzione e cenni storici; b) classificazione degli oncogeni sulla base delle funzioni dei loro prodotti; c) il gene src e la famiglia delle tirosinachinasi; d) un oncogene che codifica un recettore di membrana: erb-B-1; e) la famiglia degli oncogeni ras; f) oncogeni con prodotti a localizzazione nucleare: fos, myc, myb, jun. 5. Geni oncosoppressori o antioncogeni: a) definizione; b) l'esempio paradigmatico del retinoblastoma; c) altre sindromi tumorali ereditarie legate alla inattivazione di antioncogeni; d) ulteriore scoperta di geni coinvolti anche nello sviluppo di tumori sporadici. 6. Conclusioni e prospettive. □ Bibliografia.
1. Basi biologiche dell'oncologia
a) Le colture cellulari.
L'oncologia sperimentale è una scienza di base in auge da più di un secolo, ma soltanto negli ultimi decenni ha registrato progressi straordinari, che si sono rapidamente riflessi nelle sue applicazioni cliniche e terapeutiche, cioè nell'oncologia clinica. Uno dei momenti chiave per il suo sviluppo, che ha preluso alle straordinarie scoperte degli ultimi decenni, è rappresentato dalle nuove e fondamentali conoscenze sulle cellule, acquisite nel momento in cui si è passati dallo studio dei tumori sperimentali ottenuti in vivo, per lo più nei Roditori, allo studio delle cellule coltivate in vitro. Lo studio delle colture cellulari, pertanto, rappresenta il punto di inizio di una nuova oncologia sperimentale, che nasce su basi totalmente differenti da quelle su cui si era sviluppata per tutta la prima metà del XX secolo.
Pur essendo le colture cellulari, in realtà, note fin dagli inizi del secolo, è soltanto dai primi anni cinquanta che i ricercatori hanno cominciato a utilizzarle come un metodo corrente di laboratorio: il merito di questo enorme progresso metodologico spetta indubbiamente, tra gli altri, a un ricercatore italiano, Renato Dulbecco, premio Nobel per la fisiologia e la medicina nel 1975, il quale introdusse nel 1952 il metodo cosiddetto del plaque assay, basato sull'impiego di cellule in coltura per misurare il numero di particelle virali infettanti presenti in un dato campione biologico (v. Dulbecco, 1952).
Le colture cellulari vanno distinte in primarie, secondarie, ceppi e linee. Per colture primarie si intendono le colture originate direttamente da un espianto di organo o tessuto messo in vitro in opportune condizioni. Per colture secondarie si intendono le colture primarie che sono andate incontro in vitro a un certo numero di duplicazioni e ‛passaggi': tali colture, in genere, sono destinate a morire, soprattutto nel caso di cellule derivate da tessuti normali.
Durante il periodo dei passaggi in vitro, la maggior parte dei tipi cellulari va incontro a morte e finisce per prevalere un solo tipo cellulare, quello ‛fibroblastico', definito ‛ceppo' cellulare; anch'esso è generalmente destinato a morire. Una coltura cellulare con una capacità di sopravvivenza infinita viene detta ‛linea cellulare'. La possibilità di ottenere linee cellulari immortali varia a seconda della specie animale considerata, e nell'uomo tale possibilità esiste soltanto relativamente alle cellule tumorali: è questo il caso, ad esempio, della linea cellulare cosiddetta HeLa, che per anni ha rappresentato uno straordinario strumento di ricerca. Le cellule di pollo muoiono dopo pochi passaggi e, anche se si tratta di cellule tumorali, quasi mai diventano cellule immortali. Colture cellulari ottenute da embrioni di Roditori, invece, danno frequentemente origine a linee cellulari continue.
Linee cellulari continue, adatte soprattutto per gli studi di virologia, furono ottenute nel laboratorio di W. R. Earle (v. Sanford e altri, 1948). Le tappe più importanti per lo sviluppo e la messa a punto delle tecniche per le colture cellulari sono state: 1) l'uso di antibiotici, non dannosi per le colture, ma capaci di prevenire la contaminazione batterica; 2) l'isolamento da un carcinoma umano della linea cellulare HeLa (v. Gey e altri, 1952; v. Scherer e altri, 1953); 3) lo sviluppo, da parte di Eagle (v., 1960), di un mezzo semplice e definito per la crescita, e non solo per la sopravvivenza, di molti tipi di colture cellulari; 4) l'introduzione, da parte di T. T. Puck, di tecniche per il clonaggio cellulare (v. Puck e altri, 1956).
Malgrado i pionieri degli studi sulle colture cellulari avessero già messo in evidenza l'importanza per la crescita cellulare dell'aggiunta al mezzo di coltura del siero di sangue, tuttavia il fatto che tale crescita dipendesse strettamente dalla concentrazione del siero fu nitidamente messa a fuoco solo negli anni cinquanta.
Le caratteristiche principali di alcune tra le più conosciute linee cellulari sono illustrate nella tab. I; le caratteristiche morfologiche e di crescita di varie linee cellulari sono illustrate nelle figg. 1, 2 e 3.
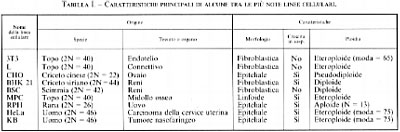
b) La trasformazione neoplastica.
La possibilità di ottenere cellule in coltura continua, appartenenti ai più vari istiotipi cellulari, ha permesso di disporre di sistemi utili come modelli per lo studio della trasformazione neoplastica in vitro. Si sono quindi rapidamente sviluppati sistemi - inizialmente soprattutto di fibroblasti, successivamente anche di altri tipi differenziati di cellule - grazie ai quali era possibile disporre da un lato del fenotipo cellulare normale, dall'altro del corrispondente fenotipo neoplastico. La chiave di volta per questo tipo di studi fu fornita da un gruppo di ricercatori all'avanguardia tra i quali ancora una volta Dulbecco, il quale individuò una categoria di virus animali che, pur contenendo una quantità relativamente piccola di informazione genetica, avevano tuttavia la capacità di trasformare le cellule normali in cellule con caratteristiche chiaramente anomale, simili, per molti versi, a quelle delle cellule presenti all'interno di una massa tumorale. È stato così possibile, paragonando le proprietà di crescita delle cellule normali con quelle delle corrispondenti cellule trasformate, mettere in evidenza una serie abbastanza numerosa di differenze che sono elencate nella tab. II.

Alcune delle caratteristiche differenziali tra cellule normali e trasformate - soprattutto quelle relative alla densità di saturazione, alla perdita della capacità di arresto della crescita, alla perdita della dipendenza dall'ancoraggio per la crescita e ai cambiamenti di morfologia e patterns di crescita - sono meglio illustrate nella fig. 4. Nella fig. 5 sono illustrati alcuni esempi delle alterazioni del citoscheletro presenti nelle cellule trasformate: i filamenti di actina e miosina, perfettamente conservati ed evidenti nelle cellule normali, sono praticamente scomparsi nelle cellule trasformate, pur essendo actina e miosina presenti in maniera diffusa all'interno delle cellule; la fibronectina, una proteina largamente rappresentata all'esterno della membrana cellulare nelle cellule normali, è scomparsa o fortemente ridotta nelle corrispondenti cellule trasformate. Nella fig. 6 è illustrato l'effetto del rilascio di fattori di crescita di tipo autocrino da parte delle cellule trasformate, fenomeno inesistente nelle cellule normali. Un esempio tipico di secrezione autocrina è fornito dalle linee cellulari ottenute da carcinomi polmonari cosiddetti ‛a piccole cellule', che secernono dei polipeptidi appartenenti alla famiglia dei Bombesin-Like Polypeptides (BLP), il cui capostipite, la bombesina, prodotto dalla cute della rana Bombina bombina, stimola la secrezione degli enzimi pancreatici. Nei Mammiferi, i polipeptidi correlati con la bombesina, tra cui il polipeptide che fa rilasciare la gastrina (gastrin releasing polypeptide), sono responsabili di varie risposte fisiologiche, tra le quali iperplasia delle cellule secernenti gastrina, aumento del contenuto in DNA delle cellule pancreatiche, stimolo alla proliferazione in coltura delle cellule della linea 3T3.
2. Cancerogenesi chimica
a) Premessa.
La storia della scoperta degli agenti cancerogeni chimici e dello sviluppo delle conoscenze sulla loro natura e sul loro meccanismo d'azione è stata esaurientemente trattata in precedenza (v. neoplasie: Oncologia sperimentale, vol. IV). Ci limiteremo qui a ricordare che tra le sostanze organiche potenzialmente in grado di esercitare attività carcinogenetica sono state più recentemente individuate le nitrosammine, in particolare la dimetilnitrosammina - la cui utilizzazione industriale è stata causa di lesioni tossiche a carico del fegato dei lavoratori esposti e rivelatasi un potente cancerogeno epatico per i Roditori - e la dialchilnitrosammina. È stato inoltre dimostrato che alcuni componenti della dieta in presenza di elevati livelli di nitriti (sali dell'acido nitroso) possono dare origine, nello stomaco degli animali da esperimento o negli alimenti che vengono loro somministrati, a nitrosammine o nitrosammidi dotate di spiccata azione mutagena e potenzialmente carcinogene per il tratto gastrointestinale. Tutte le sostanze chimiche fin qui ricordate hanno in comune la caratteristica che per poter esplicare la loro attività cancerogena debbono essere trasformate in forme biologicamente attive: sono cioè agenti cancerogeni indiretti, o procancerogeni (v. trasformazione delle cellule, vol. VII). Sono invece cancerogeni diretti (non necessitano, cioè, di attivazione biochimica) i composti acilanti e alchilanti, quali mostarde azotate, lattoni, epossidi (v. fig. 7). Ricordiamo ancora che è stato da tempo dimostrato che numerosi elementi inorganici sono dotati di potenziale attività cancerogena (v. fig. 8).
b) Metabolismo dei cancerogeni chimici e loro interazione con il DNA.
Come si è già detto, gli agenti alchilanti e acilanti, intrinsecamente elettrofili, sono in grado di esplicare la loro attività carcinogenetica senza bisogno di venire preventivamente metabolizzati. Nel processo di metabolizzazione che gli agenti cancerogeni indiretti devono subire per essere trasformati nella loro forma attiva, il passaggio dal procancerogeno al cancerogeno ultimo può avvenire attraverso la formazione di composti intermedi, chiamati cancerogeni prossimi. Il metabolismo può anche portare a forme non reattive (prodotti di detossicazione) che possono essere escrete. La potenza del cancerogeno è determinata dal bilancio tra attivazione e inattivazione (v. fig. 9). È ampiamente dimostrato che la maggior parte dei cancerogeni è mutagena ed è inoltre evidente che il DNA è il bersaglio molecolare degli agenti carcinogeni sia diretti che indiretti. Le interazioni tra agenti carcinogeni e DNA sono di tre tipi: reazione non covalente, reazione covalente, alchilazione.
La reazione non covalente può aver luogo in due modi: o con il processo detto ‛di intercalazione', consistente nell'inserzione di un gruppo planare tra le coppie di basi del DNA; oppure attraverso la formazione di un legame esterno, o con il fosfato o con i siti delle basi non coinvolte nel legame idrogeno. La reazione covalente, di gran lunga più frequente, avviene generalmente a livello degli atomi di azoto delle purine, che sono i gruppi più reattivi, in particolare quelli in posizione 3 e 7: il più reattivo è l'N-7 della guanina, seguono l'N-3 e l'N-7 dell'adenina; meno frequente è il legame con l'ossigeno in posizione 6 della guanina (v. fig. 10).
L'alchilazione è l'interazione più conosciuta. Gli agenti alchilanti sono una classe eterogenea di composti (nitrosammine, nitrosammidi, idrazine, lattoni, mostarde) che hanno la proprietà di cedere un gruppo alchilico - un metile (CH3) o un etile (C2H5) - a un azoto o a un ossigeno di una base del DNA: la reazione di alchilazione più frequente dà luogo alla formazione di N-7-alchilguanina, mentre il più potente derivato oncogeno è l'O-6-metilguanina che, essendo complementare con la timina, è responsabile della comparsa nel filamento di DNA di una mutazione da GC ad AT (v. fig. 11).
c) La cancerogenesi come processo multifasico.
Dopo la scoperta dei cancerogeni chimici, furono identificate alcune sostanze, dette ‛cocancerogeni' o agenti promoventi - la cui azione si discostava dall'azione dei cancerogeni chimici classici, pur contribuendo in qualche modo al processo di cancerogenesi -, che intervengono nel processo di cancerogenesi nella fase cosiddetta di promozione tumorale (v. fig. 12; v. neoplasie: Oncologia sperimentale, vol. IV). Tali sostanze - a differenza delle sostanze inizianti a cui appartengono i cancerogeni chimici precedentemente descritti, che presiedono appunto alla tappa di inizio della cancerogenesi - non sono in grado, da sole, di dare origine a un processo tumorale; sono invece in grado di ‛aiutare' il processo di cancerogenesi se somministrate ripetutamente dopo l'azione di una sostanza iniziante fatta assumere in dosi tanto basse da non favorirne la possibile capacità promovente (che è intrinseca in alcuni agenti cancerogeni).
Il primo composto utilizzato negli esperimenti di cocancerogenesi è stato l'olio di croton tiglio, una sostanza vegetale i cui componenti essenziali per l'azione promovente sono rappresentati da una categoria di composti chimici denominati esteri del forbolo. La struttura chimica del più conosciuto estere del forbolo, il 12-O-tetradecanoilforbolo acetato (TPA), è mostrata nella fig.13 insieme con le strutture di alcuni altri agenti promoventi noti.
Nella tab. III sono anche riportate alcune delle principali differenze tra azione iniziante e azione promovente.
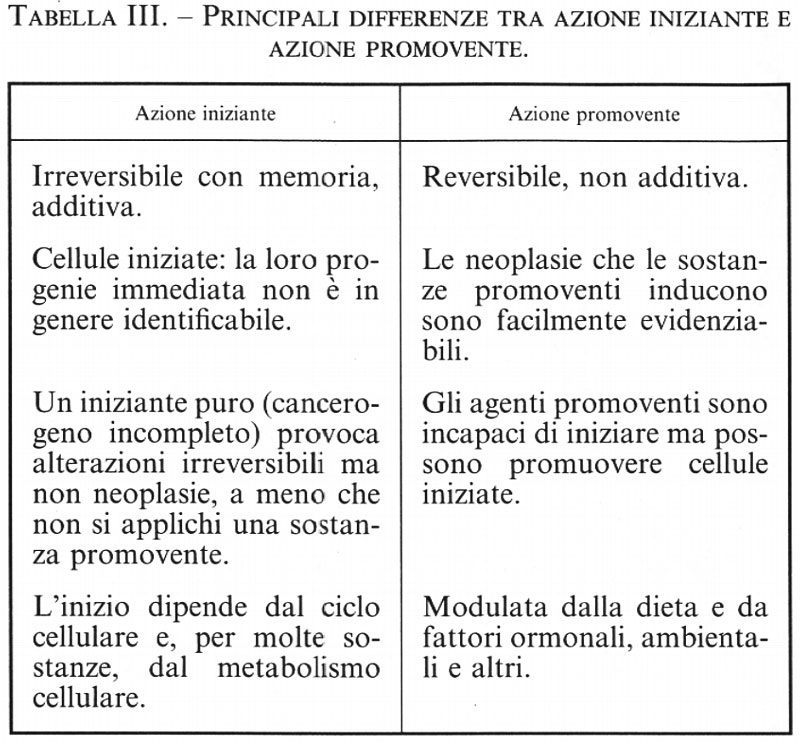
d) Meccanismo d'azione degli agenti cocancerogeni.
Poiché il TPA o i suoi analoghi si legano a tutte le cellule nucleate animali, è lecito ipotizzare l'esistenza di un recettore per il TPA sulle cellule eucariotiche.
Nel 1982 è stato dimostrato che il TPA attiva una proteinchinasi di cervello, Ca2+- e fosfolipidi-dipendente, in seguito identificata nella proteinchinasi C: si tratta di un enzima ubiquitario che per la sua attività richiede, oltre ai fosfolipidi, anche il diacilglicerolo, prodotto dal fosfatidil-inositolo in risposta a una varietà di messaggi extracellulari. Il TPA può sostituire il diacilglicerolo nell'attivazione della proteinchinasi C, e pertanto il diacilglicerolo è l'analogo endogeno del TPA.
È possibile che gli esteri del forbolo esercitino i loro effetti inizialmente mediante legame a siti di alta affinità sulla proteinchinasi C, con un aumento dell'attività chinasica e la successiva alterazione delle fosfoproteine cellulari: questo effetto (aumentata fosforilazione di proteine specifiche di membrana) è comune a molti altri agenti, quali virus oncogeni, ormoni e fattori di crescita.
3. Cancerogenesi virale
a) Premessa.
La grande attenzione rivolta ai virus oncogeni nel ruolo di agenti cancerogeni dipende in maniera determinante dal fatto che l'apparente semplicità di alcuni agenti virali induce a sperare che uno studio dettagliato dell'assetto genetico da essi conferito alla cellula infettata faciliti la comprensione dei meccanismi responsabili della conversione della cellula normale in cellula neoplastica. Infatti, il caso più semplice di un virus oncogeno che introduca nella cellula normale un singolo gene il cui prodotto è capace di iniziare e mantenere lo stato trasformato è oggi ben documentato per un numero limitato di virus studiati e sembra avere, almeno in parte, fornito le risposte ad alcune fondamentali domande che riguardano la provenienza dei geni che causano tumori (oncogeni), quali: le modalità secondo le quali l'oncogene viene introdotto, riprodotto ed espresso; il genere di proteina trasformante da esso prodotta; l'azione di questa proteina, diretta o indiretta, nel metabolismo di una cellula ospite. I virus oncogeni, come altre categorie di virus, vengono convenzionalmente classificati secondo la composizione del loro genoma in virus oncogeni a DNA e a RNA.
Fino ad alcuni anni fa gli studi di oncogenesi virale venivano ritenuti estremamente promettenti e di grande interesse per la ricerca sui meccanismi molecolari di cancerogenesi, ma scarsamente connessi con la spiegazione dell'origine dei tumori umani, la cui epidemiologia, anzi, sembrava orientare le indagini in direzioni completamente opposte.
Recentemente, tuttavia, due scoperte fondamentali hanno mutato radicalmente questa impostazione, collocando i virus oncogeni, e in particolare i virus a RNA, in una posizione di primo piano non solo nelle ricerche di biologia molecolare, ma anche negli studi relativi alla comprensione dei meccanismi di cancerogenesi umana. La prima è rappresentata dall'individuazione di categorie di Retrovirus implicati nell'eziologia di alcune malattie umane, sia francamente neoplastiche, quali le leucemie a cellule T, sia non neoplastiche, come la sindrome da immunodeficienza acquisita (AIDS), ma a prognosi altrettanto infausta. La seconda scoperta consiste nella dimostrazione che tutti i geni onc, o oncogeni, noti e studiati da tempo e presenti in una particolare categoria di Retrovirus detti acuti, sono versioni alterate di alcuni geni normalmente presenti nelle cellule di quasi tutte le specie viventi, dagli Invertebrati all'uomo, detti protoncogeni. Si ritiene attualmente che i protoncogeni siano stati acquisiti dai Retrovirus durante l'infezione di cellule di organismi eucariotici e modificati attraverso meccanismi di trasduzione e ricombinazione genetica con il genoma delle cellule ospiti. La maggior parte degli oncogeni è stata scoperta per la loro associazione con uno dei differenti tipi di Retrovirus acuti conosciuti; tuttavia, il recente isolamento di oncogeni non presenti in Retrovirus, realizzato con la tecnica della introduzione mediante trasfezione di DNA in cellule in coltura, induce a ritenere possibile l'individuazione di altri geni cellulari implicati nel processo di cancerogenesi.
Le scoperte delineate hanno avuto un impatto eccezionale sulle attuali conoscenze di cancerogenesi umana, e non è escluso che i risultati di tali ricerche possano rivelarsi importanti non solo per la comprensione dei meccanismi di cancerogenesi, ma anche per la diagnostica e, in prospettiva, per la terapia oncologica.
Per quanto riguarda i virus a DNA, ogni loro famiglia comprende generi che manifestano alcune proprietà specifiche per le quali possono essere considerati virus oncogeni: essi infatti inducono tumori in alcuni ospiti, sono capaci di alterare le proprietà morfologiche e di crescita delle cellule in coltura e presentano associazioni epidemiologiche con tumori a eziologia incerta.
I virus oncogeni a RNA rappresentano un unico gruppo tassonomico in quanto, benché siano stati isolati da animali di specie differenti e siano in grado di provocare negli animali suscettibili una vasta gamma di condizioni patologiche attraverso un largo spettro di meccanismi differenti, sono unificati dalle seguenti proprietà: 1) presentano tutti un genoma molto simile; 2) infettano le cellule in maniera tipica; 3) posseggono composizione e struttura chimica unitarie; 4) si replicano tutti utilizzando una molecola intermedia di DNA tramite l'enzima trascrittasi inversa (reverse transcriptase, da cui il nome Retrovirus; v. virus, vol. VII), una DNA-polimerasi RNA-dipendente.
b) Virus oncogeni a DNA.
Dal punto di vista didattico, i virus oncogeni a DNA sono suddivisi in cinque gruppi principali: i Poliomavirus, che comprendono il virus Polioma e il virus vacuolante della scimmia o SV (Simian Virus) 40; i virus del papilloma; gli Adenovirus; i virus dell'epatite B; gli Herpesvirus.
1. Virus Polioma e SV40. - La storia della scoperta di questi virus oncogeni è già stata ben delineata (v. neoplasie: Oncologia sperimentale, vol. IV). Ricorderemo qui che agenti virali di questo tipo sono stati isolati in alcuni casi di patologie umane: nel cervello di un paziente affetto da leucoencefalopatia progressiva multifocale (PML), in pazienti con sindrome di Wiskott-Aldrich (una malattia eterocromosomica recessiva consistente in un difetto dell'immunità cellulare e umorale e, in parte, delle resistenze aspecifiche; v. immunologia clinica e immunopatologia, vol. VIII), e nelle urine di pazienti immunosoppressi. Questi virus tipo Polioma, molto simili all'SV40 e designati con le iniziali delle persone dalle quali sono stati isolati (per es., BK, JC), sono capaci, specialmente il virus JC, di provocare tumori in criceti e di trasformare le cellule di criceto in coltura; inoltre, poiché il 70% della popolazione umana possiede anticorpi contro di essi, è possibile che questi virus rappresentino l'equivalente umano dell'SV40 delle scimmie Rhesus, oppure del virus Polioma dei topi. Il loro virione è un piccolo icosaedro senza involucro esterno, del diametro di 45 nm, con 72 capsomeri. Esso contiene un DNA circolare a doppio filamento di circa 5.000 paia di basi, associato con ottameri di istoni cellulari, quali H2a, H2b, H3, H4, per formare nucleosomi simili a quelli presenti nella cromatina eucariotica (v. tab. IV). I virus Polioma e SV40 sono molto resistenti al calore e alla formalina: l'SV40 (proveniente dalle colture di rene di scimmia Rhesus) è sopravvissuto al trattamento con formalina in alcune preparazioni di vaccino di virus della poliomielite.

Negli anni ottanta i virus Polioma e SV40 hanno rappresentato i sistemi virali più adoperati e meglio studiati: si tratta infatti di piccoli virus contenenti un'informazione genetica limitata e dotati della straordinaria capacità di indurre tumori in vivo nell'animale da esperimento e di trasformare in senso neoplastico colture cellulari in vitro, alla stessa stregua degli Adenovirus, il cui contenuto di informazione genetica è peraltro di gran lunga maggiore. Questa circostanza ha fatto nascere la speranza di poter comprendere in tempi relativamente brevi i meccanismi con cui alcuni geni virali (presumibilmente pochi) possono indurre la trasformazione neoplastica di cellule eucariotiche. I progressi della biologia molecolare hanno reso possibile la delucidazione dell'intera mappa genetica sia del virus Polioma che dell'SV40. Dal confronto delle due mappe si deduce l'esistenza di una profonda similarità e di alcune differenze tra questi virus: infatti, essi posseggono entrambi sia regioni genomiche denominate early e late, come gli Adenovirus, sia regioni di regolazione; impiegano, però, strategie differenti per la loro espressione, perché mentre il virus SV40 utilizza una sola proteina, nota come T large, sintetizzata dall'RNA messaggero precoce per ottemperare alla funzione della replicazione virale e per indurre la trasformazione neoplastica, il virus Polioma utilizza una proteina T large per la replicazione virale, e una proteina detta T middle (inesistente nell'SV40) per la funzione trasformante. Le proteine T large dei virus Polioma e SV40 sono entrambe fosfoproteine. Un'altra proprietà che accomuna questi due virus, rendendoli simili anche agli Adenovirus, è la non associazione a eventi tumorali spontanei nell'ospite naturale; in altre parole, non si è mai riscontrato in natura un topo con neoplasie indotte da virus Polioma, così come non si sono riscontrati tumori indotti da SV40 nella scimmia Rhesus o tumori indotti da Adenovirus nell'uomo. Con il virus Polioma, invece, è possibile indurre sperimentalmente, in topi neonati, la formazione di tumori di varia natura.
La proteina T large di SV40, la meglio conosciuta, possiede vari dominî funzionali: a) un dominio di legame alla regione ‛ori' (regione di origine della replicazione) del DNA virale; b) un dominio con attività ATPasica nella porzione COOH terminale della proteina; c) un dominio con capacità di legare la proteina prodotta dal gene oncosoppressore p53; d) un dominio che le conferisce la capacità di traslocare nel nucleo.
Le cellule trasformate sia da SV40 che da Polioma hanno sempre stabilmente integrata nel loro DNA una funzione early di uno di questi due virus; inoltre, non è mai stata messa in evidenza l'unicità del sito di integrazione, né è stata dimostrata l'attivazione di una funzione cellulare a opera di uno dei due virus: queste osservazioni fanno escludere l'esistenza di analogie fra i meccanismi di trasformazione indotta dai virus Polioma e SV40 e, rispettivamente, da alcuni Retrovirus, quali i Retrovirus cronici (v. sotto). Le tecnologie della biologia molecolare hanno permesso di dimostrare in maniera inequivocabile che la trasformazione indotta da SV40 e dal virus Polioma è il risultato dell'azione di un vero e proprio oncogene: infatti il cDNA che codifica per l'antigene T middle di Polioma è sufficiente a provocare la trasformazione cellulare, mentre la presenza dell'antigene T large garantisce la sintesi di un gran numero di copie di DNA virale, facendo sì che la trasformazione avvenga in modo efficiente. Inoltre, l'antigene T large di Polioma non è necessario per la trasformazione di linee cellulari stabili, ma è importante per la funzione di immortalizzazione di cellule in colture primarie. La proteina T middle di Polioma si associa con la proteina SRC cellulare e questa associazione facilita il meccanismo di trasformazione cellulare (v. cap. 4). La proteina T large di SV40 è capace sia di immortalizzare cellule primarie, sia di trasformare cellule in coltura continua; essa è complessata nelle cellule trasformate con la proteina prodotta dal gene oncosoppressore p53, così che si ritiene che all'origine della trasformazione cellulare indotta dal virus vi sia un'inibizione della funzione della proteina p53 normale. Il meccanismo della trasformazione, pertanto, presenta analogie con quello proprio degli Adenovirus, che tramite la proteina E-1A inibiscono l'azione di un altro prodotto di un gene oncosoppressore, cioè la proteina RB. Per quel che riguarda la localizzazione cellulare della proteina T large, una piccola parte si trova nella membrana cellulare, la maggior parte nel nucleo (v. Watson e altri, 19874).
2. Papillomavirus. - I virus del papilloma sono strutturalmente simili ai virioni del virus Polioma ma leggermente più grandi (diametro di circa 55 nm), in quanto è più grande il loro DNA. Per le similitudini che essi presentano, i virus del papilloma, Polioma e SV40 sono stati riuniti in un unico gruppo denominato Papovavirus, dalle iniziali dei loro nomi: pa(pilloma), po(lioma) e virus delle scimmie va(cuolating agent). Anche i Papillomavirus posseggono un DNA bicatenario circolare chiuso, di peso molecolare di circa 5 × 106 dalton, complessato con istoni cellulari e condensato in nucleosomi, incapsidato in virioni icosaedrici. I genomi dei virus del papilloma differiscono da quelli del virus tipo Polioma nell'organizzazione delle ORF (Open Reading Frames, cornici di lettura): infatti, per esempio, nel Polioma e nell'SV40 la trascrizione avviene su entrambi i filamenti del DNA, in direzione opposta, mentre nel caso dei virus del papilloma tutte le ORF più importanti sono trascritte su un solo filamento. I virus del papilloma si replicano come episomi e pertanto costituiscono un buon modello non solo di replicazione virale, ma anche di espressione di geni estranei in cellule di Mammifero.
Esistono almeno 42 tipi diversi di virus del papilloma umano, dei quali quelli con tropismo per la cute in generale non hanno tropismo per le mucose e viceversa. La loro moltiplicazione è possibile soltanto nei cheratinociti differenziati degli epiteli di rivestimento. Nell'epidermide, che matura in 10-14 giorni, cellule proliferanti sono presenti solo nello strato basale; le cellule figlie, soprabasali, non si dividono, si differenziano e producono una serie di cambiamenti nell'espressione dei geni delle citocheratine. Il processo ha termine con la formazione di legami crociati delle citocheratine attraverso ponti disolfuro per formare le microfibrille dello strato corneo; lo strato superficiale è anucleato e non consente la replicazione del Papillomavirus. L'epitelio delle mucose esprime differenti tipi di citocheratine ma è privo di strato corneo. I virus del papilloma possono infettare tramite accesso diretto alle cellule basali, il cui strato costituisce così la riserva del DNA virale, dando origine, in vari mammiferi incluso l'uomo, a verruche a decorso benigno chiamate papillomi. Nel coniglio e nei bovini le verruche, se persistono per lungo tempo o sotto l'influenza di piccole dosi di cancerogeni chimici, possono diventare maligne; Shope (v., 1937) isolò per primo un virus del papilloma, dimostrando che estratti di verruche provenienti da conigli selvatici (cotton tail) producevano verruche quando inoculati sotto cute sia nei conigli d'allevamento che in quelli selvatici; i virus delle verruche furono poi isolati da estratti di verruche sia umane che di altre specie animali. I virus del papilloma umano (HPV) e del coniglio inducono lo sviluppo di papillomi sulla cute e su alcune mucose in individui della stessa specie o di specie correlate, mentre il virus bovino (BPV) può dare origine anche a tumori mesenchimali nei cavalli o nei criceti. Questi virus non crescono in colture cellulari, ma cellule cutanee umane in coltura possono essere trasformate da virus del papilloma umano, così come il virus del papilloma bovino è capace di trasformare cellule fetali di vitello in coltura.
Numerose evidenze epidemiologiche e molecolari indicano che i Papillomavirus sono correlati a varie malattie dell'uomo, in particolare ai carcinomi delle mucose orali e genitali. Lo studio dello sviluppo dei papillomi nell'uomo dimostra che l'iperplasia può avere origine sia da un'aumentata velocità di divisione cellulare, sia da una ritardata maturazione dei cheratinociti che impiegano più tempo per raggiungere la superficie. Alcuni tipi di papillomi rimangono benigni, altre lesioni diventano displastiche, altre ancora diventano carcinomi in situ: in quest'ultimo caso la successiva penetrazione delle cellule attraverso la membrana basale dà origine al carcinoma invasivo. I virus del papilloma umano sono stati associati con il carcinoma della cervice uterina della donna, la cui incidenza mondiale è di 450.000 casi/anno, mentre quella di altre neoplasie della mucosa genitale e orale è stimata in 150.000 casi/anno. Le ricerche epidemiologiche suggeriscono che i Papillomavirus siano trasmessi per via sessuale per quanto riguarda le patologie a carico delle vie genitali (maggiore incidenza in soggetti a precoce inizio di attività sessuale o nei casi di eccessiva promiscuità sessuale, ecc.) e che rappresentino probabilmente per l'uomo una concausa nello sviluppo di papillomi, displasie e carcinomi della mucosa orale, più frequenti nei fumatori che nei non fumatori. Il primo virus del papilloma umano associato a lesioni genitali è stato identificato nei condilomi acuminati (v. Dunn e Ogilvie, 1968).
I ceppi HPV-6 e HPV-11 sono stati clonati a partire da lesioni benigne genitali e laringee. Molti carcinomi primari, tumori metastatici e linee cellulari derivate da carcinomi cervicali (come HeLa, Siha e Caski) contengono HPV-16, HPV-18 o tipi correlati di HPV DNA, spesso integrati in uno o più siti cromosomici. Ricerche dello stesso tipo condotte su carcinomi della laringe hanno consentito di rivelare la presenza di sequenze integrate di HPV-16 o HPV-30. Esistono due tipi differenti di lesioni delle mucose associate a virus del papilloma: quelle associate a HPV-6, che generalmente rimangono benigne, e quelle correlate con HPV-16 e HPV-18 che pongono un serio rischio di conversione maligna. L'osservazione che solo una piccola percentuale di queste lesioni progredisce verso la malignità suggerisce che la presenza di HPV-16 o HPV-18 possa non essere sufficiente a provocare il passaggio da lesione media a carcinoma, e che pertanto debbano esistere altri fattori coinvolti nella patogenesi di tali condizioni a livello sia genitale che orale. Il coinvolgimento di HPV è stato dimostrato anche in un altro gruppo di neoplasie, quelle che si sviluppano su una malattia cutanea rara, denominata epidermodisplasia verruciforme: in questa condizione, la progressione delle lesioni verso carcinomi si osserva in 1/3 dei pazienti portatori di HPV-5, 8, 12, 17 e 20, e generalmente in aree esposte all'irradiazione solare. L'irradiazione favorisce anche la trasformazione dei papillomi laringei in carcinomi laringei.
Tra i virus umani soltanto HPV-1 è produttivo in vivo: la trasformazione di cellule delle linee NIH-3T3 e C-127 è possibile, ma molto più complessa rispetto a quella indotta da BPV. Molti genomi di HPV sono stati interamente sequenziati ed è stato così possibile dimostrarne la notevole somiglianza con gli altri Papillomavirus animali: la maggiore omologia di sequenza, variabile dal 45 all'85%, si è riscontrata nei geni E-1 e L-1, che sono i geni più conservati.
L'RNA messaggero virale è difficilmente purificabile. Le cellule C-127 infettate sia da BPV che da HPV producono RNA messaggero della regione E: tutta la trascrizione avviene su di un solo filamento di DNA e vengono utilizzati vari promotori.
Parecchi quesiti rimangono aperti circa i meccanismi di replicazione degli HPV: per esempio, perché si verifica una riproduzione di tipo episomico nelle verruche e invece si ha integrazione del DNA nei carcinomi? E ancora, come fa a replicarsi un virus in una cellula che non si divide? Le proteine virali sono presentate al sistema immunitario dell'ospite? L'integrazione del DNA nei cromosomi della cellula ospite equivale sempre alla progressione verso il carcinoma? L'esatta identificazione delle funzioni di tutte le proteine virali consentirà probabilmente sia di rispondere a molte di queste domande, sia di interferire con l'infezione virale a livello della replicazione episomica tipica di questi virus.
3. Adenovirus. - Nel tentativo di stabilizzare in vitro colture cellulari da tonsille e da tessuto adenoideo di bambini, Wallace Rowe notò che un agente infettivo virale provocava degenerazione e morte di cellule epiteliali (v. Rowe e altri, 1953). Agenti virali simili furono successivamente isolati a partire da materiale faringeo ottenuto da personale militare affetto da malattie respiratorie lievi (v. Hilleman e Werner, 1954), e furono poi descritti vari sierotipi virali correlati tra loro da antigeni cosiddetti gruppo-specifici identificati da anticorpi fissanti il complemento (v. Huebner e altri, 1963). Nel 1962 Trentin dimostrò che l'adenovirus 12 provoca tumori quando inoculato in criceti neonati: fu questa la prima dimostrazione che un virus patogeno umano è in grado di indurre tumori maligni in animali da esperimento (v. Trentin e altri, 1962).
Gli Adenovirus sono stati associati a diverse manifestazioni patologiche umane, tra cui sindromi respiratorie acute osservate in reclute militari, nonché una congiuntivite epidemica; tuttavia, al di là della loro importanza nella patologia umana, gli Adenovirus hanno rappresentato un modello ideale per gli studi di biochimica e, soprattutto, di oncologia sperimentale. Poiché, infatti, l'infezione cellulare da Adenovirus è seguita rapidamente dall'inibizione della sintesi proteica della cellula ospite, cellule infettate da Adenovirus hanno rappresentato un'utile sorgente per la purificazione di proteine virali che, al contrario, vengono prodotte in grande quantità dalle cellule infettate. Per quanto riguarda la biologia molecolare, poi, le cellule infettate dagli Adenovirus hanno rappresentato i primi modelli eucariotici in cui siano stati dimostrati fenomeni quali il trasporto dell'RNA dal nucleo ai poliribosomi citoplasmatici e la maturazione dell'RNA messaggero (splicing dell'RNA) e in cui siano state impiegate le tecniche di mappaggio degli RNA sul DNA genomico.
Gli Adenovirus hanno la struttura di icosaedri regolari, non provvisti di membrana, a 20 facce triangolari con 12 vertici. Il capside virale è costituito da 252 capsomeri, dei quali 240 vengono detti esoni (exons) e 12 pentoni (pentons), localizzati sui 12 vertici: ciascun pentone possiede una base e una fibra che si proietta all'esterno. Ognuna delle basi dei pentoni è circondata da 5 esoni, mentre ciascun esone è a sua volta circondato da 6 esoni. Il DNA virale è costituito da un doppio filamento di 24 × 106 dalton, pari a circa 36 kilobasi. I due filamenti di DNA posseggono a ciascuna estremità 5′ una catena polipeptidica di 52 kilodalton (kDa) legata covalentemente a una molecola di desossi-cAMP; inoltre, il DNA è caratterizzato da una ridondanza terminale invertita.
Le proteine degli Adenovirus si distinguono in virioniche e non virioniche. A loro volta le proteine virioniche si distinguono in proteine del capside, cioè dell'involucro esterno del virus, e del core, cioè della porzione interna della particella virale. Tra le proteine del capside esterno ci sono: il polipeptide II, che costituisce l'esone, il cui monomero ha un peso molecolare di 120 kDa e il cui trimero ha un peso molecolare di 360 kDa; i polipeptidi VI, VIII e IX, che sono polipeptidi associati all'esone; il polipeptide III, che rappresenta la proteina base del pentone, di peso molecolare di 85 kDa; il polipeptide IV, che rappresenta la fibra del pentone, di 62 kDa. Tra le proteine del core, ci sono il polipeptide V, di 48,5 kDa, e il polipeptide VII, di 18,5 kDa; entrambi questi polipeptidi sono legati in maniera non covalente al DNA e sono di natura basica: si tratta, cioè, di proteine ricche in arginina, come gli istoni cellulari.
Per quanto riguarda le funzioni delle proteine virioniche, i determinanti antigenici presenti all'interno della superficie dell'esone costituiscono un antigene comune che determina la formazione di anticorpi presenti in tutti gli individui umani sieropositivi per gli Adenovirus. Altri determinanti antigenici, presenti sia sul pentone che sull'esone, sono antigeni cosiddetti tipo-specifici e danno origine agli anticorpi neutralizzanti. Uno dei determinanti antigenici presenti sulle fibre è l'emoagglutinina virale. Infine, la base del pentone è tossica per le cellule e provoca l'effetto citopatico tipico degli Adenovirus.
Le proteine non virioniche sono essenzialmente tre: a) una proteina di 100 kDa, necessaria per l'assemblaggio dei trimeri della proteina esone; b) una proteina di 72 kDa, che è una proteina legante il DNA; c) una proteina di 140 kDa, con funzione di DNA-polimerasi.
Gli Adenovirus esercitano un effetto profondo sulle sintesi macromolecolari delle cellule ospiti, come si è accennato in precedenza. Infatti sia la sintesi del DNA cellulare che quella delle proteine cellulari vengono fortemente inibite dall'infezione virale.
Meno dello 0,19% dell'RNA totale presente nelle cellule infettate dagli Adenovirus è rappresentato da RNA virale cosiddetto precoce (early messenger RNA); tuttavia, dal 15 al 18% dell'RNA polisomiale è complementare al DNA di Adenovirus. Entrambi i filamenti di DNA vengono trascritti. La complementarità dell'RNA si ha con 4 regioni definite, non contigue, del DNA virale.
I geni early 1 e 3 (E-1 ed E-3) vengono trascritti dal filamento cosiddetto r (right), ovvero il filamento per convenzione denominato ‛destro' del DNA virale; il gene E-1, ulteriormente suddiviso in E-1A e in E-1B, codifica per la funzione trasformante degli Adenovirus. Il gene E-1A codifica per 5 RNA messaggeri che posseggono regioni 5′ e 3′ comuni, ma differenti tipi di splicing. Gli RNA messaggeri hanno dimensioni corrispondenti a costanti di sedimentazione 13S, 12S, 11S, 10S e 9S. Tutti gli RNA messaggeri utilizzano la stessa cornice di lettura, e pertanto le proteine che vengono sintetizzate da questi diversi RNA messaggeri sono strettamente correlate tra di loro. Si distinguono almeno tre funzioni differenti per le proteine codificate da E-1A: funzione di trans-attivazione, funzione di trans-repressione e funzione di trasformazione cellulare. L'esistenza di una funzione di trans-attivazione è stata dimostrata da esperimenti effettuati con mutanti virali contenenti mutazioni puntiformi ovvero delezioni di questa regione degli Adenovirus: cellule infettate con tali mutanti fanno registrare l'accumulo di RNA messaggeri provenienti da altre unità di trascrizione virale, il che lascia supporre che l'unità di trascrizione virale sotto il controllo di E-1A risenta della funzione di attivazione in trans delle stesse proteine sintetizzate da E-1A. Geni simili a E-1A sono stati descritti nei Papovavirus, negli Herpesvirus e nei Retrovirus. I prodotti di E-1A attivano non solo la trascrizione di geni virali, ma anche quella di geni cellulari. Quanto al meccanismo d'azione della funzione trans-attivante, sembra che le proteine sintetizzate da E-1A non interagiscano direttamente con il DNA, ma provochino un aumento della concentrazione di alcuni fattori trascrizionali. La funzione di trans-repressione di E-1A consiste nella possibilità di inibire la trascrizione di geni che codificano la sintesi di proteine appartenenti alla famiglia delle β-globine.
La funzione trasformante fu individuata nel 1980, quando venne dimostrato che un frammento dell'adenovirus del ceppo 5, contenente il gene E-1A, era capace di immortalizzare ma non di trasformare fibroblasti in coltura primaria. Le proteine codificate da E-1A cooperano con quelle codificate da E-1B per trasformare sia cellule in coltura primaria che cellule in coltura continua. Inoltre, le proteine codificate da E-1A cooperano con la proteina RAS per la trasformazione di cellule in coltura primaria. Due sono le proteine principali codificate dal gene E-1A: una di 289 e una di 243 amminoacidi, entrambe con gli stessi effetti di cooperazione. Il meccanismo dell'azione trasformante delle proteine codificate da E-1A consiste nella loro capacità di formare un legame stabile con la proteina prodotto del gene cellulare ad attività oncosoppressiva, detta del retinoblastoma (RB). Il legame con la proteina RB determina l'inattivazione di quest'ultima e la conseguente stimolazione della proliferazione cellulare.
A ridosso di E-1A si trova il gene E-1B, che codifica a sua volta 5 RNA messaggeri i quali, anche in questo caso, condividono regioni 5′ e 3′ terminali. I due mRNA più grandi hanno costanti di sedimentazione 22S e 13S, quelli più piccoli rispettivamente 14,5S, 14S e 11S. Tutti i messaggeri, a eccezione di 11S, codificano un polipeptide di 175 amminoacidi e inoltre gli mRNA 22S, 14,5S, 13S e 11S codificano polipeptidi di 495, 92, 155, 82 e 162 amminoacidi, rispettivamente, tradotti su una differente cornice di lettura. Sia la proteina di 175 amminoacidi che quella di 495 amminoacidi contengono un dominio conservato e uno variabile; le regioni variabili cadono in regioni di sovrapposizione. Parte della proteina di 495 amminoacidi è complessata con la proteina p53.
L'osservazione che E-1B non esplica attività trasformante in assenza di E-1A e che le cellule immortalizzate soltanto da E-1A esprimono quest'ultimo gene a livelli più bassi rispetto a cellule trasfettate sia con E-1A che con E-1B, dimostra che E-1B è in grado di far aumentare la trascrizione di E-1A.
4. Virus dell'epatite B. - L'epatite di tipo B rappresenta un problema importante per la salute pubblica: infatti nell'Est asiatico e in Africa i portatori cronici di epatite B rappresentano il 10% della popolazione attiva, mentre l'epatite cronica attiva, la cirrosi e l'epatocarcinoma primitivo sono cause importanti di mortalità in molte zone del mondo. Oltre al virus dell'epatite B umana, esistono altre specie correlate di virus simili in vari animali: il Woodchuck Hepatitis Virus (WHV, virus della marmotta), trovato nel siero di animali con epatite ed epatocarcinoma (v. Summers e altri, 1978); il Ground Squirrel Hepatitis Virus (GSHV, virus dello scoiattolo terricolo americano), trovato nel siero di scoiattoli selvatici in California (v. Marion e altri, 1980); il Duck Hepatitis Virus (DHV, virus dell'anatra), trovato nel siero di anatre domestiche cinesi che andavano incontro a carcinoma epatocellulare (v. Mason e altri, 1980). Questo gruppo di virus, insieme con il virus dell'epatite umana (HBV), costituisce il gruppo degli Hepadnavirus: si tratta di virus costituiti da piccole molecole circolari di DNA, parzialmente a singolo filamento, con una DNA-polimerasi capace di rendere il DNA completamente bicatenario. Caratteristiche di questi virus sono la replicazione attraverso un RNA intermediario e lo straordinario tropismo per il fegato: molto frequente è il riscontro di casi di infezione persistente con antigeni virali e virus infettivo circolante nel sangue ad alte concentrazioni.
La scoperta dell'antigene HBsAg fu effettuata da Blumberg nel corso di ricerche sul polimorfismo di oligoproteine del siero (v. Blumberg e altri, 1965), ma soltanto dopo parecchi anni fu possibile dimostrare la connessione dell'HBsAg con l'epatite B. L'HBsAg, che a tutt'oggi rappresenta il marker più utile durante l'infezione acuta da HBV, compare nel siero come un componente virale, ovvero come forme virali particolate incomplete. Non si trova come antigene solubile e si può presentare in forma o di piccole particelle sferiche, di diversa grandezza (diametro 16-25 nm), o di particelle a bastoncino (filamenti larghi 22 nm e lunghi parecchi nm): queste ultime, che rappresentano la forma più numerosa di particelle trovate nel siero dei pazienti, consistono di proteine, carboidrati e lipidi e non contengono DNA. Nel 1970 Dane descrisse particelle più grandi, del diametro di 42 nm, contenenti HBsAg; tali particelle, che con ogni probabilità rappresentano il virus completo, sono provviste di un involucro lipidico e di un core elettrondenso di 28 nm, detto nucleocapside (v. Dane e altri, 1970); la rimozione dell'involucro esterno, possibile con un detergente come NP40, dà luogo al rilascio di particelle contenenti l'antigene del core, detto HBcAg, antigenicamente distinto da HBsAg. Il core virale contiene il DNA attaccato covalentemente a un polipeptide, una proteinchinasi e un terzo antigene, detto HBeAg. Virioni con core vuoti si possono trovare in tutte le preparazioni virali. L'infettività è altissima, tanto che diluizioni del siero fino a 108 sono capaci di infettare lo scimpanzé, che rappresenta l'unico ospite, oltre l'uomo, suscettibile all'infezione virale. L'antigene HBsAg, contenuto in concentrazioni fino a 10.000 volte più elevate del virus, non è capace di infettare; l'infettività virale è conservata dopo congelamento a - 20° per 30 anni ed è annullata se il virus è tenuto a una temperatura di 100 °C per 20 minuti.
Si possono trovare cinque specificità antigeniche nelle particelle HBsAg: ‛a', che si trova in tutte le preparazioni; ‛d' o ‛y', mutualmente esclusivi, che si comportano come alleli; ‛W' o ‛r', a loro volta mutualmente esclusivi e comportantisi come alleli.
L'immunizzazione con HBsAg conferisce protezione contro l'infezione da HBV. Le particelle HBsAg di 22 nm contengono pochi polipeptidi virali, i cui due componenti quantitativamente più importanti hanno peso molecolare 25.000 dalton e 29.000 dalton; ambedue questi componenti potrebbero in realtà essere identificati nello stesso polipeptide, perché hanno composizione amminoacidica identica, identica sequenza dei primi 19 amminoacidi N terminali e identica sequenza di 3 amminoacidi al COOH terminale: poiché il polipeptide di 29.000 dalton è glicosilato, è possibile che il suo più elevato peso molecolare sia dovuto alla presenza dei carboidrati. Le sequenze amminoacidiche del polipeptide di 25.000 dalton posseggono un alto contenuto in prolina (10%), in cisteina (6%; al centro della molecola sono presenti ponti disolfuro), di serina e treonina, e a questi amminoacidi si legano i residui glicosidici.
La cross-reattività tra HBsAg e gli antigeni di superficie di WHV e di GSHV è bassa.
Nel nucleocapside, oltre ad HBsAg, sono presenti vari polipeptidi (uno di questi, di 19.000 dalton, è probabilmente identificabile nell'antigene ‛e'), proteinchinasi, DNA e DNA-polimerasi. Il DNA si trova sotto forma di piccole molecole parzialmente a doppia catena, poiché mentre una delle due eliche è completa, l'altra è interrotta ed è a filamento singolo per una porzione che varia tra il 15 e il 60% del totale; il filamento lungo, long strand (L), è di 3.200 basi, il filamento corto, short strand (S), di 1.700-2.800 basi. La DNA-polimerasi ripara la regione a filamento singolo e la rende completamente a doppia catena (i due filamenti completi sono costituiti da 3.200 paia di basi): la reazione inizia al 3′ terminale del filamento S e la sintesi termina quando è raggiunto il 5′ terminale dello stesso filamento S. Il filamento L non è un circolo chiuso, ma possiede un vuoto di circa 300 basi, e al suo 5′ terminale è attaccato un polipeptide.
Il filamento L, che è stato completamente clonato, possiede quattro regioni, o cornici di lettura (ORF), nelle quali codificano quattro geni, S e pre-S, C, P, X (v. Tiollais e altri, 1985; v. Ganem e Varmus, 1987). Nella ORF-2 S e pre-S, contigui, codificano le glicoproteine di superficie: il gene S contiene la sequenza di 678 amminoacidi che specifica HBsAg, una proteina di 25.000 dalton; la sequenza a monte pre-S potrebbe dare origine a un precursore di HBsAg, ma non si sa se questo precursore venga realmente sintetizzato in vivo. Nella ORF-3 il gene C contiene le sequenze codificanti la proteina strutturale del core presente nel nucleocapside, di 183 amminoacidi. La ORF-1 si sovrappone completamente alla ORF-2 e contiene il gene P, che codifica un polipeptide di 95.000 dalton corrispondente alla polimerasi virale, la quale esplica attività di trascrittasi inversa. Infine, la ORF-4 contiene il gene X, il più piccolo, che sembra codificare un polipeptide basico di 1.600 dalton la cui capacità di attivatore della trascrizione in trans può regolare in senso positivo l'attività dei promotori degli Hepadnavirus (v. Spandau e Lee, 1988; v. Colgrove e altri, 1989).
L'esistenza di una correlazione tra infezioni da Hepadnavirus ed epatocarcinomi primitivi è suggerita da una serie di osservazioni: a) la coincidenza geografica di elevati livelli di infezione cronica da virus dell'epatite B con alta incidenza di epatocarcinoma; b) i portatori di HBsAg sono esposti a un rischio 100 volte maggiore di ammalarsi di epatocarcinoma rispetto ai non portatori (rilievi effettuati tra i lavoratori delle poste dell'isola di Taiwan; v. Beasley e altri, 1981); c) la comparsa di epatocarcinomi in animali infettati cronicamente con Hepadnavirus (100% di individui dopo 2 anni nelle marmotte, percentuali inferiori negli scoiattoli terricoli e nelle anatre); d) la presenza, in una elevata percentuale di tumori e linee cellulari derivate da tumori umani, di DNA integrato, che può trovarsi allo stato subgenomico con nessuna o una sola ORF rimasta intatta e i cui siti di integrazione possono anche trovarsi su cromosomi diversi.
La presenza di oncogeni negli Hepadnavirus, non dimostrata sperimentalmente, è messa in dubbio da alcune evidenze: l'esistenza di un periodo (medio) di latenza virale di 40 anni nelle regioni in cui l'infezione è contratta alla nascita; la dimostrazione che la trasfezione con Hepadnavirus non dà luogo a trasformazione cellulare; l'accertata mancanza di omologia di sequenza tra geni virali e geni cellulari.
È possibile ipotizzare un'attivazione di oncogeni cellulari indotta dall'integrazione del DNA di HBV? Finora in un unico caso è stata riscontrata la fusione della sequenza pre-S del DNA di HBV con una sequenza cellulare che codifica la proteina dell'oncogene erb-A (v. cap. 4; v. Dejean e altri, 1986). È allora possibile ipotizzare una mutazione recessiva, del tipo di quelle che si riscontrano nei geni oncosoppressori (v. cap. 5), indotta dall'integrazione del DNA di HBV? L'integrazione potrebbe in effetti inattivare geni cellullari: in un caso di epatocarcinoma l'inserzione del DNA di HBV ha provocato la delezione di 12 kb nel cromosoma 11, in vicinanza della regione p13, ma tali sequenze sono lontane dalla regione implicata nel tumore di Wilms. Per quel che riguarda la possibilità di altre mutazioni, è stata osservata l'inserzione del DNA di WHV nell'interno del protoncogene N-myc nel 20% dei tumori di marmotte analizzati: tuttavia, un'esauriente ricerca di situazioni simili nel caso degli epatocarcinomi ha dimostrato che nei tumori umani questo fenomeno avviene molto raramente (v. Wang e altri, 1990).
Le convincenti argomentazioni epidemiologiche circa l'associazione tra virus dell'epatite B ed epatocarcinomi non escludono un ruolo eziologico importante di altri fattori (ad esempio le aflatossine), dei quali, al contrario, è stata riconosciuta tutta l'importanza nel processo di cancerogenesi, anche nei casi nei quali è dimostrabile la responsabilità di HBV; inoltre, alcuni indizi fanno ritenere che l'epatite cronica provocata dal virus dell'epatite C (HCV), un virus a RNA con notevoli somiglianze con i Flavivirus, abbia avuto un ruolo rilevante in alcuni casi di epatocarcinoma con negatività di HBV. Rimane peraltro da chiarire come l'HBV provochi l'epatocarcinoma. La proteina virale X, per la sua proprietà di attivatore della trascrizione, potrebbe, direttamente o indirettamente, contribuire a una deregolazione della crescita come fattore di trascrizione: in effetti alcuni lavori hanno dimostrato che topi transgenici, che iperesprimono la proteina X, possono sviluppare tumori (v. Kim e altri, 1991); il possibile ruolo indiretto della proteina X nella genesi di questi tumori sembra invece confermato dall'osservazione che anche topi transgenici che iperesprimono la forma più grande dell'antigene di superficie dell'HBV sviluppano tumori (v. Chisari e altri, 1989). In quest'ultimo caso il ruolo indiretto di HBV consiste nell'indurre un insulto epatico immuno-mediato, che a sua volta provoca la rigenerazione epatocellulare in grado di espandere il pool delle cellule a rischio per lesioni genetiche addizionali: cellule con appropriate mutazioni possono allora andare incontro all'espansione clonale e, infine, progredire verso lo stadio di epatocarcinoma.
5. Virus di Epstein-Barr. - Il virus di Epstein-Barr (EBV) fu scoperto in seguito a studi su un linfoma riscontrato in bambini dell'Africa tropicale, già da tempo identificato; nel 1958 esso fu però descritto da Dennis Burkitt come un'entità a sé stante, con proprie caratteristiche cliniche, patologiche ed epidemiologiche. Già nei suoi studi iniziali Burkitt (v., 1958 e 1962) considerò la possibilità che il linfoma - data la sua distribuzione geografica in una fascia che attraversava l'Africa equatoriale, identificabile con l'area di distribuzione del virus della febbre gialla - fosse indotto da un virus. Nel 1964 Epstein e Barr descrissero particelle virali della famiglia degli Herpesvirus in cellule linfoblastoidi di pazienti affetti da linfoma di Burkitt (BL; v. Epstein e Barr, 1964; v. Epstein e altri, 1964). La presenza di particelle virali in cellule linfoidi non risultò tuttavia limitata a tessuti provenienti da pazienti con BL: essa, infatti, fu dimostrata anche in linee cellulari di pazienti con altri tipi di tumori o affetti da mononucleosi infettiva e addirittura in alcuni individui sani. Ciò nonostante EBV è stato il primo virus al quale sia stato attribuito il ruolo di virus oncogeno umano.
EBV è un virus contenente DNA a doppio filamento appartenente alla famiglia degli Herpesvirus umani, della quale fanno parte anche HSV-1 e HSV-2, il virus della varicella-zoster, il citomegalovirus e i tre Herpesvirus 6, 7 e 8, identificati di recente. La particella di EBV è essenzialmente indistinguibile dalle particelle virali degli altri virus erpetici: sono tutti virus piuttosto grandi, che misurano 150-180 nm di diametro, il cui patrimonio di informazione genetica è contenuto in un genoma di DNA a doppio filamento di circa 170.000 paia di basi, con uno strato capsidico fatto di capsomeri di forma icosaedrica e uno strato esterno di lipoproteine. L'EBV, a causa del suo tropismo per le cellule linfoidi sia in vivo che in vitro, è considerato un membro degli Herpesvirus γ, che generalmente infettano solo i linfociti B o T, e che comprendono fra gli altri anche il virus di Marek dei polli, nonché due Herpesvirus che infettano le scimmie, ateles e saimiri.
La complessa organizzazione del genoma di EBV è caratterizzata dalla presenza di regioni con sequenze nucleotidiche ripetitive e un numero variabile di copie in tandem (da 6 a 12) di un'unità di 500 paia di basi localizzate nella porzione terminale. Quando il virus è allo stato latente si trova in forma circolare nel nucleo delle cellule: si pensa che la capacità del genoma virale a circolarizzare richieda la ricombinazione omologa attraverso queste sequenze di DNA terminale ripetute. La determinazione della completa sequenza nucleotidica del DNA di EBV (v. Bear e altri, 1984) ha consentito l'identificazione delle ORF e dei geni relativi, e ha permesso di operare nell'ultimo decennio una ‛dissezione' molecolare del virus.
Nelle cellule infettate e trasformate da EBV sono stati identificati e descritti vari antigeni. Nelle cellule che producono particelle virali si trovano gli antigeni capsidici (VCA; v. Hummel e Kieff, 1982) e gli antigeni di membrana (MA); questi ultimi sono antigeni tardivi, in quanto la loro espressione si verifica dopo la sintesi del DNA nel ciclo vitale del virus; agli antigeni di membrana è attribuita la responsabilità di stimolare la produzione degli anticorpi neutralizzanti. Nell'infezione produttiva i primi geni espressi, quelli ‛immediatamente precoci', stimolano l'espressione dei geni successivi o ‛geni precoci'; particolarmente studiato è stato il gene immediatamente precoce BZLF-1, un attivatore della trascrizione che può interrompere la latenza facendo iniziare l'espressione di geni virali necessari per la replicazione virale. Gli antigeni definiti ‛antigeni precoci indotti' (EA) sono sintetizzati prima dell'inizio della replicazione del DNA. Tutte le cellule contenenti il genoma di EBV allo stato latente esprimono un antigene del nucleo, l'antigene nucleare indotto da EBV (EBNA; v. Reedman e Klein, 1973) che, come è stato dimostrato, consiste di un gruppo di almeno 6 proteine codificate dall'EBV: due di queste proteine, EBNA-2 e LMP-1, svolgono un ruolo importante nei processi di latenza virale e di immortalizzazione dei linfociti umani. LMP-1, una delle proteine di membrana latenti (LMP), attraversa la membrana 6 volte e contiene sia il carbossi-terminale che l'ammino-terminale nel citoplasma; essa assomiglia alla proteina P-170 implicata nel meccanismo della resistenza ai farmaci antiblastici (Multidrug Resistance, MDR).
Il linfoma di Burkitt africano - una neoplasia maligna dei linfociti B che si sviluppa molti anni dopo l'infezione primaria con EBV - è un linfoma monoclonale (a differenza della mononucleosi infettiva, che è una malattia policlonale causata da EBV) caratterizzato dalla rapida crescita in siti non linfoidi, come la mandibola o il retroperitoneo, e costituito da elementi cellulari morfologicamente simili alle cellule presenti nei follicoli linfoidi normali. Nei campioni bioptici prelevati da portatori africani è sempre dimostrabile il genoma dell'EBV e la positività per EBNA, mentre soltanto nel 15-20% dei casi di BL non africano è presente il genoma dell'EBV.
La constatazione che l'EBV è un virus comune (più del 90% della popolazione mondiale ne è stata infettata) ha indotto a ritenere che la concentrazione del BL nella fascia equatoriale dell'Africa orientale non possa essere correlata soltanto all'infezione da EBV, e che nella genesi del linfoma possano svolgere un ruolo importante anche disordini del sistema immunitario, come l'iperstimolazione dovuta alla malaria, endemica in quelle regioni. In individui che provengono da questa regione, in effetti, è stata dimostrata una diminuzione dell'attività T citotossica, mentre è proprio la risposta T citotossica all'infezione da EBV che limita la proliferazione delle cellule B da questo stimolata: è stato quindi ipotizzato che tale meccanismo possa rappresentare il primo stadio di un processo che favorisce altre mutazioni, la trasformazione neoplastica e la linfomagenesi. Le cellule dei linfomi di Burkitt presentano sempre alterazioni cromosomiche, spesso in regioni che contengono i geni delle immunoglobuline, in particolare nei cromosomi 2, 14 e 22: in oltre il 90% dei linfomi è dimostrabile una traslocazione cromosomica del braccio lungo del cromosoma 14, che codifica le catene pesanti delle immunoglobuline, sul cromosoma 8, che contiene l'oncogene c-myc (v. Manolov e Manolova, 1972); traslocazioni meno frequenti interessano il cromosoma 2 (catene leggere k) e il cromosoma 22 (catene leggere λ), ma anche queste comportano in genere una traslocazione reciproca sul braccio distale del cromosoma 8, che contiene l'oncogene c-myc (v. Leder e altri, 1983). Si pensa che a questa traslocazione faccia seguito una espressione deregolata dell'oncogene c-myc, probabilmente dovuta alla vicinanza di tale oncogene ai controlli trascrizionali dei geni delle immunoglobuline (v. Erikson e altri, 1982; v. Leder e altri, 1983). Klein ha proposto il seguente meccanismo per spiegare il ruolo dell'EBV nell'eziologia del BL africano: la prima tappa consisterebbe in un'immortalizzazione dei linfociti B in seguito all'infezione primaria; la seconda nella proliferazione delle cellule B EBV-positive, facilitata, nelle zone geografiche dove il BL è endemico (probabilmente a causa della malaria), dalla stimolazione delle cellule B e dalla soppressione delle cellule T che sono coinvolte nel controllo della proliferazione delle cellule infettate da EBV. Il pool delle cellule infettate da EBV aumenterebbe così di dimensioni e funzionerebbe da popolazione di cellule bersaglio per riarrangiamenti cromosomici a caso; la terza e ultima tappa consisterebbe nella traslocazione reciproca che interessa un locus deputato alla sintesi delle immunoglobuline e l'oncogene c-myc per lo sviluppo del clone maligno e la comparsa della massa tumorale (v. Klein, 1979).
Correlato con EBV è anche il carcinoma naso-faringeo (NPC; v. Henle e Henle, 1985), che si sviluppa in genere in adulti di età compresa tra 20 e 50 anni, ma in alcune parti dell'Africa anche in età infantile. Benché i tassi mondiali di questo tumore siano relativamente bassi, vi sono aree della Cina (come le province meridionali) nelle quali l'incidenza della neoplasia è di 10 casi per 100.000 abitanti per anno. Sono stati chiamati in causa alcuni fattori ambientali, come l'ingestione di fumi, l'azione di sostanze chimiche, il fumo di sigaretta e l'assunzione di pesce affumicato. Genomi di EBV sono dimostrabili nelle cellule epiteliali in quasi tutte le biopsie di NPC provenienti da tutto il mondo (v. Raab-Traub e altri, 1987), e nei pazienti con NPC sono presenti elevati livelli di anticorpi contro gli antigeni capsidici virali e contro gli EA. In effetti, l'associazione tra EBV e NPC fu suggerita per la prima volta proprio sulla base di evidenze sierologiche di livelli anticorpali più alti contro le proteine virali (v. Old e altri, 1966), tanto che la diagnosi precoce di questo tumore mediante test sierologici potrebbe essere di utilità nelle zone dove NPC è endemico, rendendo possibili interventi terapeutici in una fase precoce di sviluppo del tumore.
c) Virus oncogeni a RNA (Retrovirus).
I virus oncogeni a RNA rappresentano la categoria di virus oncogeni di gran lunga più numerosa, comprendente anche virus oncogeni umani. Sono stati isolati virus a RNA da ratti, cavie, criceti, bovini e recentemente anche da Rettili e da Mammiferi superiori (Primati). Poiché, a differenza di quanto si osserva nel caso dei virus Papova e degli Adenovirus, ne è evidente l'associazione causale diretta con alcuni tumori ‛spontanei' degli animali, questi virus per anni sono stati l'oggetto di studi sempre più numerosi. Negli animali i virus a RNA provocano fondamentalmente leucemie e sarcomi, con l'eccezione dell'MMTV (virus del tumore mammario murino) che provoca un tumore di natura epiteliale. A differenza dei Papovavirus, degli Adenovirus e degli Herpesvirus, i virus a RNA non determinano la lisi delle cellule infettate: difatti sono capaci di infettare le cellule e di replicarsi, fuoriuscendone senza provocare fenomeni citopatici di rilievo; oppure, sono capaci di indurre solo la trasformazione cellulare senza che a questa si accompagni replicazione virale.
I virus a RNA sono particelle di circa 100 nm di diametro, consistenti di una parte interna, il nucleocapside, e di una doppia membrana di rivestimento. Si moltiplicano nel citoplasma della cellula ospite (ma il loro ciclo comprende una fase nucleare) e vengono riversati nell'ambiente extracellulare per gemmazione.
1. Virus animali. - I virus oncogeni a RNA possono essere classificati in vari modi, in base alla specie che infettano, alla loro morfologia, o alle alterazioni patologiche che inducono. Secondo il primo di questi criteri, si distinguono virus aviari, murini, felini, bovini e virus dei Primati. In base poi alla loro morfologia, quale appare al microscopio elettronico, si distinguono quattro classi di particelle virali, A, B, C, D: le particelle A rappresentano forme immature intracellulari, verosimilmente precorritrici delle particelle C; nelle particelle B il nucleocapside si trova in posizione eccentrica rispetto alle membrane; le particelle C sono le forme extracellulari mature del virus, il cui nucleocapside è in posizione centrale; le particelle D sono tipiche di alcuni virus di scimmia.
A seconda delle alterazioni patologiche che producono negli animali suscettibili, i virus oncogeni a RNA possono poi essere classificati in virus cronici, acuti e apatogeni. I virus cronici sono caratterizzati da una lunga fase di latenza tra l'infezione e il manifestarsi della malattia: negli animali delle varie specie che possono essere infettate da questi virus (scimmie, polli, topi, gatti, ecc.) e che sono quindi cronicamente portatori di virus (viremici), le neoplasie indotte appaiono dopo parecchi mesi. Analisi delle cellule infettate effettuate con enzimi di restrizione del DNA rivelano, nonostante gli animali siano cronicamente portatori di virus, che queste neoplasie sono di origine clonale. Caratteristica dei virus cronici è quella di infettare un certo numero di cellule bersaglio oltre quella che darà poi origine al tumore; pertanto, la tumorigenicità sembra essere una rara conseguenza dell'infezione a livello cellulare, anche se la possibilità teorica che un animale sviluppi la leucemia è molto alta. L'oncogenesi indotta dai virus cronici sembra essere un processo a più stadi, fenomeno tipico dei tumori di origine non virale.
Sono stati spesso isolati, da animali che presentavano linfomi o leucemie, virus della leucemia con bassa potenzialità oncogena, in grado di replicarsi in differenti tipi cellulari, sia in vivo che in vitro, senza causare trasformazione cellulare e contenenti nel loro genoma tutti i geni responsabili per la replicazione. Questi virus sono capaci di trasformare solo una piccola parte delle loro specifiche cellule bersaglio (rappresentate per lo più da cellule di derivazione linfatica) e, dopo ricombinazione con il genoma delle cellule ospiti, possono generare virus derivati o difettivi.
I virus della leucemia murina sono stati isolati e caratterizzati nel corso di studi condotti su particolari ceppi puri di topi: alcuni di questi erano stati selezionati perché presentavano un'alta incidenza di leucemia spontanea nei primi mesi di vita (ceppi AKR, o C58), altri perché presentavano leucemia allo stadio adulto (tipo Balb/c, C57 BL, C3 H/He), altri ancora (tipo NIH SWISS) perché non presentavano alcun tipo di leucemia. Gross (v., 1951) trovò che gli estratti filtrati di cellule leucemiche di topi AKR ad alta incidenza di leucemia potevano trasmettere leucemia a topi neonati del ceppo che presentava una bassa frequenza di leucemia i quali, essendo immunologicamente immaturi, non erano in grado di innescare una reazione di rigetto né nei confronti dei virus né delle cellule tumorali: da questa prima serie di esperimenti fu isolato un virus chiamato virus di Gross, o virus AKR, e successivamente fu possibile correlare la differente frequenza di produzione spontanea di virus leucemogeno nella milza o nel timo del topo AKR. Le cellule bersaglio della leucemia sono quelle del timo, come risulta evidente non solo dalla dimostrazione che le prime lesioni spontanee dei topi AKR hanno localizzazione timica, ma anche dal fatto che la rimozione del timo alla nascita è in grado di prevenire lo sviluppo della leucemia. Gross dimostrò inoltre che nei ceppi ad alta incidenza di leucemia non è necessario, per provocare la malattia, infettare gli animali subito dopo la nascita: infatti le uova di tali ceppi, impiantate nell'utero di una femmina di ceppo a bassa incidenza leucemica, sviluppano ugualmente individui con leucemia di tipo T. È evidente che questa malattia viene trasmessa verticalmente. In seguito furono isolati altri virus di tale tipo, come quello della leucemia di Moloney, che è molto più virulento del virus di Gross. Appartengono a questa categoria molti virus aviari, alcuni dei quali inducono leucemie di tipo B. Leucemie causate da virus cronici simili a quelli aviari o murini sono riscontrabili in svariate specie; quello responsabile della leucemia felina, a differenza degli altri, si trasmette orizzontalmente, cioè mediante contagio tra gatti infetti.
I virus acuti, in grado di causare tumori maligni dopo pochi giorni dall'infezione, inducono varie specie di leucemie o di tumori solidi negli animali: tipici rappresentanti di questa categoria sono i virus del sarcoma di Kirsten e del sarcoma di Rous, che possono indurre tumori di 10 mm di diametro dopo solo una settimana. Questi virus, poiché contengono nel loro genoma un frammento di materiale genetico proveniente dalla cellula ospite che conferisce loro l'elevato potere di indurre neoplasie, sono anche definiti virus ad alto potenziale oncogeno. In molti casi l'incorporazione del genoma virale nel materiale genetico cellulare è accompagnata dalla perdita di geni virali che sono essenziali per la replicazione: i virus acuti sono quindi difettivi, incapaci di replicarsi autonomamente ma in grado di farlo in presenza di un virus helper, appartenente cioè alla classe dei virus cronici. Sono inoltre in grado di trasformare le cellule in coltura.
Tra i virus murini, quello di Friend produce un'eritroleucemia con foci nella milza e un ingrossamento sia della milza che del fegato; il virus di Rausher, che produce una neoplasia simile, può infettare un maggior numero di ceppi murini; il virus di Abelson, isolato a partire dal virus della leucemia di Moloney, produce linfomi di tipo B e trasforma anche fibroblasti in vitro. Tra i ceppi aviari, il virus dell'eritroblastosi aviaria (AEV) e il virus della mielocitomatosi (MC29) producono questi tipi di leucemia quando inoculati nei polli; alcuni però sono capaci di produrre anche sarcomi (AEV) o carcinomi del fegato o del rene (MC29). Questi virus, a differenza di quelli cronici, presentano in vivo la proprietà di infettare un ampio spettro di cellule, una maggiore capacità di trasformazione e un periodo di latenza molto più breve tra infezione e induzione della neoplasia. Quasi tutti i virus acuti possono essere titolati in vitro mediante un saggio di ‛formazione dei foci' su cellule suscettibili. I virus che infettano le cellule del sistema ematopoietico presentano una specificità per determinati stadi della differenziazione ematopoietica.
Ceppi di questi virus contengono di solito due tipi di particelle e quindi due RNA di differente grandezza: quello appartenente al virus difettivo, trasformante, e quello appartenente al virus helper della classe dei virus cronici non difettivi, il quale fornisce le funzioni essenziali per la replicazione. Le cellule trasformate che non producono virus sono cellule infettate solamente dalla componente difettiva, ma non contengono il virus helper. La separazione delle proteine dei virus a RNA mediante elettroforesi su gel di poliacrilammide in dodecilsolfato di sodio consente di distinguere almeno 7 principali specie proteiche nei virus murini e 8 negli aviari. Le proteine vengono designate con la lettera ‛p' seguita dalle prime due cifre del peso molecolare in dalton: così, nel caso dei virus murini, si distinguono una proteina p30 (p.m. 30.000), quantitativamente preponderante, una p15, una p12, una p10, e almeno due proteine a peso molecolare più elevato contenenti glicidi, denominate gp (glicoproteine) 69 e 71. Circa la funzione dei vari componenti proteici, la proteina p30 possiede la proprietà cosiddetta di antigene gruppo-specifico, ha cioè struttura identica per tutti i tipi di virus murini, tanto del sarcoma che della leucemia. La proteina virale di gran lunga più importante, la trascrittasi inversa, non ben distinguibile all'analisi elettroforetica, è una polimerasi che utilizza l'RNA virale come stampo: quella dei virus aviari, la meglio studiata, risulta costituita da una subunità α (p.m. 65.000) e una subunità β (p.m. 110.000), quella dei virus murini è invece costituita da una sola unità di p.m. 70.000 caratterizzata anche da un'attività ribonucleasica H, capace cioè di degradare gli ibridi DNA-RNA. La reazione di polimerizzazione presenta essenzialmente le seguenti fasi: 1) sintesi di un filamento unico di DNA (single stranded, ssDNA) sullo stampo dell'RNA 70S virale, il quale inoltre contiene anche molecole di RNA 4S che vi sono legate in maniera non covalente e agiscono da innesco (primer) della reazione di sintesi del DNA; 2) il ssDNA, mentre è ancora associato all'RNA 70S, serve ora da stampo per la sintesi del DNA bicatenario (double stranded, dsDNA), cosicché in tempi brevi il prodotto della reazione consiste di ssDNA, dsDNA e ibridi DNA-RNA; 3) il DNA sintetizzato, oltre a essere legato all'RNA di stampo con ponti idrogeno, è anche attaccato covalentemente al 3′ terminale di una piccola molecola di RNA primer (4S tRNA); 4) alla fine della reazione vi sarà prevalentemente dsDNA e poco ssDNA, mentre gli ibridi DNA-RNA saranno stati degradati dalla ribonucleasi H presente nella stessa DNA-polimerasi. La molecola a doppio filamento di DNA lineare viene trasportata nel nucleo della cellula ospite e qui circolarizzata e successivamente integrata nel genoma come provirus (v. fig. 14).
Tutti i virus oncogeni a RNA capaci di riprodursi (tipo cronico) posseggono tre geni che codificano per differenti proteine virioniche: il gene gag (group-specific antigen gene), che contiene l'informazione per quattro proteine strutturali non glicosilate le quali costituiscono i cosiddetti antigeni gruppo-specifici del virus (p12, p10, p30, p15); il gene pol (polimerasi), che contiene l'informazione per la sintesi dell'enzima trascrittasi inversa (DNA-polimerasi RNA-dipendente); il gene env (envelope), che contiene le informazioni per le glicoproteine di membrana (gp 69/71, gp 15E). Queste tre funzioni sono necessarie e sufficienti per la replicazione e l'espressione virale e sono sempre presenti nel genoma dei virus capaci di replicarsi, come appunto i virus cronici. Analisi dettagliate del genoma dei virus di tipo acuto hanno dimostrato: 1) che essi perdono una parte dei geni gag, pol, env, cosicché per replicarsi necessitano della presenza di un virus competente per la replicazione, detto virus helper, ma conservano invece inalterate le regioni genomiche alle estremità 5′ e 3′, dette LTR (long terminal repeats, lunghe sequenze terminali ripetute), necessarie per l'attività regolatrice sia dell'espressione che della replicazione virale; 2) che per questi motivi i virus di tipo acuto hanno genoma più piccolo di quello dei virus competenti per la replicazione, dai quali derivano; 3) che al posto delle sequenze eliminate vi è di solito una sequenza non correlata con il genoma dei virus helper, che viene oggi denominata oncogene; 4) che tale sequenza è omologa a un gene unico presente nel DNA cellulare dell'animale da cui il virus è stato isolato, identificabile in un normale gene eucariotico.
Numerose osservazioni in vivo e in vitro, condotte con virus mutanti per delezione o sensibili alla temperatura, hanno provato che responsabili della trasformazione neoplastica delle cellule infettate sono le sequenze acquisite dei virus oncogeni a RNA di tipo acuto, sequenze definite oncogene per diverse ragioni: 1) sono associate a virus capaci di trasformare le cellule in coltura e indurre rapidamente tumori nell'animale infettato; 2) non sono necessarie per la replicazione virale e non sono correlate con le sequenze presenti nei genomi dei virus di tipo cronico competenti per la replicazione, che non hanno geni trasformanti; 3) nei casi in cui è stato possibile effettuare questo studio, è risultato che le mutazioni che alterano la capacità trasformante del virus risiedono proprio in queste regioni.
Come già si è detto, nella maggioranza dei casi l'acquisizione di sequenze cellulari che servono poi come sequenze trasformanti virali comporta la perdita di regioni virali necessarie per la replicazione (gene pol, parte del gene env), per cui i virus acuti, per potersi replicare, cosa che in genere sono incapaci di fare autonomamente, richiedono l'aiuto del virus helper: l'unica eccezione è costituita dal virus del sarcoma di Rous, che è competente per la replicazione e possiede anche una sequenza capace di causare una trasformazione cellulare sia in vivo che in vitro (oncogene src). È ragionevole ipotizzare che i virus di tipo acuto abbiano avuto origine da un evento di ricombinazione tra il DNA della cellula ospite e il DNA di un provirus di tipo cronico: questo evento di ricombinazione per delezione avrebbe portato un normale gene cellulare sotto il controllo virale, permettendone l'espressione e la traduzione a livelli elevati.
Da differenti virus trasformanti di tipo acuto sono state isolate indipendentemente numerose sequenze codificanti l'oncogene, differenti le une dalle altre e in grado di codificare proteine con proprietà piuttosto diverse tra loro, pur provocando tutte lo stesso effetto, cioè la trasformazione di una cellula normale in una cellula maligna; a ognuna di queste sequenze è stato attribuito un nome diverso, in genere derivato dal virus acuto che la contiene. Le sequenze virali codificanti l'oncogene vengono indicate con il prefisso v (per es., v-onc, v-src, v-mos); le loro omologhe cellulari sono indicate dalla stessa sigla preceduta dalla lettera c (per es., c-onc, c-src, c-mos). Il virus dell'eritroleucemia di Friend è l'unico virus di tipo acuto che non possiede nel suo genoma sequenze trasformanti omologhe a geni cellulari.
La struttura della maggior parte delle proteine trasformanti espresse dai virus di tipo acuto è ibrida, caratterizzata dalla presenza all'estremità NH2-terminale di una sequenza di origine virale che deriva da una parte del gene gag o dai geni gag e pol, e all'estremità COOH-terminale delle sequenze derivate dall'oncogene; altre proteine trasformanti, invece, vengono codificate interamente dalla sequenza trasformante e non posseggono sequenze derivate dai geni virali non trasformanti.
Le proteine ibride generate dalla fusione delle sequenze di geni trasformanti e non trasformanti sono state maggiormente studiate, in quanto possono essere rivelate anche mediante immunoprecipitazione con anticorpi rivolti contro le proteine strutturali del gene gag. Un'analisi dettagliata delle proteine trasformanti dei Retrovirus acuti verrà fornita nel seguito (v. cap. 4).
2. Retrovirus umani. - Il primo isolamento di un retrovirus umano, l'HTLV-I (Human T-cell Leukemia Virus tipo I), è stato ottenuto nel 1980 nel laboratorio di R. C. Gallo a Bethesda, negli Stati Uniti (v. Poiesz e altri, 1981). Il virus non ha cross-reattività antigenica né omologia di sequenza con i Retrovirus animali, a eccezione del virus della leucemia bovina (BLV) con il quale è abbastanza correlato, e non è dimostrabile con i metodi convenzionali. Un contributo importante per la sua scoperta è stata l'individuazione dell'interleuchina-2 (IL-2), che ha permesso la coltura in vitro per lunghi periodi delle cellule T mature utilizzate per la trasmissione del virus. Ben presto è stata dimostrata una correlazione eziologica tra il virus HTLV-I e una forma di leucemia dell'adulto, descritta per la prima volta in Giappone nel 1977 (v. Uchiyama e altri, 1977) e denominata Adult T-cell Leukemia-lymphoma (ATL; v. fig. 15): clusters del virus che provoca la malattia, caratterizzata da particolare aggressività e dalla abituale associazione con ipercalcemia, si osservano nelle isole sudoccidentali del Giappone e tra gli immigrati di colore dei Caraibi. Screenings per anticorpi anti-HTLV-I hanno consentito di dimostrare la diffusione del virus anche in Africa, in America Centrale e Meridionale e negli Stati Uniti, particolarmente negli immigrati di colore dei Caraibi; alta frequenza di infezione da HTLV-I è stata anche riscontrata nei membri di una stessa famiglia. La storia clinica del paziente dal quale è stato isolato il virus HTLV-I è stata allora rivalutata: non si trattava, come si pensava in un primo momento, di micosi fungoide né della sindrome di Sézary, ma di ATL. Studi di infettività in vitro hanno dimostrato il tropismo del virus per i linfociti T4 (marker CD4) che, in seguito a infezione, divengono immortalizzati. Parecchie caratteristiche delle cellule leucemiche vengono acquisite dalle cellule infettate in vitro, come l'indipendenza da IL-2. Alti livelli del recettore per IL-2 sono presenti sulle cellule infettate, così come sulle cellule tumorali.
Un altro retrovirus umano, denominato HTLV-II, è stato isolato da un paziente affetto da hairy cell leukemia (v. sangue: Leucemie, vol. VI), malattia che pure interessa le cellulle T4 (v. Kalayanaraman e altri, 1982). Il virus HTLV-II è stato clonato (v. Gelmann e altri, 1984): il pattern di restrizione enzimatica è risultato essere completamente differente da quello dell'HTLV-I. Tuttavia, esiste un'omologia di sequenza nucleotidica nella regione 3′ dei due genomi virali. È stato possibile isolare il virus HTLV-II anche da un paziente emofiliaco trasfuso con il fattore VIII e, nel 1986, da un differente caso di hairy cell leukemia.
La determinazione delle sequenze nucleotidiche complete di HTLV-I e HTLV-II ha consentito di dimostrare che le strutture dei due genomi sono simili a quelle dei classici Retrovirus cronici, cioè contengono i geni gag, pol ed env. In questi due virus umani esiste inoltre una regione di 1,6 kb tra il gene env e la sequenza LTR all'estremità 3′, denominata inizialmente regione X, che codifica fattori di trans-regolazione coinvolti nell'attivazione della trascrizione e nel trasporto di mRNA dal nucleo al citoplasma della cellula ospite, e due geni regolatori detti tax e rex. A differenza dei geni onc dei Retrovirus di tipo acuto queste sequenze non hanno omologia con geni umani e pertanto non sembra che possiedano le caratteristiche tipiche dei geni onc virali. La più grande ORF, il gene pol, codifica una proteina di 99 kb con una omologia di sequenza tra i due virus del 61% nella regione della polimerasi e dell'82% nella regione della ribonucleasi H. Il gene env codifica una proteina precursore, prodotto di traduzione di un mRNA di 3,5 kb, la cui proteolisi dà origine a due glicoproteine, dette gp45 e gp21: tra HTLV-I e HTLV-II esiste un'alta omologia di sequenze in questa regione e in un'altra di circa 1.000 nucleotidi nella porzione 3′ terminale. Il gene tax funziona da gene principale per l'attivazione della trascrizione a partire dalla LTR virale; il gene rex è coinvolto nel trasporto di molecole di RNA messaggeri specifici dal nucleo al citoplasma. Il prodotto del gene tax funziona incrementando l'attività trascrizionale del promotore virale situato nella LTR attraverso elementi che rispondono alla stimolazione indotta dal prodotto di tax (TRE, Tax Responsive Elements). Questi elementi TRE, d'altra parte, sono anche siti di legame per fattori di trascrizione cellulari, attivati in conseguenza della loro interazione con il prodotto del gene tax, il quale inoltre attiva la trascrizione di geni cellulari specifici, incluso il gene per l'interleuchina-2. La localizzazione intracellulare della proteina tax è nucleare. Un meccanismo attraverso il quale l'HTLV-I indurrebbe la proliferazione e l'immortalizzazione cellulare potrebbe coinvolgere un circuito autocrino attraverso la stimolazione sia di IL-2 che del suo recettore. L'attivazione di geni cellulari mediata da tax potrebbe coinvolgere anche meccanismi paracrini.
4. Oncogeni
a) Introduzione e cenni storici.
L'origine degli studi sugli oncogeni (geni dotati di attività oncogena) risale all'identificazione, in circa una ventina di Retrovirus acuti, di sequenze nucleotidiche di dimensioni variabili, non contenenti informazioni necessarie per la replicazione né per la protezione esterna dei Retrovirus e quindi presuntivamente interpretabili come sequenze responsabili dell'attività trasformante dei Retrovirus stessi. Il prototipo di questi Retrovirus è il virus del sarcoma di Rous, identificato da Peyton Rous come l'agente virale responsabile dell'insorgenza di un sarcoma nei polli (v. Rous, 1911), e il prototipo degli oncogeni retrovirali è src, così denominato per indicare la sequenza nucleotidica presente nel genoma del virus del sarcoma di Rous direttamente responsabile dell'insorgenza del sarcoma nei polli e dell'induzione della trasformazione maligna in fibroblasti di pollo coltivati in vitro (v. Stehelin e altri, 1976).
I risultati ottenuti con il gene src promossero gli studi volti ad analizzare tutti gli altri geni virali trasformanti (detti in questi casi genericamente geni onc od oncogeni) presenti nei Retrovirus acuti fino a quel momento isolati: così dal virus del sarcoma murino di Moloney venne isolato l'oncogene mos (da Mo-loney s-arcoma; v. Tronick e altri, 1979); dai virus dei sarcomi murini di Kirsten e di Harvey, rispettivamente K-ras (v. Tsuchida e altri, 1982) e H-ras (v. Chang e altri, 1980); dal virus della mielocitomatosi aviaria il gene myc (v. Duesberg e Vogt, 1979) e da quello della mieloblastosi aviaria il gene myb (v. Klempnauer e altri, 1982); dal virus del sarcoma della scimmia il gene sis (da si-mian s-arcoma; v. Robbins e altri, 1981); e così via, dando origine a una nomenclatura fantasiosa che richiama il nome dello scopritore del virus, o l'abbreviazione della patologia indotta, o entrambi.
Analogamente a quanto osservato a proposito del virus del sarcoma di Rous, anche questi altri oncogeni virali presentavano una controparte, per così dire, normale in tutte le cellule di animali superiori esaminati, donde la definizione di oncogeni per i geni virali (v-onc) e di protoncogeni od oncogeni cellulari (c-onc) per i geni presenti nelle cellule. A quell'epoca cominciava a essere chiaro che, al di là dell'omologia di sequenza dimostrata con esperimenti di ibridazione molecolare, dovessero esistere differenze importanti tra le due classi di geni, i quali in un caso presidiavano funzioni in cellule normali e in altri casi erano responsabili della trasformazione neoplastica. Era altresì chiaro - dal momento che i protoncogeni si ritrovavano senza grosse variazioni non solo in tutti gli organismi superiori, ma anche in quelli via via inferiori nella scala zoologica fino ai lieviti - che le loro funzioni dovessero essere importanti.
All'epoca di queste scoperte (fine anni settanta-inizi anni ottanta) giunse a immettere nuova linfa negli studi sugli oncogeni una serie di risultati ottenuti con una tecnica particolare, detta di trasfezione, messa a punto alcuni anni prima. Mediante questa tecnica, consistente nel mettere a contatto DNA ad alto peso molecolare isolato da tumori umani con fibroblasti di topo NIH 3T3, fu possibile dimostrare la presenza di geni trasformanti, e quindi di oncogeni, nel DNA umano (v. Shih e altri, 1979). Derivava da questi studi, quindi, la nozione nuova che le cellule di organismi superiori in condizioni normali posseggono i protoncogeni, cioè i geni necessari per i processi di proliferazione e differenziazione normali, e in condizioni patologiche, quali i tumori, anche una copia dello stesso gene alterato, od oncogene. La presenza di oncogeni, quindi, non è una peculiarità di alcuni virus oncogeni, ma è proprietà comune a molte cellule neoplastiche.
Con le tecniche di biologia molecolare fu possibile dimostrare l'appartenenza degli oncogeni isolati dalla grande maggioranza dei tumori umani a una famiglia di oncogeni già presente nei Retrovirus, in particolare ai geni ras (v. Parada e altri, 1982). In molti casi di tumori umani, tuttavia, venivano via via isolate sequenze di DNA oncogeniche che non presentavano omologie con oncogeni virali noti (v. Burk e altri, 1989). Derivava dall'insieme di queste osservazioni la nozione che la maggior parte dei protoncogeni cellulari era stata trasdotta da Retrovirus acuti oncogeni e che piccole o importanti modificazioni apportate a queste sequenze avevano quindi dato origine agli oncogeni virali. Tuttavia, modificazioni del tutto simili a quelle riscontrate negli oncogeni virali possono verificarsi a carico dei protoncogeni cellulari, senza che questi vengano trasdotti dai Retrovirus, ma per effetto di meccanismi normalmente attivi nei tumori. Infine, è possibile che alcuni oncogeni riscontrati nei tumori umani non siano mai stati trasdotti da Retrovirus e che pertanto siano dimostrabili soltanto con la tecnica della trasfezione (v. Burk e altri, 1989).
Poiché con l'introduzione di una o più copie del gene alterato (oncogene) in presenza di due copie del gene corrispondente normale (protoncogene) esistente nei fibroblasti NIH 3T3 di topo è possibile indurre il fenotipo neoplastico in tali cellule, agli oncogeni è stata attribuita la proprietà di geni dominanti. È importante notare che, poiché sono presenti nel DNA di tutti gli ordini di Vertebrati, queste sequenze trasformanti durante l'evoluzione debbono essere state fortemente conservate ed ereditate da individuo a individuo come un qualsiasi carattere mendeliano.
b) Classificazione degli oncogeni sulla base delle funzioni dei loro prodotti.
Lo studio delle sequenze trasformanti ha fornito ai ricercatori la possibilità di analizzare quali alterazioni biochimiche si manifestino nel processo di trasformazione di una cellula normale in una cellula neoplastica, e in che modo i prodotti degli oncogeni possano avviare tale processo.
I risultati conseguiti da queste indagini, volte a precisare la localizzazione cellulare dei prodotti degli oncogeni e le loro funzioni in condizioni normali, sono stati resi possibili dall'impiego delle tecnologie del DNA ricombinante e di antisieri specifici, ma anche, e soprattutto, da una buona dose di fortuna. All'inizio di questi studi si pensava che sarebbe stato abbastanza semplice, una volta identificati i prodotti degli oncogeni, individuare nella cellula tumorale le alterazioni a essi correlate. In pratica, le cose si sono rivelate molto più complesse: l'osservazione che gli oncogeni possiedono la capacità di modificare drammaticamente la crescita cellulare e l'evidenza che tutti i protoncogeni sono stati perfettamente conservati durante l'evoluzione suggeriscono che nelle cellule normali questi geni possano rivestire funzioni importanti nel controllo della crescita. È noto, infatti, che la maggior parte dei protoncogeni studiati codifica proteine implicate nei complessi meccanismi che regolano la trasmissione del segnale, che hanno cioè ruoli nelle vie che portano impulsi mitogenici dall'esterno della cellula all'interno del citoplasma, fino al nucleo. Fino a oggi sono stati isolati molti oncogeni, ma le funzioni normali di molti di questi sono ancora sconosciute; tuttavia, gli studi in questo campo sono attivissimi e hanno già permesso di realizzare notevoli progressi.
In seguito al chiarimento della funzione del prodotto del gene src, negli ultimi dieci anni è risultato chiaro che i prodotti degli oncogeni e dei protoncogeni sono elementi di una rete di segnali cellulari diversi, da ligandi extracellulari e fattori di crescita, a proteinchinasi, a GTPasi e proteine leganti il GTP (guanosintrifosfato), a fattori di trascrizione nucleari (v. Cantley e altri, 1991). Si possono quindi classificare gli oncogeni e i protoncogeni sulla base delle funzioni dei loro prodotti: nella tab. V la loro classificazione è ordinata secondo il sito di azione dei prodotti che essi esprimono, da quelli che agiscono al di fuori della cellula a quelli che agiscono al suo interno.

Gli oncogeni della prima classe codificano proteine che si comportano in modo analogo ai fattori di crescita: il prodotto di sis, ad esempio, è identico alla catena β del PDGF (Platelet-Derived Growth Factor), e quello di hst appartiene alla famiglia degli FGF (Fibroblast Growth Factors). È forse anche possibile, ma non ancora dimostrato, che essi stessi siano fattori di crescita (come int-1). È probabile che queste oncoproteine agiscano attraverso un circuito autocrino stimolando la crescita cellulare.
Nella seconda classe sono compresi geni che codificano proteine con attività tirosinachinasica. Un certo numero di tali oncogeni codifica forme mutate di recettori di membrana per fattori di crescita noti (per esempio, il gene v-erb-B-1 codifica una forma mutata del recettore per l'EGF, Epidermal Growth Factor). Queste oncoproteine trasformano inviando un segnale mitogeno continuo indipendente dalla presenza del ligando. A questa classe appartengono anche oncogeni come src, che codificano proteine aventi attività tirosinachinasica ma prive di attività recettoriale: si tratta di proteine mutate, localizzate nella parte interna della membrana cellulare. Una delle tirosinachinasi della famiglia delle proteine prodotte da src, pp56lck, si lega alla porzione citoplasmatica delle molecole di superficie CD4 e CD8 presenti sui linfociti T. Si pensa che pp56lck funzioni da subunità catalitica di queste proteine di superficie delle cellule T, che viene attivata in seguito al legame del ligando a CD4 e CD8. È probabile che tutte le tirosinachinasi della famiglia di src abbiano funzioni analoghe, e che le versioni mutate di queste proteine mandino un segnale continuo piuttosto che un segnale regolato dalla presenza del ligando. Esperimenti di mutagenesi effettuati sul segnale di miristilazione situato in posizione N-terminale di queste proteine, segnale richiesto per l'adesione delle proteine alla membrana, hanno dimostrato che la sua abolizione elimina anche l'attività trasformante.
Alla terza classe appartiene l'oncogene mas, il cui prodotto è un'oncoproteina ad attività recettoriale per l'angiotensina ma priva di attività tirosinachinasica.
Anche la quarta classe di oncogeni codifica proteine mutate associate con la parte interna della membrana citoplasmatica, ma si tratta in questo caso di proteine aventi attività di legame per il GTP e GTPasica. Appartengono a questa classe gli oncogeni ras, che hanno subito mutazioni che li rendono attivi in maniera costitutiva mantenendo le proteine nello stato di legame con il GTP.
Alla quinta classe - che comprende oncogeni che codificano oncoproteine a localizzazione citoplasmatica, per la maggior parte solubili, ad attività serina/treonina chinasica - appartengono 4 oncogeni: mos, cot, pim-1 e raf. Il prodotto di c-raf è associato con i recettori attivati di PDGF e di EGF e, poiché se ne è dimostrata la traslocazione nel nucleo dopo uno stimolo, sembra che il suo ruolo sia quello di trasmettere il segnale dalla membrana al nucleo. La perdita della sequenza N-terminale dell'oncogene raf, che si ritiene abbia una funzione regolatrice, porterebbe a un'attivazione permanente di questo gene.
Nella sesta classe è compreso l'oncogene crk, che codifica proteine con attività citoplasmatica contenenti regioni SH2 e SH3 (v. sotto, § c).
La settima classe di oncogeni codifica proteine nucleari, molte delle quali sono fattori di trascrizione, e in almeno tre casi (jun, fos e myb) questi geni hanno mutazioni che conducono alla perdita degli elementi regolatori negativi. È interessante notare che molti di questi oncogeni corrispondono a geni cellulari che vengono rapidamente indotti quando cellule a riposo sono trattate con mitogeni (per es., myc, jun, fos e rel). Si presume che questi geni codifichino proteine necessarie per iniziare la cascata di eventi che porta la cellula dalla fase G1 alla fase S (v. cellula: Fisiologia della cellula, vol. I) e che la loro espressione costitutiva debba pertanto indurre le cellule a entrare continuamente in ciclo: in effetti, in molti sistemi cellulari l'espressione del gene c-myc è repressa quando le cellule differenziano ed escono dal ciclo, per cui si ritiene che la sua espressione continuata sia responsabile della trasformazione neoplastica di queste cellule.
Esistono almeno altri due tipi di oncoproteine citoplasmatiche, codificate da dbl e bcl-2. L'oncogene dbl è stato isolato per la prima volta da un linfoma B-linfocitico diffuso: esso codifica una proteina la cui funzione sembra consistere nella capacità di agire da exchange factor per proteine simili alla proteina prodotta da ras. Data l'omologia tra la porzione C-terminale dell'oncogene dbl e il gene che codifica la proteina CDC 24 del lievito, avente una funzione stimolatrice del ciclo cellulare, è stata avanzata l'ipotesi che dbl sia l'omologo nelle cellule superiori del gene cdc24 del lievito, ma gli studi sono ancora lontani dall'aver chiarito questo punto (v. Ron e altri, 1991).
Nei paragrafi che seguono verranno riportati alcuni esempi di prodotti di oncogeni, cercando di chiarirne il ruolo funzionale nella cellula normale e, soprattutto, l'importanza nei meccanismi di cancerogenesi.
c) Il gene src e la famiglia delle tirosinachinasi.
Il virus del sarcoma di Rous, uno dei primi virus oncogeni isolati, si è rivelato uno dei più interessanti dal punto di vista sia genetico, sia biochimico: è l'unico virus oncogeno a RNA che possiede un oncogene ed è contemporaneamente in grado di replicarsi autonomamente. L'isolamento delle sue sequenze trasformanti specifiche, v-src, ne ha consentito l'impiego come sonda molecolare: fu subito evidente che questa sonda v-src poteva ibridare con il DNA di tutti i Vertebrati (v. Stehelin e altri, DNA related..., 1976).
Il paragone tra la sequenza codificante del virus e il gene presente nel DNA dei polli dimostra che le due sequenze sono praticamente identiche. Tuttavia si possono notare due differenze importanti: 1) il gene portato dal virus non ha introni, dal che si deduce che nel processo di trasduzione di sequenze cellulari nell'interno del genoma virale gli introni vengono eliminati, e poiché questo fenomeno si verifica anche per gli altri oncogeni virali, deve esistere un meccanismo comune di trasduzione dal DNA cellulare al genoma virale; 2) gli ultimi 12 amminoacidi presenti nella porzione COOH-terminale del prodotto dell'oncogene virale sono differenti da quelli presenti nel prodotto dell'oncogene cellulare: infatti il gene cellulare c-src è più lungo di sette amminoacidi rispetto a v-src, e ciò dimostra che la trasduzione di queste sequenze cellulari porta a eventi di ricombinazione, riarrangiamenti e mutazioni che modificano la struttura originaria del gene, fenomeno questo che, d'altra parte, si verifica in quasi tutti i virus oncogeni a RNA che contengono oncogeni.
Il momento successivo più importante nella storia di src è stata la scoperta di un antisiero in grado di reagire specificamente con il prodotto proteico dell'oncogene, ottenuto da conigli portatori di un tumore indotto dal virus del sarcoma di Rous. Il prodotto del gene src, immunoprecipitato da questo antisiero, è la proteina SRC, localizzata al livello della parte interna della membrana citoplasmatica e con un peso molecolare di circa 60.000 dalton. Poiché mediante marcatura delle cellule con fosforo radioattivo 32P è stato dimostrato che sia il prodotto dell'oncogene virale v-src sia quello del gene cellulare c-src sono fosforilati, la proteina SRC viene comunemente indicata con la sigla pp60 (phosphoprotein 60 kDa) o pp60src. Di particolare importanza è risultata l'osservazione che questa proteina è non solo responsabile della propria fosforilazione (autofosforilazione), ma è anche in grado di fosforilare le immunoglobuline presenti nell'antisiero utilizzato per immunoprecipitarla. In studi successivi, grazie all'analisi dei siti fosforilati, è stato possibile dimostrare che ambedue le proteine, normale e trasformante, erano fosforilate nei residui di tirosina (v. Hunter e Sefton, 1980) e identificare quindi in questo gene una nuova proprietà enzimatica. Prima della scoperta dell'attività di src si conoscevano enzimi cellulari in grado di fosforilare in treonina e in serina, ma non in tirosina, cosicché i ricercatori furono sorpresi di trovare questo tipo di attività in un prodotto genico con capacità trasformante. La capacità trasformante della proteina codificata da v-src risiede nella sua attività fosforilante: infatti, in mutanti temperatura-sensibili l'attività della tirosinachinasi è ridotta alla temperatura alla quale non trasformano, mentre è elevata nelle cellule trasformate alla temperatura cosiddetta ‛permissiva per la trasformazione'.
Il prodotto pp60 del gene cellulare c-src è espresso a bassi livelli nelle cellule normali. Queste osservazioni hanno costituito la prima dimostrazione dell'esistenza e dell'espressione di oncogeni in cellule normali; ma la malignità del prodotto del gene virale non risiede solo nei suoi elevati livelli di espressione: infatti, quando il gene c-src viene inserito in una cellula normale sotto il controllo di forti promotori che ne elevano l'espressione a livelli simili a quelli virali, ma non ne fanno aumentare l'attività chinasica, la cellula non risulta trasformata. È quindi evidente che l'aumento in vivo dell'attività chinasica della proteina SRC, importante per la trasformazione di una cellula normale, deve dipendere da qualche altra mutazione. La scoperta che una proteina, come pp60src, possiede una funzione di chinasi ha destato un grande interesse, dal momento che la fosforilazione proteica è uno dei meccanismi di regolazione cellulare di maggior importanza. La fosforilazione, infatti, può indurre effetti pleiotropici nelle cellule e sembra quindi ben correlata con la presenza di una proteina capace di provocare le molteplici modificazioni associate al passaggio da uno stato normale a uno neoplastico.
Quando furono isolati altri oncogeni o dai virus oncogeni a RNA o dal DNA cellulare, si è potuto constatare che alcuni di questi, come erb-B-1, abl, fes, fgr, fms, yes e altri, erano capaci di esprimere proteine con funzioni di tirosinachinasi, ognuna delle quali rappresenta il prodotto di un singolo, distinto gene cellulare che mappa su di un preciso locus in un determinato cromosoma. Anche l'analisi della loro sequenza nucleotidica rivela le differenze tra i vari geni, le cui omologie appaiono limitate alla codificazione di una piccola regione della proteina che possiede l'attività enzimatica tirosinachinasica. Questo gruppo di oncogeni che codificano funzioni biochimiche simili viene definito della ‛famiglia delle tirosinachinasi', e si può dividere in due sottogruppi: quelli che codificano proteine con funzioni di recettori di membrana (v. sotto, § d) e quelli che codificano chinasi citoplasmatiche, il cui prototipo è la pp60src che è anche certamente la tirosinachinasi maggiormente caratterizzata (v. tab. VI). Tutte le proteine oncogeniche ad attività tirosinachinasica hanno omologia di sequenza in una zona di 300 amminoacidi (a volte interrotta), denominata ‛dominio tirosinachinasi' (v. anche neuroscienze: Basi molecolari della comunicazione neuronale, vol. XI).
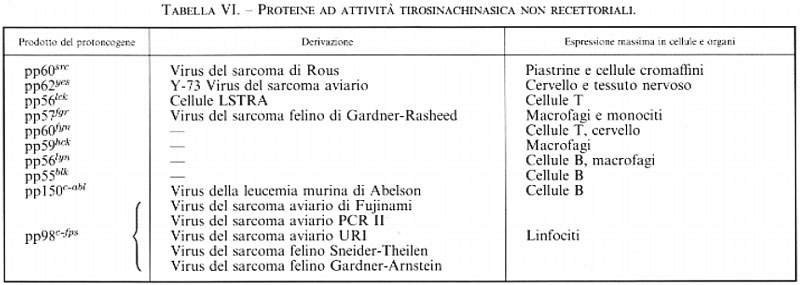
La famiglia delle proteine SRC possiede altre regioni di omologia che non si trovano nella famiglia delle tirosinachinasi recettoriali, e includono una corta sequenza ammino-terminale per l'aggiunta di acido miristico e due regioni denominate SH2 e SH3 (SRC Homology regions, regioni di omologia con SRC). Tali regioni sono state dimostrate anche in altre proteine che non hanno attività tirosinachinasica, come la fosfolipasi C-γ (PLC-γ), rasGAP e la fosfatidilinositolo-3-chinasi (PI-3-chinasi). Tutte le proteine di questo gruppo si associano con altre proteine con funzione tirosinachinasica attivate. I membri della famiglia SRC hanno bassa attività chinasica, e pur non avendo un dominio extracellulare, sono attivati da fattori di crescita e da altri attivatori.
d) Un oncogene che codifica un recettore di membrana: erb-B-1.
Benché si conoscano molti fattori di crescita, il PDGF e i membri della famiglia dell'FGF rimangono gli unici esempi finora trovati di fattori di crescita omologhi a prodotti di oncogeni. Tuttavia una cellula può essere trasformata da normale in maligna non solo per azione diretta dei fattori di crescita, ma anche per meccanismi agenti sui recettori con i quali tali fattori interagiscono. Infatti, altri oncogeni sono stati posti in relazione con i recettori per fattori di crescita, i quali, se alterati nel livello di espressione o nella loro struttura, acquistano potenziale trasformante tanto da poter essere stimolati anche in assenza del ligando.
Con l'impiego di adeguati sistemi computerizzati è stato possibile dimostrare gli alti livelli di omologia tra il prodotto dell'oncogene v-erb-B-1, isolato dal virus della eritroblastosi aviaria, e la sequenza amminoacidica derivata dal recettore per il fattore di crescita dell'EGF-R (Epidermal Growth Factor-Receptor). In questo caso la trasduzione del gene cellulare ha portato all'eliminazione di una parte del gene trasdotto, il cui prodotto, di conseguenza, è un recettore con struttura alterata a livello sia della parte NH2-terminale sia di quella COOH-terminale della molecola. Il recettore per l'EGF possiede tre dominî funzionali: 1) una porzione extracellulare altamente glicosilata che lega il fattore di crescita; 2) una sequenza corta localizzata nella membrana; 3) un dominio citoplasmatico che possiede attività di tirosinachinasi, nonché una tirosina sito di autofosforilazione. Il prodotto codificato dall'oncogene v-erb-B-1 perde una gran parte della regione ammino-terminale, così che non è più capace di legare l'EGF, e presenta alterazioni anche nella parte COOH-terminale che è all'interno del citoplasma: quindi questa molecola potrebbe funzionare come recettore continuamente attivato anche in assenza di ligando. È interessante notare che la sequenza responsabile dell'attività tirosinachinasica è omologa al dominio con identica funzione del prodotto del gene src; ma, a differenza del recettore per l'EGF, pp60src è localizzata interamente nell'interno della cellula a livello della faccia interna della membrana citoplasmatica e non ha corrispondente dominio transmembrana o extracellulare.
Un altro oncogene con struttura simile è erb-B-2, detto anche neu, che codifica una glicoproteina transmembrana di peso molecolare di 185 kDa con attività tirosinachinasica. L'oncogene erb-B-2, isolato per la prima volta in una linea di carcinoma mammario ove è stato trovato iperespresso, si ritrova frequentemente amplificato a livello di DNA e anche iperespresso a livello di RNA e di proteina in tumori mammari umani per i quali spesso viene considerato come marker di una maggiore aggressività della neoplasia.
La tab. VII presenta un elenco degli oncogeni più importanti aventi attività tirosinachinasica con funzione recettoriale, insieme con l'elenco dei rispettivi ligandi extracellulari. La stimolazione cellulare con fattori di crescita che legano e attivano recettori ad attività tirosinachinasica può avviare una serie di risposte cellulari condizionate dalla struttura recettoriale e dalla natura della cellula: l'attivazione di tirosinachinasi, quale si verifica a opera dell'insulina, può indurre proliferazione e differenziazione cellulare o successiva attivazione di vie metaboliche; l'iperespressione di tirosinachinasi recettoriali o di loro mutazioni può provocare la crescita tumorale; la perdita della funzione di tirosinachinasi può portare a malattie, quali il diabete insulino-resistente, il piebaldismo e il morbo di Hirschsprung. Per questi motivi i meccanismi attraverso i quali le tirosinachinasi recettoriali attivano le vie di trasduzione del segnale sono di grandissimo interesse sia per comprendere il controllo della crescita normale, il metabolismo e lo sviluppo, sia per individuare alterazioni nella trasduzione del segnale che si verificano in una cellula con attività tirosinachinasica aberrante.
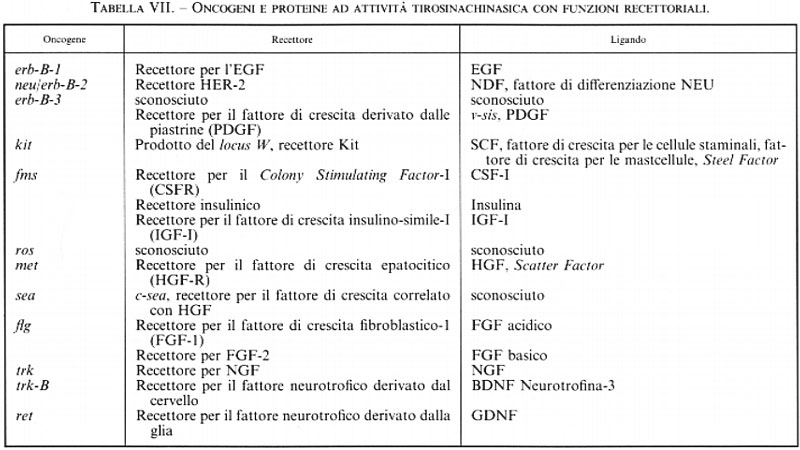
e) La famiglia degli oncogeni ras.
L'oncogene ras è stato scoperto per la prima volta come oncogene virale nei virus del sarcoma di Kirsten e del sarcoma di Harvey. Le sequenze di questi due oncogeni, K-ras e H-ras, localizzati su cromosomi differenti, non sono omologhe, mentre i loro prodotti genici sono virtualmente identici. Questi stessi geni sono stati isolati mediante trasfezione del DNA di vari tumori umani. Infatti i geni della famiglia ras sono quelli che vengono riscontrati più frequentemente dopo trasfezione di DNA tumorale umano.
Dal DNA di cellule di neuroblastoma si è potuto identificare, mediante esperimenti di trasfezione, un altro oncogene simile a questi, detto N-ras: questo gene ras-simile mappa su di un altro cromosoma. Alla famiglia degli oncogeni ras appartengono anche alcuni pseudogeni presenti nel genoma cellulare, ma non trascritti.
Il prodotto degli oncogeni H-ras e K-ras è la proteina p21: si tratta di una proteina di tipo G, di peso molecolare 21.000 dalton, che appartiene, cioè, a quel gruppo di proteine capaci di legare e di idrolizzare il GTP; esso, infatti, agisce come una GTPasi, in quanto converte il GTP in GDP. Le proteine che legano e idrolizzano il GTP costituiscono una vasta famiglia con strutture primarie fortemente conservate durante l'evoluzione e possono essere considerate interruttori molecolari il cui stato di ‛acceso' o ‛spento' è dovuto al legame e all'idrolisi del GTP. Questa famiglia di proteine è coinvolta in numerose funzioni cellulari, come la sintesi di proteine ribosomiali, la mediazione di segnali transmembrana generati dagli ormoni e dalla luce, la traslocazione delle proteine nascenti nel reticolo endoplasmico e il controllo della differenziazione e della proliferazione cellulare (v. anche farmacologia molecolare, vol. X). La principale caratteristica delle proteine prodotte da ras capaci di provocare la trasformazione neoplastica consiste nella perdita della funzione GTPasica, in quanto, secondo l'ipotesi più accreditata, la mancata idrolisi del GTP mantiene la proteina in uno stato di costante attivazione: il legame del GTP, infatti, è certamente necessario per l'attivazione delle proteine G, e di conseguenza l'attività GTPasica è un elemento regolatore della proliferazione.
Un'altra caratteristica della proteina p21 codificata dal gene virale consiste nella sua attività autochinasica, essa può cioè trasferire un gruppo fosfato dal GTP su una treonina in posizione 59, mentre la proteina codificata dal gene c-ras ha un residuo di alanina nella posizione corrispondente e non possiede attività autochinasica: tuttavia, la presenza di una treonina al residuo 59 sembra non essere essenziale per la conversione del prodotto dell'oncogene in una proteina in grado di avviare il processo di trasformazione. I geni ras, in tutti i casi in cui siano stati trasdotti da virus a RNA, sono risultati integri, non troncati, ma costantemente contrassegnati da modificazioni nucleotidiche responsabili delle sostituzioni di amminoacidi, e quindi di alterazioni della struttura primaria della proteina virale. Mutazioni in siti specifici sono state rilevate anche nei geni ras isolati dai tumori umani mediante trasfezioni (v. Reddy e altri, 1982) e nei geni N-ras isolati da neuroblastoma.
Con il clonaggio molecolare dei geni ras, isolati dal DNA tumorale o dal DNA cellulare normale, è stato possibile dimostrare che quelli isolati dai tumori umani o sperimentali (quindi gli oncogeni ras) presentavano un'alterazione tipica della sequenza proteica codificata: infatti, al posto della glicina in posizione 12 e 13 e della glutammina in posizione 61, presenti nel prodotto del protoncogene, si trovavano altri amminoacidi (v. Reddy e altri, 1982). Sorprendentemente, un'analoga sostituzione era stata riscontrata negli amminoacidi in posizione 12 e 59 della proteina codificata dai geni ras isolati da Retrovirus (v. tab. VIII). Sia i risultati degli studi sui geni virali, che quelli ottenuti mediante trasfezione o da mutazioni sito-specifiche in vitro, hanno mostrato che le modificazioni nella proteina prodotta da ras all'altezza del residuo amminoacidico 12 o 13, oppure al residuo 61, portano invariabilmente a cambiamenti nel potenziale oncogeno del prodotto genico: la più probabile spiegazione di questo fenomeno è che queste mutazioni comportino alterazioni nella struttura tridimensionale della proteina, le quali, a loro volta, provocano una diminuita attività GTPasica (v. Watson e altri, 19874).
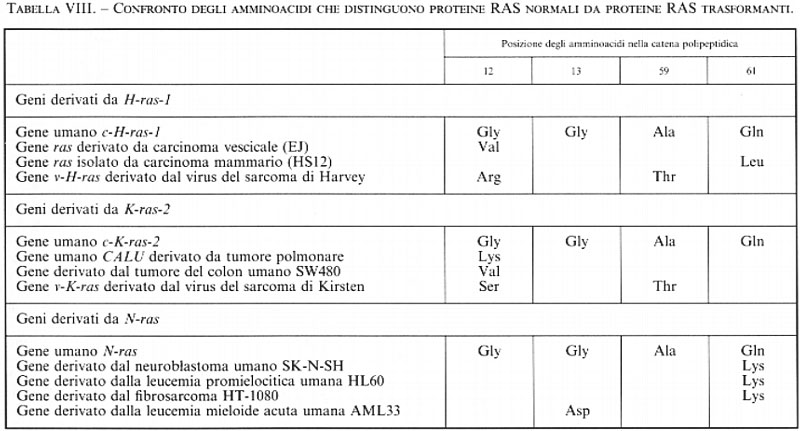
Nella specie Saccharomyces cerevisiae i prodotti dei geni ira (Inhibitors of Ras Activity) stimolano l'attività GTPasica intrinseca del prodotto dei geni ras di lievito e il prodotto del gene cdc25 fa aumentare il rilascio di GDP. Nelle cellule di Mammiferi i prodotti di due geni correlati con i geni ira, e cioè GAP (GTPase Activating Protein) e NF1 (prodotto del gene della neurofibromatosi di tipo 1), agiscono come regolatori negativi delle proteine prodotte da c-ras. Recentemente è stato clonato molecolarmente un cDNA che codifica una proteina rilasciante i nucleotidi guaninici, detta anche Guanine Nucleotide releasing Factor (GNF), la quale ha un peso molecolare di 140 kDa e la porzione carbossi-terminale omologa a quella del prodotto del gene cdc25 di lievito.
Poiché anticorpi anti-ras bloccano la crescita di fibroblasti trasformati da prodotti degli oncogeni della famiglia delle tirosinachinasi, ras si trova evidentemente a valle delle tirosinachinasi attivate.
f) Oncogeni con prodotti a localizzazione nucleare: fos, myc, myb, jun.
Poiché per far passare una cellula da uno stato di quiescenza a uno di proliferazione devono avvenire cambiamenti nell'espressione dei geni, è ragionevole supporre che l'obiettivo finale dell'azione degli oncogeni sia quello di indurre modificazioni nella trascrizione genica: infatti, parecchie proteine codificate da oncogeni agiscono direttamente nel nucleo, dove probabilmente controllano l'espressione di importanti geni preposti al controllo della proliferazione cellulare. Di solito le proteine di questo tipo sono capaci di legare il DNA e probabilmente agiscono come fattori che regolano la trascrizione (v. tab. IX). Una delle caratteristiche di questi oncogeni è che la loro espressione è altamente regolata: geni come fos, myc, rel o jun sono espressi a livelli piuttosto bassi nelle cellule quiescenti, ma i livelli di espressione aumentano molto in risposta a stimoli mitogenici; infatti, in varie neoplasie è stato messo in evidenza che oncogeni nucleari, come myc, sono soggetti ad alterazioni nei loro livelli di espressione o sono espressi in tempi non appropriati.
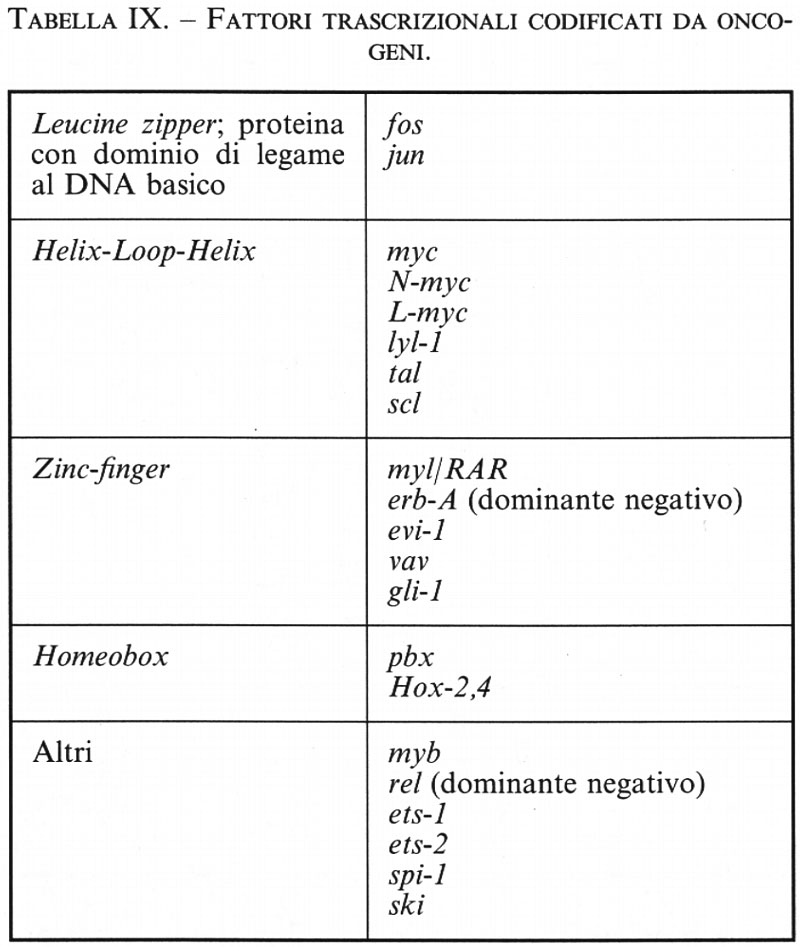
Il primo oncogene di questa famiglia è proprio l'oncogene myc, individuato come gene virale trasformante in quattro virus aviari, isolati indipendentemente, in grado di causare sarcomi, carcinomi o leucemie mieloidi; questo stesso oncogene, inoltre, si trova attivato nelle leucemie causate da virus oncogeni a RNA di tipo cronico, che non possiedono oncogeni, ed è coinvolto in alcune traslocazioni specifiche del linfoma di Burkitt (v. cap. 3, § b, punto 5).
Esistono altri due geni simili al gene myc: l'oncogene N-myc, che si trova frequentemente amplificato a livello di DNA nei retinoblastomi e nei neuroblastomi, e l'oncogene L-myc, trovato amplificato nei carcinomi polmonari a piccole cellule (microcitomi). L'espressione di questo gene, osservata in molti tipi cellulari, è di solito correlata a uno stato di elevata proliferazione. Nelle cellule quiescenti, indotte alla proliferazione mediante stimolo esterno, questo oncogene viene indotto dopo l'oncogene fos, ma prima della sintesi di DNA. È importante notare a questo proposito che il gene è costituito da tre esoni, di cui il primo non è tradotto: è stata avanzata l'ipotesi che la presenza di questo esone sia necessaria per una corretta espressione del gene, data la sua costante assenza in tutti i casi in cui il gene è stato trasdotto da virus oncogeni a RNA e ha quindi acquisito un potenziale trasformante.
L'oncogene fos è stato isolato da due virus oncogeni che provocano osteosarcomi nei topi, benché la sua espressione nel tessuto osseo sia molto bassa. Questo gene viene espresso molto rapidamente dalle cellule, in risposta ai più svariati stimoli esterni: la sua espressione è indotta dopo pochi minuti dallo stimolo e successivamente diminuisce. La regolazione di tale espressione, non mediata da nuova sintesi proteica, è stata studiata in dettaglio: sono stati individuati, nelle regioni di controllo a monte del gene, siti di attacco di fattori proteici regolatori, sia come attivatori che come repressori. Sembra che proprio la proteina codificata dal gene fos agisca come repressore della sua stessa trascrizione.
Un altro oncogene a localizzazione nucleare è l'oncogene jun, isolato dal virus del sarcoma aviario 17, il cui prodotto proteico è capace di legare il DNA in una regione specifica di circa 30 coppie di basi che ha proprietà di enhancer: questa sequenza nucleotidica è presente nelle regioni di controllo dell'espressione sia del virus SV40, sia di altri geni di Vertebrati, come il gene umano per la sintesi della metallo-tioneina. Dati recenti hanno potuto dimostrare che il prodotto dell'oncogene c-jun è identico a un fattore nucleare (AP-1) che, legandosi alle regioni specifiche del DNA summenzionate, stimola la trascrizione genica.
Analisi delle sequenze amminoacidiche delle proteine a localizzazione nucleare codificate da questi oncogeni hanno potuto mettere in rilievo particolari analogie di alcune regioni specifiche. Sono state identificate due piccole regioni proteiche (di circa 20 amminoacidi), importanti per il legame di queste proteine al DNA, una con prevalenza di amminoacidi basici e una, immediatamente successiva, con una particolare sequenza caratterizzata dall'alternanza di varie leucine ogni sette amminoacidi: questa struttura, detta leucine zipper, sembra essere necessaria all'interazione delle varie proteine nucleari. I due oncogeni nucleari fos e jun possono formare un complesso proteico che lega il DNA con maggiore efficienza, interagendo tra loro proprio attraverso la regione leucine zipper.
L'oncogene myb, isolato da due differenti virus della leucemia mieloide aviaria, codifica anch'esso una proteina a localizzazione nucleare e la sua espressione è stata correlata all'attivazione della sintesi di DNA: infatti si trovano livelli elevati di questo prodotto durante la fase G1 del ciclo cellulare.
5. Geni oncosoppressori o antioncogeni
a) Definizione.
I tumori si formano in conseguenza di alterazioni del controllo della proliferazione e della differenziazione cellulare, molte delle quali derivano da mutazioni che attivano i protoncogeni in oncogeni. Recenti osservazioni sperimentali hanno portato a definire un altro gruppo di geni direttamente coinvolti nel processo neoplastico, ma in modo totalmente diverso dagli oncogeni; dato il modo peculiare con cui contribuiscono allo sviluppo dei tumori, a tale gruppo è stato dato il nome di antioncogeni, oncogeni recessivi o geni oncosoppressori. Il termine di antioncogene, seppure impreciso, è ormai entrato nell'uso comune. A differenza degli oncogeni, che sono coinvolti nel processo neoplastico quando modificati nel senso di una attivazione, i geni oncosoppressori devono essere inattivati per consentire lo sviluppo di un tumore: infatti, essi sono presenti nel corredo genico di ogni cellula normale, ed è la loro inattivazione o addirittura delezione che fa sì che la neoplasia possa svilupparsi.
La scoperta dei geni oncosoppressori è stata resa possibile dallo studio di alcune neoplasie ereditarie delle quali ha contribuito a chiarire le cause, e successivamente è stato documentato il ruolo altrettanto importante che essi svolgono anche in tumori non ereditari. La scoperta dei geni oncosoppressori ha chiarito il meccanismo patogenetico di alcuni virus oncogeni a DNA, tra i quali il virus Polioma, l'SV40, gli Adenovirus e i Papillomavirus: si è visto infatti che gli oncogeni di questi virus interagiscono con alcune proteine prodotte dai geni oncosoppressori, inattivandone la funzione di repressori della proliferazione neoplastica e, quindi, indirettamente, favorendo la proliferazione stessa.
b) L'esempio paradigmatico del retinoblastoma.
Il retinoblastoma è una rara malattia neoplastica che colpisce 1 bambino su 20.000 e può manifestarsi in una forma ereditaria a insorgenza precoce, con tumori spesso bilaterali e multifocali, e in una forma sporadica a insorgenza più tardiva e in genere unilaterale e unifocale. La forma ereditaria è caratterizzata da una modalità di trasmissione di tipo autosomico dominante (il 50% dei figli di un genitore affetto è malato) con penetranza superiore all'80%.
Per spiegare l'insorgenza delle forme unilaterali e bilaterali del retinoblastoma ereditario A. Knudson (v., 1971), in uno studio che ne analizzava la frequenza relativa, formulò la teoria dei due hits, ossia dei due ‛colpi', confermata poi da numerose evidenze sperimentali: i due hits sono, secondo Knudson, due alterazioni genetiche in grado di provocare l'inattivazione di due alleli di un gene; nel caso del retinoblastoma ereditario, delle due mutazioni una verrebbe ereditata e l'altra acquisita (ossia si verificherebbe in una cellula retinica nel corso dello sviluppo). Viceversa, nella forma sporadica tutti e due gli alleli del gene bersaglio verrebbero inattivati a livello somatico (v. fig. 16).
A distanza di 15 anni dall'epoca della formulazione dell'ipotesi, giunsero le conferme sperimentali che avvalorarono completamente le osservazioni di Knudson, e il gene coinvolto, chiamato RB, fu isolato e caratterizzato (v. Friend e altri, 1986). L'identificazione di tale gene si è avvalsa di precedenti osservazioni citogenetiche che dimostravano come pazienti affetti da retinoblastoma ereditario presentassero costitutivamente, in tutte le cellule somatiche - quali per esempio i linfociti del sangue periferico - una delezione del braccio lungo di uno dei due cromosomi 13, in particolare della porzione 13q14 (v. Yunis e Ramsey, 1978). Utilizzando dei markers genetici si vide inoltre che le cellule tumorali di tali pazienti presentavano alterazioni anche della porzione corrispondente dell'altro cromosoma 13. È stato successivamente dimostrato che le alterazioni del secondo cromosoma possono essere mutazioni puntiformi, oppure microdelezioni, o ancora delezioni dell'intero cromosoma causate da non-disgiunzione mitotica, seguite o meno da duplicazione dell'altro cromosoma. Fu così possibile procedere al clonaggio molecolare e alla caratterizzazione del gene RB, localizzato appunto in posizione 13q14, il quale dimostrò di rispecchiare fedelmente le caratteristiche previste per il gene della predisposizione al retinoblastoma: era infatti espresso in tutti i tessuti normali ed eliminato, o comunque non più espresso, nelle cellule provenienti da tutti i casi studiati di retinoblastoma, sia ereditari che non. Dunque il gene RB è stato il primo esempio di gene oncosoppressore, ossia di un gene che funziona nel tessuto normale e la cui inattivazione comporta la trasformazione neoplastica.
La dimostrazione definitiva delle sue proprietà di gene soppressore del fenotipo neoplastico è stata poi ottenuta con esperimenti in vitro; è stato infatti dimostrato che l'introduzione sperimentale di una copia normalmente funzionante di RB in cellule di retinoblastoma comportava la reversione delle loro caratteristiche tumorali (v. fig. 17; v. Huang e altri, 1988).
L'analisi della sequenza nucleotidica del gene RB ha consentito di dimostrare che esso codifica una proteina di peso molecolare di 105 kDa, chiamata p105RB, che è localizzata nel nucleo e probabilmente in grado di esercitare una funzione di controllo sull'espressione genica. In particolare si è visto che tale proteina, quando è attiva, rappresenta un efficace inibitore della proliferazione cellulare: di conseguenza nei retinoblastomi, per la perdita di questa funzione, la crescita cellulare non è più controllata (v. Kaye e altri, 1990).
c) Altre sindromi tumorali ereditarie legate alla inattivazione di antioncogeni.
Il retinoblastoma non è l'unico caso di sindrome tumorale familiare in cui sia stato possibile identificare l'associazione con la delezione di una precisa regione cromosomica: utilizzando lo stesso approccio, infatti, si sta via via chiarendo l'origine di altri tipi di tumore trasmessi ereditariamente in maniera autosomica dominante (v. tab. X).
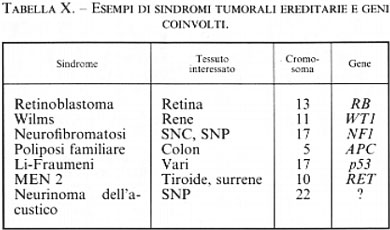
Il tumore di Wilms è un tumore embrionale del rene (nefroblastoma) che si può presentare in forma ereditaria o sporadica. In alcuni casi esso è parte di più complesse sindromi: la WAGR, in cui possono manifestarsi, oltre al tumore di Wilms, aniridia, anomalie genitourinarie e ritardo mentale; o la sindrome di Beckwith-Wiedemann, in cui il tumore di Wilms si associa ad altri disordini dello sviluppo. Sembra che nel caso del nefroblastoma di Wilms possano essere coinvolti più loci genici, almeno 3: per uno di questi, localizzato in posizione 11p13, è stato isolato il gene interessato, chiamato WT1 (v. Riccardi e altri, 1978), che codifica una proteina con probabile funzione di regolazione dell'espressione genica (contiene infatti delle sequenze zinc-finger, caratteristiche di proteine che legano il DNA; v. Call e altri, 1990; v. anche chimica bioinorganica, vol. X). Normalmente questa proteina è espressa nel corso dello sviluppo embrionale del rene, mentre è inattiva in alcuni tumori di Wilms.
Un altro esempio classico di tumore ereditario è la neurofibromatosi di tipo 1, descritta nel 1882 da von Recklinghausen, che, con la sua incidenza di 1 su 3.000, è uno dei più comuni disordini ereditari umani di tipo autosomico dominante. Si tratta di un complesso disordine proliferativo caratterizzato da macchie color caffellatte sulla cute, neurofibromi e tumori benigni del sistema nervoso centrale; raramente si osservano neurofibrosarcomi, tumori maligni delle cellule di Schwann. Il gene la cui espressione è responsabile della neurofibromatosi 1 è stato localizzato sul cromosoma 17, in posizione q11, ed è stato chiamato NF1 (v. Wallace e altri, 1990). Anche tale gene sembra essere coinvolto nei processi di controllo della proliferazione cellulare, in quanto interagisce con proteine che regolano la trasmissione dei segnali generati da fattori di crescita.
Infine, recentemente, è stata chiarita la patogenesi di un'altra complessa sindrome neoplastica ereditaria, descritta da Li e Fraumeni (v. Li e altri, 1988), caratterizzata da un alto rischio di sviluppo di una serie di neoplasie come sarcomi, tumori del cervello, carcinomi della mammella e del surrene: è stato dimostrato che un altro gene oncosoppressore chiamato p53, e localizzato sul braccio corto del cromosoma 17, è direttamente coinvolto in tale patogenesi.
d) Ulteriore scoperta di geni coinvolti anche nello sviluppo di tumori sporadici.
Lo studio di queste sindromi tumorali ereditarie è stato di estremo interesse non solo perché ha contribuito al chiarimento della loro patogenesi, ma anche perché ha portato alla scoperta e alla caratterizzazione dei geni oncosoppressori. Infatti, subito dopo la loro scoperta, numerosi gruppi di ricerca in tutto il mondo si sono chiesti se questi geni, o per lo meno meccanismi analoghi, potessero essere coinvolti anche nella patogenesi di forme neoplastiche più frequenti, quali il cancro del colon, del polmone e della mammella. È stato così dimostrato che il gene RB è frequentemente inattivato in sarcomi, in microcitomi del polmone e in carcinomi della mammella, mentre l'inattivazione del gene p53 è stata spesso dimostrata in carcinomi della mammella e nelle forme più maligne di cancro del colon.
Infine, le affinate metodologie genetiche impiegate nello studio dei tumori ereditari sono state successivamente applicate allo studio di tumori non ereditari, allo scopo di evidenziare l'inattivazione di antioncogeni non ancora descritti: il gruppo di Vogelstein ha così caratterizzato il gene DCC (Delected Colon Carcinoma; v. Vogelstein e altri, 1988), localizzato sul cromosoma 18, che viene eliminato nelle forme più avanzate di cancro del colon.
6. Conclusioni e prospettive
Il processo di cancerogenesi è sempre stato considerato un processo multistep, cioè a più stadi, soprattutto sulla base di osservazioni epidemiologiche. Negli ultimi anni è stato dimostrato che alcuni di questi stadi si verificano a livello genetico ed è iniziato il chiarimento delle basi molecolari di questo processo. I tumori deriverebbero dall'attivazione di alcuni geni, gli oncogeni, e dalla inattivazione di altri geni, gli antioncogeni, che funzionano nei tessuti normali. L'inattivazione di alcuni specifici membri di tale famiglia rappresenta l'unica alterazione presente in alcuni rari tumori ereditari. Successivamente si è visto che tali geni, che presumibilmente giocano un ruolo molto importante nel controllo della proliferazione e del differenziamento cellulare, sono implicati anche nella patogenesi di tumori con un più rilevante impatto epidemiologico, quali ad esempio i carcinomi del polmone, del colon e della mammella.
Queste osservazioni aprono nuove prospettive diagnostiche, prognostiche e terapeutiche. Avere a disposizione markers molecolari per valutare il coinvolgimento di tali geni nelle neoplasie umane significa, infatti, poter sfruttare metodiche estremamente sensibili per accertare la predisposizione a sviluppare tumori di tipo ereditario o il rischio di dare alla luce figli che, con alta probabilità, contrarranno uno di questi tumori.
Forse più proiettate verso il futuro, che comunque si spera sia non tanto remoto, sono invece le ambizioni di poter stabilire la prognosi di un paziente neoplastico a seconda di quali geni siano alternativamente attivati o inattivati. Si può addirittura prospettare la possibilità, una volta accertato che un tumore è legato all'inattivazione di uno specifico gene oncosoppressore, di reintegrarne la funzione soppressa.
BIBLIOGRAFIA
Bear, R., Bankier, A. T., Biggin, M. D. e altri, DNA sequence and expression of B95-8 Epstein-Barr virus genome, in ‟Nature", 1984, CCCX, pp. 207-211.
Beasley, R. P., Hwang, L. Y., Lin, C. C. e altri, Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan, in ‟The lancet", 1981, n. 2, pp. 1129-1132.
Blumberg, B. S., Alter, H. J., Visnich, S., A ‟new" antigen in leukemia sera, in ‟Journal of the American Medical Association", 1965, CXCI, pp. 541-546.
Burk, K. B., Liu, E. J., Larrick, J. N., Additional oncogenes, in Oncogenes (a cura di J. Cooper), New York-Berlin 1989, pp. 222-232.
Burkitt, D., A sarcoma involving the jaws in African children, in ‟British journal of surgery", 1958, XLVI, pp. 218-223.
Burkitt, D., Determining the climatic limitations of a children's cancer common in Africa, in ‟British medical journal", 1962, II, pp. 1019-1023.
Call, K. M., Glases, T., Ito, C. Y. e altri, Isolation and characterization of a zinc finger polypeptide gene at the human 11 Wilms' tumor locus, in ‟Cell", 1990, LX, pp. 509-520.
Cantley, L. C., Auger, K. R., Carpenter, C., Duckworth, B., Graziani, A., Kapeller, R., Soltoff, S., Oncogenes and signal transduction, in ‟Cell", 1991, LXIV, pp. 281-302.
Chang, H. W., Garon, C. F., Chang, E. H., Lowy, D. R., Hager, G. L., Scolnick, E. M., Repake, R., Martin, M. A., Molecular cloning of the Harvey sarcoma virus circular DNA intermediates. II. Further structural analyses, in ‟Journal of virology", 1980, XXXIII, pp. 845-855.
Chisari, F. V., Klopchin, K., Moriyama, T. e altri, Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice, in ‟Cell", 1989, LIX, pp. 1145-1156.
Colgrove, R., Simon, G., Ganem, D., Transcriptional activation of homologous and heterologous genes by the hepatitis B virus X gene product in cells permissive for viral replication, in ‟Journal of virology", 1989, LXIII, pp. 4019-4026.
Dane, D. S., Cameron, C. H., Briggs, M., Virus-like particles in serum of patients with Australia-antigen-associated hepatitis, in ‟The lancet", 1970, n. 1, pp. 695-698.
Dejean, A., Bougueleret, L., Grzeschik, K. H. e altri, Hepatitis B virus DNA integration in a sequence homologous to v-Erb-A and steroid receptor genes in hepatocellular carcinoma, in ‟Nature", 1986, CCCXXII, pp. 70-72.
Duesberg, P. H., Vogt, P. K., Avian acute leukemia viruses MC 29 and MH 2 share specific RNA sequences: evidence for a second class of transforming genes, in ‟Proceedings of the National Academy of Sciences", 1979, LXXVI, pp. 1633-1637.
Dulbecco, R., Production of plaques in monolayer tissue cultures by single particles of an animal virus, in ‟Proceedings of the National Academy of Sciences", 1952, XXXVIII, pp. 747-757.
Dunn, A. E., Ogilvie, M. M., Intranuclear virus particles in human genital wart tissue: observation on the ultrastructure of epidermal layer, in ‟Journal of ultrastructure research", 1968, XXII, pp. 282-295.
Eagle, H., Metabolic studies with normal and malignant human cells in the culture, in ‟Harvey lectures", 1960, LIV, pp. 156-175.
Epstein, M. A., Achong, B. G., Barr, Y. M., Virus particles in cultured lymphoblasts from Burkitt's lymphoma, in ‟The lancet", 1964, n. 1, pp. 702-703.
Epstein, M. A., Barr, Y. M., Cultivation in vitro of human lymphoblasts from Burkitt's malignant lymphoma, in ‟The lancet", 1964, n. 1, pp. 252-253.
Erikson, J., Finam, J., Nowell, P. C. e altri, Translocation of immunoglobulin VH genes in Burkitt lymphoma, in ‟Proceedings of the National Academy of Sciences", 1982, LXXIX, pp. 5611-5615.
Friend, S. H., Bernards, R., Rogelj, S. e altri, A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma, in ‟Nature", 1986, CCCXXIII, pp. 643-646.
Ganem, D., Varmus, H. E., The molecular biology of the hepatitis B viruses, in ‟Annual reviews of biochemistry", 1987, LVI, pp. 651-693.
Gelmann, E. P., Franchini, G., Manzari, V. e altri, Molecular cloning of a unique human T cell leukemia virus (HTLV-II Mo), in ‟Proceedings of the National Academy of Sciences", 1984, LXXXI, pp. 993-997.
Gey, G. O., Coffman, W. D., Kubieck, M. T., Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium, in ‟Cancer research", 1952, XII, pp. 264-265.
Gross, L., Pathogenic properties and vertical transmission of the mouse leukemia agent, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1951, LXXVIII, 78, pp. 342-348.
Henle, W., Henle, G., Epstein-Barr virus and human malignancies, in Advances in viral oncology (a cura di G. Klein), New York 1985, vol. V, pp. 201-238.
Hilleman, M. R., Werner, I. H., Recovery of new agents from patients with acute respiratory illness, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1954, LXXXV, pp. 183-188.
Huang, H. J. S., Yee, J. K., Shew, J. Y. e altri, Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells, in ‟Science", 1988, CCXLII, pp. 1563-1566.
Huebner, R. J., Rowe, W. P., Turner, H. C., Lane, W. T., Specific adenovirus complement fixing antigenes in virus-free hamster and rat tumors, in ‟Proceedings of the National Academy of Sciences", 1963, L, pp. 379-389.
Hummel, M., Kieff, E., Mapping of polypeptides encoded by the Epstein-Barr virus genome in productive infection, in ‟Proceedings of the National Academy of Sciences", 1982, LXXIX, pp. 5698-5702.
Hunter, T., Sefton, B. M., Transforming gene product of Rous sarcoma virus phosphorylates tyrosine, in ‟Proceedings of the National Academy of Sciences", 1980, LXXVII, pp. 1311-1315.
Kalayanaraman, V. S., Sarngadharan, M. G., Robert-Guroff, M. e altri, A new subtype of human T-cell leukemia virus (HTLV-II) associated with a T-cell variant of hairy cell leukemia, in ‟Science", 1982, CCXVIII, pp. 571-573.
Kaye, F. J., Kratzche, R. A., Gerster, J. L. e altri, A single aminoacid substitution results in a retinoblastoma protein defective in phosphorylation and oncoprotein binding, in ‟Proceedings of the National Academy of Sciences", 1990, LXXXVII, pp. 6922-6926.
Kenneway, E. L., Hieger, I., Carcinogenic substances and their fluorescence spectra, in ‟British medical journal", 1930, I, pp. 1044-1046.
Kim, C.-M., Koike, K., Saito, I. e altri, HBX gene of hepatitis B virus induces liver cancer in transgenic mice, in ‟Nature", 1991, CCCLI, pp. 317-320.
Klein, G., Lymphoma development in mice and humans. Diversity of initiation is followed by convergent cytogenetic evolution, in ‟Proceedings of the National Academy of Sciences", 1979, LXXVI, pp. 2442-2446.
Klempnauer, K. H., Gonda, T. J., Bishop, J. M., Nucleotide sequence of the retroviral leukemia gene V-myb and its cellular progenitor c-myo: the architecture of a transduced oncogene, in ‟Cell", 1982, XXXI, pp. 453-463.
Knudson, A. G. Jr., Mutation and cancer: statistical study of retinoblastoma, in ‟Proceedings of the National Academy of Sciences", 1971, LXVIII, pp. 820-823.
Leder, P., Battey, J., Lenoir, G. e altri, Translocation among antibody genes in human cancer, in ‟Science", 1983, CCXXII, pp. 765-771.
Li, F. P., Fraumeni, J. F. Jr., Mulvihill, J. J., Blattner, W. A., Dreyfus, M. G., Tucker, M. A., Miller, R. W., A cancer family syndrome in twenty-four kindreds, in ‟Cancer research", 1988, XLVIII, pp. 5358-5362.
Manolov, G., Manolova, Y., Marker band in one chromosome 14 from Burkitt lymphomas, in ‟Nature", 1972, CCXXXVII, pp. 33-34.
Marion, P. L., Oshiro, L., Regenery, D. C. e altri, A virus in Beechey ground squirrels that is related to hepatitis B virus of humans, in ‟Proceedings of the National Academy of Sciences", 1980, LXXVII, pp. 2941-2945.
Mason, W. S., Seal, G., Summers, J. W., Virus of Peking ducks with structural and biological relatedness to human hepatitis B virus, in ‟Journal of virology", 1980, XXXVI, pp. 829-836.
Old, L. J., Boyse, A. E., Ottgen, H. F. e altri, Precipitation antibody in human serum to an antigen present in cultured Burkitt's lymphoma cells, in ‟Proceedings of the National Academy of Sciences", 1966, LVI, pp. 1699-1704.
Parada, L. F., Tabin, C. J., Shih, C., Weiberg, R. A., Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma ras virus gene, in ‟Nature", 1982, CCXCVII, pp. 474-478.
Poiesz, B. J., Ruscetti, F. W., Reitz, M. S. e altri, Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sézary T-cell leukaemia, in ‟Nature", 1981, CCXCIV, pp. 268-271.
Puck, T. T., Marcus, P. I., Cieciura, S. J., Clonal growth of mammalian cells in vitro. Growth characteristics of colonies from single HeLa cells with and without a feeder layer, in ‟Journal of experimental medicine", 1956, CIII, pp. 273-284.
Raab-Traub, N., Flynn, K., Pearson, G. e altri, The differentiated form of nasopharyngeal carcinoma contains Epstein-Barr virus DNA, in ‟International journal of cancer", 1987, XXXIX, pp. 25-29.
Reddy, E. P., Reynolds, R. K., Santos, E., Barbacid, M., A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene, in ‟Nature", 1982, CCC, pp. 149-152.
Reedman, B. M., Klein, G., Cellular localization of an Epstein-Barr virus associated complement-fixing antigen in producer and non-producer lymphoblastoid cell lines, in ‟International journal of cancer", 1973, XI, pp. 499-520.
Riccardi, V. M., Sujansky, E., Smith, A. C., Francke, U., Chromosomal imbalance in the Aniridia-Wilms' tumor association: 11p interstitial deletion, in ‟Pediatrics", 1978, LXI, pp. 604-610.
Robbins, K. C., Devare, S. G., Aaronson, S. A., Molecular cloning of integrated simian sarcoma virus. Genome organization of infectious DNA clones, in ‟Proceedings of the National Academy of Sciences", 1981, LXXVIII, pp. 2918-2922.
Ron, D., Zannini, M. S., Lewis, M., Wickner, R. B., Hunt, L. T., Graziani, G., Tronick, S. R., Aaronson, S. A., Eva, A., A region of proto-dbl essential for its transforming activity shows sequence similarity to a yeast cell cycle gene, CDC24 and the human breakpoint cluster gene, bcr, in ‟The new biologist", 1991, III, pp. 372-379.
Rous, P., A sarcoma of the fowl transmissible by an agent separable from the tumor cells, in ‟Journal of experimental medicine", 1911, XIII, pp. 397-411.
Rowe, W. P., Huebner, R. I., Gilmore, L. K., Parrot, R. H., Word, T. G., Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture, in ‟Proceedings of the Society for Experimental Medicine", 1953, LXXXIV, pp. 570-573.
Sanford, K. K., Earle, W. R., Likely, G. D., The growth in vitro of single isolated tissue cells, in ‟Journal of the National Cancer Institute", 1948, IX, pp. 229-246.
Scherer, W. F., Syverton, J. T., Gey, G. O., Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix, in ‟Journal of experimental medicine", 1953, XCVII, pp. 695-709.
Seeger, C., Ganem, D., Varmus, H. E., Biochemical and genetic evidence for the hepatitis B virus replication strategy, in ‟Science", 1986, CCXXXII, pp. 477-484.
Shih, C., Shih, B. Z., Goldfarb, M. P., Donnenberg, A., Weinberg, R. A., Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin, in ‟Proceedings of the National Academy of Sciences", 1979, LXXVI, pp. 5714-5718.
Shope, R. E., A filterable virus causing tumor-like condition in rabbits and its relationship to virus myxomatosum, in ‟Journal of experimental medicine", 1937, LVIII, pp. 607-624.
Spandau, D., Lee, C., Trans-activation of viral enhancers by the hepatitis B virus X protein, in ‟Journal of virology", 1988, LXII, pp. 427-434.
Stehelin, D., Guntaka, R. V., Varmus, H. E., Bishop, J. M., Purification of DNA complementary to nucleotide sequences required for neoplastic transformation of fibroblasts by avian sarcoma viruses, in ‟Journal of molecular biology", 1976, CI, pp. 349-365.
Stehelin, D., Varmus, H. E., Bishop, J. M., Vogt, P. K., DNA related to the transforming genes of avian sarcoma viruses is present in normal avian DNA, in ‟Nature", 1976, CCLX, pp. 170-173.
Stewart, S. E., Eddy, B. E., Gochenour, A. M., Borgese, N. G., Grubbs, G. E., The induction of neoplasms with a substance released from mouse tumor by tissue culture, in ‟Virology", 1957, III, pp. 380-400.
Summers, J., Smolec, J. M., Snyder, R., A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks, in ‟Proceedings of the National Academy of Sciences", 1978, LXXXIV, pp. 4533-4537.
Tannock, J., Hill, R., The basic science of oncology, New York 19922 (tr. it.: Le basi scientifiche dell'oncologia, Milano 19922).
Tiollais, P., Pourcel, C., Dejean, A., The hepatitis B virus, in ‟Nature", 1985, CCCXVII, pp. 489-495.
Trentin, J. J., Yabe, Y., Taylor, G., The quest for human cancer viruses, in ‟Science", 1962, CXXXVII, pp. 835-841.
Tronick, S. R., Robbins, K. C., Canaani, E., Devare, S. G., Andersen, P. R., Aaronson, S. A., Molecular cloning of Moloney murine sarcoma virus: arrangement of virus-related sequences within the normal mouse genome, in ‟Proceedings of the National Academy of Sciences", 1979, LXXVI, pp. 6313-6318.
Tsuchida, N., Ryder, T., Ohtsubo, E., Nucleotide sequence of the oncogene encoding the p21 transforming protein of Kirsten murine sarcoma virus, in ‟Science", 1982, CCXVII, pp. 937-939.
Uchiyama, T., Yodoi, J., Sagawa, K. e altri, Adult T-cell leukemia: clinical and hematologic features of 16 cases, in ‟Blood", 1977, L, pp. 481-492.
Vogelstein, B., Fearon, E. R., Hamilton, S. R. e altri, Genetic alterations during colorectal tumor development, in ‟New England journal of medicine", 1988, CCCXIX, 9, pp. 525-532.
Wallace, M. R., Marchunck, D. A., Anderson, L. B. e altri, Type 1 Neurofibromatosis gene: identification of a large transcript disrupted in three NF-1 patients, in ‟Science", 1990, CCXLIX, pp. 181-186.
Wang, J., Chenivesse, X., Henglein, B. e altri, Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma, in ‟Nature", 1990, CCCXLIII, pp. 555-557.
Watson, J. D., Hopkins, N. H., Roberts, J. W., Steitz, J. A., Weiner, A. M., Molecular biology of the gene, Menlo Park, Cal., 19874, vol. II (tr. it.: Biologia molecolare del gene, Bologna 19894, vol. II).
Yunis, J. J., Ramsey, N., Retinoblastoma and sub-band deletion of chromosome 13, in ‟American journal of diseases of childhood", 1978, CXXXII, pp. 161-165.