
Genetica
GENETICA
Genetica di Marcello Siniscalco
Sommario: 1. Premessa. 2. Un decennio di scoperte dionisiache e progressi apollinei: a) la reazione polimerasica a catena (PCR) e il suo impatto sull'analisi della variabilità genetica; b) una vendemmia di polimorfismi genetici multiallelici; c) regioni cromosomiche instabili e variabilità genetica normale e patologica; d) nuovi strumenti a disposizione del ‛cacciatore di geni'. 3. Il Progetto Genoma: a) lo stato dell'arte: progressi e problemi; b) considerazioni etiche.
□ Bibliografia.
1. Premessa
Questo aggiornamento dei precedenti articoli genetica - pubblicati nei voll. III e VIII - si rende quanto mai necessario perché non è esagerato affermare che le conoscenze acquisite in questo campo nell'ultimo decennio superano di gran lunga quelle accumulate durante l'intero secolo che ci separa dalla riscoperta delle leggi di Mendel ai primi del Novecento. A determinare questa rapida evoluzione di conoscenze genetiche hanno contribuito essenzialmente tre circostanze, maturate appunto nel corso dell'ultimo decennio: 1) il lancio del Progetto Genoma per identificare la sequenza totale dei tre miliardi di basi nucleotidiche che compongono il bagaglio ereditario della nostra specie (v. tav. I); 2) la fortunata e casuale scoperta della reazione polimerasica a catena (PCR, Polymerase Chain Reaction); 3) la massiccia e diretta partecipazione del mondo degli affari alla ricerca competitiva di soluzioni biotecnologiche per i problemi medici, ecologici e industriali del nostro tempo (v. anche biotecnologie; v. genoma).
Albert Szent-Györgyi (premio Nobel per la scoperta della vitamina C) usava dire che il progresso della conoscenza umana è il risultato di scoperte ‟dionisiache" e di progressi ‟apollinei", ovvero di scoperte dell'imprevedibile - che ovviamente non possono essere oggetto di pianificazione - e dei continui progressi di informazione ottenuti attraverso l'applicazione sistematica di strategie sperimentali sempre più efficienti allo scopo di colmare le lacune delle nostre conoscenze attuali. La scoperta della PCR e il lancio del Progetto Genoma costituiscono validi esempi di come due modi così diversi di acquisire conoscenze risultino ambedue indispensabili all'evoluzione della scienza.
2. Un decennio di scoperte dionisiache e progressi apollinei
a) La reazione polimerasica a catena (PCR) e il suo impatto sull'analisi della variabilità genetica
Kary B. Mullis, che scoprì la PCR mentre era ricercatore della Cetus Corporation in California, racconta con comprensibile compiacimento le circostanze fortuite della sua scoperta. Da anni egli lavorava sulla sintesi di piccole catene di basi nucleotidiche, dette ‛oligonucleotidi', ottenute mediante l'enzima DNA-polimerasi (isolato da Arthur Kornberg a Stanford, negli Stati Uniti) secondo il classico protocollo dell'inglese Fred Sanger, generalmente usato fin dagli inizi degli anni settanta per il sequenziamento delle basi nucleotidiche del DNA (v. biotecnologie; v. genetica: Applicazioni della genetica). Per lungo tempo la strategia per l'isolamento di geni di potenziale importanza medica e farmacologica si era basata appunto sull'uso di miscele di sequenze oligonucleotidiche (dedotte dalla composizione amminoacidica dei prodotti finali di specifici geni, in base alla ben nota corrispondenza tra le sequenze nucleotidiche di un gene e quelle amminoacidiche della proteina da esso codificata) così da poter individuare e isolare il gene ricercato tra i numerosi frammenti di DNA presenti nelle cosiddette ‛librerie genomiche' di DNA umano. Si trattava tuttavia di una strategia tutt'altro che semplice, in quanto ogni singolo gene contenuto in una libreria genomica totale di DNA umano, ottenuta per digestione parziale, è frammentato in numerosi segmenti di DNA genomico di lunghezza e composizione nucleotidica diverse; per ricostruire la sequenza nucleotidica univoca del gene desiderato, ogni frammento deve essere pazientemente analizzato, senza che sia peraltro possibile evitare che porzioni di esso, fortuitamente assenti dalla libreria genomica utilizzata, sfuggano all'analisi. Pertanto, l'obiettivo primario della ricerca di Mullis (la pianificazione ‛apollinea') era quello di modificare opportunamente la tecnica di sequenziamento di Sanger, in modo che funzionasse anche su preparazioni di DNA totale molto complesse, come il DNA genomico umano, e non soltanto sui piccoli frammenti di DNA clonabili nei plasmidi batterici. Una delle più significative modifiche introdotte da Mullis fu quella di usare due oligonucleotidi (detti primers), invece dell'unico prescritto dal protocollo originario di Sanger, in modo da riuscire ad avviare la reazione di sequenziamento da entrambe le opposte estremità complementari delle singole catene del frammento di DNA da sequenziare; in tal modo sarebbe stato possibile avere un controllo interno sull'attendibilità della sequenza ottenuta. Fu questo che dette inizio a quella singolare esperienza che portò Mullis alla scoperta - inaspettata, e pertanto ‛dionisiaca' - di quanto egli non aveva minimamente cercato: la tanto semplice quanto straordinaria PCR, che permette di amplificare una singola molecola di DNA per miliardi di volte in un solo pomeriggio
La vivace narrazione di questa scoperta, fatta dallo stesso Mullis (v., 1990), si conclude con un interrogativo che sembra un ammonimento rivolto a quanti sostengono l'opportunità di finanziare soltanto ricerche dai risultati prevedibili e di sicuro successo: come mai un'applicazione tanto semplice della DNA-polimerasi era sfuggita alle migliaia di ricercatori che ne avevano fatto uso continuo per un quarantennio dalla sua scoperta? ‟Nessuno saprebbe dare una risposta, meno che mai io - dice modestamente Mullis - che mi sono imbattuto per caso nella PCR durante una lunga notte di guida al volante della mia auto attraverso le montagne della California".
A circa un decennio dalla scoperta della PCR, il bilancio dei contributi che questa nuova tecnologia ha portato nei settori più disparati della ricerca biologica è letteralmente straordinario: essa ha infatti reso possibile isolare geni senza bisogno di ricorrere alle laboriose e costose tecniche di clonaggio, descrivere la variabilità genetica individuale a livello del DNA in modo rapido ed economico, identificare innumerevoli nuovi esempi di polimorfismi multiallelici nelle popolazioni; ha avuto numerose applicazioni diagnostiche, farmacologiche e medico-legali; ha consentito la mappatura di geni attraverso l'ibridazione in situ di amplificati-PCR fluorescenti; ha promosso le ricerche comparative ed evoluzionistiche in genere e, in particolare, gli ambiziosi programmi di sequenziamento totale di genomi complessi. Tutte queste applicazioni sono state naturalmente favorite dall'automazione industriale che ha reso semplice la reazione, anche se essa può essere realizzata avendo a disposizione niente di più che una minuscola provetta, pochi reagenti e una sorgente di calore. Per questa ragione la PCR è persino entrata a far parte del materiale didattico per l'insegnamento della biologia molecolare nelle scuole e nei musei di informazione scientifica per il pubblico non specializzato.
b) Una vendemmia di polimorfismi genetici multiallelici
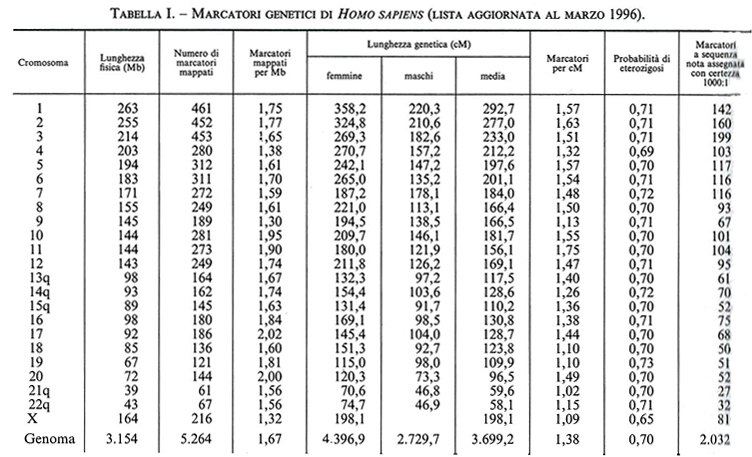
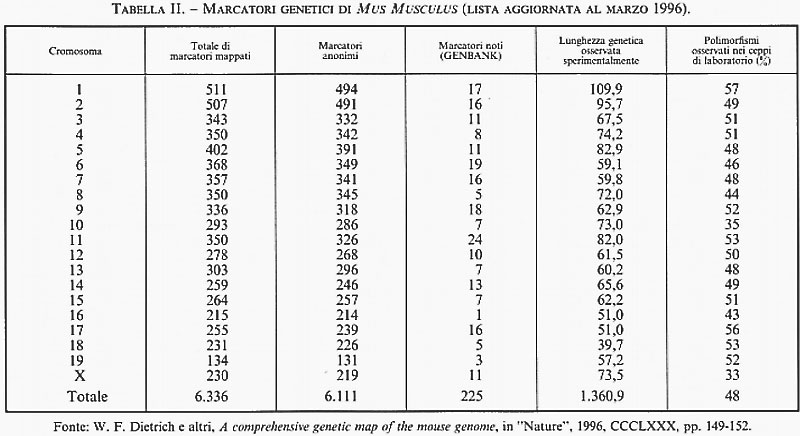
Il numero delle differenze ereditabili, riconoscibili in ciascuna specie animale o vegetale, è andato progressivamente aumentando man mano che l'analisi genetica si è spostata dal livello della classificazione diretta dei fenotipi visibili (essenzialmente qualitativa e macroscopica) a quella indiretta di fenotipi e genotipi dedotti per mezzo di analisi biochimiche, immunologiche, citogenetiche e molecolari. Quest'ultimo livello di indagine ha cominciato a essere utilizzato con successo dopo la scoperta dei polimorfismi dei frammenti di restrizione (RFLP, Restriction Fragment Length Polimorphism; v. genetica): nel giro di solo quattro anni (dal 1981 al 1985) l'analisi della diversità genomica con le tecnologie del DNA ricombinante e degli enzimi di restrizione ha portato il totale dei segmenti di DNA clonati nell'uomo da 51 a 808 e quello dei polimorfismi riconoscibili a tale livello da 24 a 333. Oggi, in due recenti rassegne (peraltro incomplete, in quanto si riferiscono soltanto ad alcune categorie di polimorfismi comuni attualmente individuabili attraverso la PCR) sono stati elencati oltre 15.000 siti multiallelici in Homo sapiens e oltre 6.000 in Mus musculus, insieme alla loro localizzazione subregionale per cromosoma e alle distanze fisiche e genetiche rispettivamente stimate in termini del numero di nucleotidi o delle frequenze di ricombinazione meiotica che li separano (v. tabb. I e II). Va subito precisato che in effetti queste nuove differenze genetiche sono per la maggior parte il risultato di meccanismi molto diversi dalle rare mutazioni puntiformi che generano varianti alleliche di uno stesso gene attraverso la sostituzione di una singola base nucleotidica. Peraltro, questi elenchi di marcatori genetici, aggiornati al 1996, sono tutt'altro che definitivi, in quanto il loro numero aumenta con il progredire delle conoscenze sulla struttura primaria del genoma e in particolare sulla composizione nucleotidica di geni strutturali, dai virus all'uomo. Per esempio, una vasta categoria di differenze molecolari è rappresentata dalle variazioni del numero di particolari sequenze nucleotidiche (da pochi nucleotidi fino a 60 e più) che si trovano all'estremità di diversi geni strutturali, tra i quali quelli che codificano per l'insulina, la mioglobina I e II, la zeta-globina, l'apoliproteina B e il collageno tipo II. I loci cromosomici cui questi geni afferiscono sono detti ‛ipervariabili' o - date le ripetizioni in tandem di specifiche sequenze nucleotidiche - VNTR (Variable Number of Tandem Repeats). Altre differenze molecolari, ancora più comuni, sono rappresentate dai cosiddetti ‛polimorfismi da microsatelliti', che consistono nell'assenza o nell'eccedenza di nucleotidi (da uno a tre/quattro) in regioni cromosomiche (chiamate introni) interposte alle sequenze codificanti (chiamate esoni) di geni strutturali. Infine, una nuova categoria di differenze ereditarie molto comuni - di solito non associate a manifestazioni patologiche e pertanto considerate espressione della normale variabilità genetica di ciascuna specie - sono quelle generate dalla perdita e/o re-inserzione di sequenze Alu, dall'integrazione di sequenze virali per retroposizione e dalla perdita e/o ridondanza di sequenze genomiche più o meno lunghe (delezioni e duplicazioni). È probabile che la maggior parte di queste ultime - e presumibilmente anche i polimorfismi VNTR - siano la conseguenza di rari eventi di ricombinazione ineguale (unequal crossing-over) tra segmenti di cromosomi omologhi non correttamente appaiati nel processo meiotico che permette di ridurre a metà il corredo cromosomico delle cellule germinali (uovo e spermio) durante la loro maturazione (v. riproduzione). Invece, i polimorfismi da microsatelliti e le amplificazioni graduali di specifiche sequenze trinucleotidiche (CAG) - le quali, al di là di una certa soglia di ridondanza, sono associate a gravi stati morbosi - sembrano piuttosto essere la conseguenza di errori nella replicazione del DNA durante il processo di divisione cellulare, sia meiotico che mitotico. È utile a questo punto fornire maggiori dettagli su una categoria di ‛differenze' che ha per denominatore comune il fatto di essere il prodotto ricorrente dell'instabilità genomica. Si tratta di differenze di cui si conosceva approssimativamente l'esistenza, ma che nessuno immaginava potessero essere così frequenti da diventare, per il biologo evoluzionista e il genetista molecolare, gli abituali ‛arnesi' di lavoro per la descrizione della diversità genetica della specie e per l'interpretazione del suo significato. La straordinaria messe di dati oggi disponibile sulle mappe genetiche dell'uomo e di altri mammiferi ha portato all'interessante scoperta che un gran numero dei gruppi di linkage stretto (ovvero di geni localizzati a una distanza genetica molto ravvicinata) individuati nell'uomo e nel topo sono tali anche in altre specie di mammiferi. L'esistenza di queste omologie dei gruppi di linkage in specie diverse - a prescindere dal fatto che esse ovviamente forniscono un forte sostegno all'ipotesi darwiniana sul meccanismo monofiletico dell'evoluzione biologica - è diventata un importante criterio strategico per la costruzione di mappe genetiche comparative e, di conseguenza, per individuare le regioni cromosomiche sulle quali concentrare lo sforzo per il clonaggio dei geni non ancora mappati in una specie, ma già noti perché parte di ben definiti gruppi di linkage in altre specie.
c) Regioni cromosomiche instabili e variabilità genetica normale e patologica
L'esistenza di regioni cromosomiche instabili nel genoma umano evoca l'immagine di una natura maligna che dissemina qua e là ‛trabocchetti' biologici tali da scompigliare la struttura primaria di importanti geni strutturali, mettendo in questo modo a rischio il normale sviluppo dei genomi portatori. In effetti, con il progressivo accumulo delle conoscenze molecolari riguardanti il nostro genoma, diviene sempre più evidente che buona parte della diversità genetica della specie è generata a livello di particolari siti genomici più facilmente soggetti a fenomeni di instabilità cromosomica e molecolare, che talvolta può essere anche causa di malattia. Peraltro, recenti studi hanno dimostrato che diversi processi di differenziamento somatico (come la generazione di milioni di cellule immunocompetenti, ognuna ‛diversa' rispetto alla costituzione genetica dei loci per le immunoglobuline) sono appunto il risultato di errori nella replicazione del DNA genomico durante il lungo iter che separa l'uovo fecondato da quel complesso organizzato di miliardi di cellule da esso derivate che costituisce l'organismo completamente sviluppato. Vista in questa luce, l'occasionale associazione tra instabilità genomica e malattie sembra quasi un modesto prezzo che la specie è costretta a pagare per assicurarsi, proprio attraverso gli errori di replicazione del DNA, quell'alto e ricorrente potenziale di variabilità genetica che garantisce la sua sopravvivenza nel tempo evolutivo. Pertanto, questo paragrafo sulle regioni cromosomiche instabili intende principalmente fornire un aggiornamento sul ruolo che esse sembrano avere nella produzione di varianti genetiche normali e patologiche, riconducibili essenzialmente ai seguenti tre tipi di instabilità genomica: 1) perdita o ridondanza di sequenze nucleotidiche più o meno lunghe (mutazioni sezionali); 2) retroinserzione di sequenze trasponibili e di genomi virali; 3) amplificazione di specifiche sequenze trinucleotidiche.
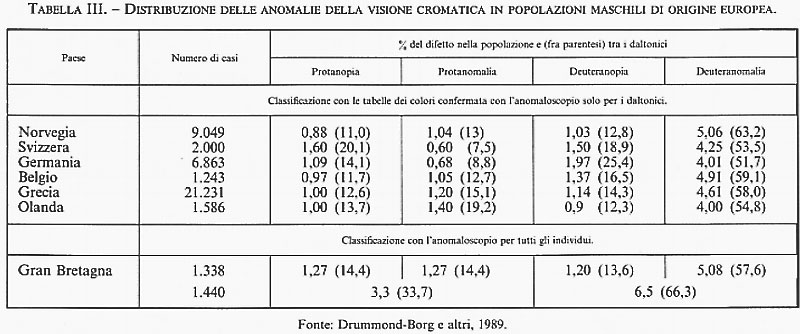
1. Mutazioni sezionali da errori della ricombinazione genetica. - Il primo esempio di mutazioni di questo tipo fu identificato nel 1914 da S. C. Tice in Drosophila melanogaster, il moscerino dell'aceto, che per oltre un quarto di secolo è stato il mezzo di studio esclusivo per la sperimentazione genetica. Si trattava di una mutazione della morfologia dell'occhio (mutazione bar) apparsa occasionalmente in un maschio e trasmessa ai discendenti secondo il meccanismo dell'eredità dominante e diaginica (dal greco dia, attraverso, e gune, femmina), ovvero trasmessa dai maschi affetti esclusivamente alla discendenza femminile e da questa ai discendenti di ambo i sessi, come ci si attende per geni localizzati sul cromosoma X nelle specie con cromosomi sessuali maschio/femmina di tipo XY/XX. L'analisi genetica di questa mutazione, realizzata attraverso lo studio di numerosi marcatori genici del cromosoma X di Drosophila, permise ad A. H. Sturtevant (già nel 1924) di intuire l'esistenza di mutazioni prodotte da occasionali errori della ricombinazione genetica che egli ipotizzò fossero conseguenza di uno scambio ineguale (crossing-over ineguale) tra cromosomi omologhi appaiati in modo sbagliato. Una decina di anni dopo (1936), questa geniale intuizione di Sturtevant fu confermata dagli studi citogenetici di C. B. Bridges sui cromosomi giganti delle ghiandole salivari, grazie ai quali era divenuto possibile visualizzare direttamente questo tipo di mutazioni - definite da Sturtevant ‛mutazioni sezionali' per distinguerle dalle molto più rare e invisibili mutazioni puntiformi. Studiando la struttura morfologica della mutazione in oggetto sui cromosomi giganti delle ghiandole salivari di moscerini con fenotipo bar, Bridges si accorse che quest'ultimo era in effetti costantemente associato con l'amplificazione più o meno estesa di una specifica sezione del cromosoma X, come ci si attendeva appunto dalla duplicazione di materiale genetico prodotta da crossing-over ineguale . Peraltro, apparve subito chiaro che nella discendenza di femmine eterozigoti per la mutazione bar si verificavano spesso eventi di ulteriore amplificazione sezionale (mutanti triplo- e infra-bar), come se la disparità di lunghezza sezionale, propria degli eterozigoti per una prima mutazione bar, favorisse il replicarsi di analoghi errori di ricombinazione nello stesso sito cromosomico. Questo meccanismo di duplicazione e/o di perdita di interi segmenti cromosomici è stato per lungo tempo considerato un fenomeno raro, se non addirittura isolato, e quindi senza alcun significato evolutivo. Fu solo all'inizio degli anni sessanta che vennero individuate, sempre in Drosophila, altre due regioni cromosomiche che presentavano esempi di polimorfismo genetico generati da frequenti e spontanei errori della ricombinazione meiotica: il locus white, anch'esso localizzato sul cromosoma X, e il locus rosy, sul cromosoma 3. Con il trasferimento dell'analisi genetica a livello molecolare, ci si è accorti che questo tipo di mutazione è certamente il più comune; tuttavia, ciò non sembra mettere in dubbio la conclusione che esse abbiano scarso significato evolutivo, in quanto la maggior parte dei prodotti di ricombinazioni sbagliate viene persa nel giro di poche generazioni per semplice effetto della segregazione, oppure eliminata per selezione naturale quando è associata a un fenotipo anormale. Come c'era da attendersi, l'estesa applicazione della PCR all'analisi delle differenze molecolari, individuate in base alla lunghezza dei frammenti nucleotidici amplificabili, ha permesso, nell'ultimo decennio, di riconoscere facilmente l'esistenza di numerosi esempi di duplicazioni/delezioni da crossing-over ineguale in vari organismi, dal batterio Escherichia coli all'uomo. In quest'ultimo, oltre alle duplicazioni e/o delezioni associate a specifiche malattie (come la neuropatia ereditaria di Charcot-Marie-Tooth, CMT, caratterizzata da una duplicazione in tandem di un frammento di 1,5 megabasi nella regione cromosomica 17p11.2-p12, o la sindrome velo-cardio-facciale, VCFS, associata a una cospicua delezione di 4-5 megabasi nella regione cromosomica 22q11.2-q12), sono ormai ben noti diversi esempi di variabilità genetica normale generati da crossing-over ineguale: tra questi, vale la pena di citare la variabilità individuale nella percezione dei colori, che recenti studi molecolari hanno direttamente messo in relazione alla presenza di duplicazioni e/o delezioni in una specifica regione del cromosoma X (Xq28), dove sono localizzati i due più importanti sistemi genetici coinvolti nella discriminazione della gamma di colori dal rosso al verde. I geni per la visione cromatica per il rosso (PROTAN) e per il verde (DEUTAN) sono entrambi localizzati nella porzione sub-terminale del braccio lungo del cromosoma X; la spiccata omologia strutturale di questi due geni autorizza la conclusione che essi siano derivati dalla duplicazione di un singolo gene ancestrale, probabilmente avvenuta in uno stadio molto precoce dell'evoluzione dei Primati, attraverso lo stesso meccanismo di crossing-over ineguale descritto per le mutazioni bar in Drosophila. Negli individui con visione cromatica normale, il gene PROTAN è sempre presente in singola dose, mentre le unità del gene DEUTAN variano da uno a cinque, e forse anche più; la cecità per il rosso non si accompagna mai alla totale perdita del gene PROTAN, ma è determinata da sue delezioni parziali che, a seconda della localizzazione ed estensione, risultano in un difetto cromatico totale (protanopia) o parziale (protanomalia), mentre la cecità totale per il verde è associata alla completa mancanza di geni DEUTAN, e la cecità parziale (deuteranomalia) è dovuta alla presenza di uno o più geni anormali prodotti da fusioni parziali di geni DEUTAN e PROTAN per crossing-over ineguale (v. tab. III; v. anche visione).
2. Retroinserzioni di sequenze trasponibili e di genomi virali. - Con la scoperta delle leggi di Mendel e del ruolo dei cromosomi come vettori fisici dell'eredità biologica, al patrimonio genetico di ciascuna specie era stata attribuita quella caratteristica di fissità implicita nella classificazione linneana della specie, malgrado il progressivo accumulo di nuove varianti genetiche prodotte da rare mutazioni geniche e cromosomiche. Questo scenario fu seriamente compromesso dall'ipotesi formulata nel 1951 da Barbara McClintock per spiegare le mutazioni instabili del genoma di mais, che ella attribuì alla trasposizione di elementi mobili del genoma (gli odierni ‛geni saltatori' o jumping genes) capaci di modificare la funzione dei geni entro i quali essi venivano a inserirsi. Com'è noto, dopo circa un quarantennio di scetticismo da parte della genetica ufficiale, la geniale ipotesi della McClintock venne premiata con il Nobel (1983) per il ruolo che essa aveva avuto nella scoperta di elementi trasponibili (‛trasposoni') nei Batteri e in Drosophila (v. tav. II). Nel solo genoma di quest'ultima specie sono state scoperte da 3.000 a 5.000 sequenze trasponibili, corrispondenti a circa il 10-15% del suo DNA totale. Nei Batteri e negli animali di laboratorio sono stati individuati diversi esempi di modificazioni genomiche attribuibili all'inserzione casuale di elementi trasponibili. Per esempio, sono stati riconosciuti come conseguenza dell'inserzione di un elemento trasponibile in seno a geni strutturali del tutto normali alcuni fenomeni di adattamento rapido alle modificazioni ambientali, come lo sviluppo della resistenza agli antibiotici nei Batteri e l'apparizione improvvisa di alcuni stati morbosi ereditari (quali l'emofilia, la distrofia di Duchenne e certe forme di cancro). Gli elementi trasponibili, che sono non soltanto numerosi ma anche diversi entro e tra le specie, vengono raggruppati in due grandi classi a seconda del meccanismo alla base della loro mobilità: i ‛trasposoni' e i ‛retrotrasposoni'.
I trasposoni sono sequenze nucleotidiche mobili disperse in gran numero nei genomi eucariotici e denominate, a seconda della loro lunghezza, LINEs o SINEs (Long, o Short, Interspersed Elements). Per dare un'idea quantitativa dell'abbondanza di questi elementi ripetitivi, basta ricordare che il tipo più comune di SINEs presente nel genoma umano - la famiglia delle sequenze Alu, di lunghezza media compresa tra 150 e 300 nucleotidi - costituisce da solo circa il 5% del DNA totale della specie, con una distribuzione pressappoco costante di un cluster Alu per ogni 4.000 nucleotidi. Negli ultimi anni l'affascinante e difficile problema dell'origine evoluzionistica degli elementi Alu è stato affrontato al livello molecolare, attraverso la comparazione delle sequenze nucleotidiche di qualche centinaio di elementi di origine umana e di analoghi SINEs derivati da specie diverse. È così emerso che, anche se non esistono due copie di sequenze Alu umane assolutamente identiche, tutti questi elementi sono caratterizzati da una generica somiglianza (in ragione dell'80% entro la stessa specie e del 50-60% tra specie diverse) suggerendo pertanto una loro unica origine ancestrale. Inoltre, questi studi hanno mostrato che non tutti gli elementi di questa famiglia contengono necessariamente un sito di restrizione Alu (come era parso in un primo tempo), neppure entro la stessa nostra specie; tuttavia, appare difficile che l'appellativo corretto di SINEs riesca a soppiantare del tutto l'espressione ‛famiglia di sequenze Alu', che si è ormai consolidata per l'uso generale che ne è stato fatto per oltre vent'anni. La vasta massa di dati prodotta da questi studi molecolari è ancora ben lungi dall'essere compiutamente analizzata, ma il suo potenziale nell'interpretazione molecolare dell'evoluzione biologica è ovvio, data la presenza ubiquitaria di famiglie Alu in tutti gli organismi animali e vegetali provvisti di un nucleo cellulare (Eucarioti) e la loro spiccata tendenza all'accumulo di varianti specie-specifiche che vengono utilizzate come marcatori delle distanze evolutive tra le specie (v. Batzer e altri, 1996). Peraltro, da tempo i biologi molecolari sostengono che l'analogia strutturale tra sequenze Alu e l'elemento 7SL RNA - una sequenza di 294 nucleotidi conservata in un largo intervallo evolutivo, dai Batteri ai Primati - è palese testimonianza dell'origine monofiletica di Procarioti ed Eucarioti. La più comune delle sequenze ripetitive LINEs (di lunghezza media da 1.000 a 5.000 nucleotidi) è la cosiddetta famiglia LINE 1, presente nel genoma umano in un numero di copie compreso tra 20.000 e 40.000. È interessante notare che LINEs e SINEs terminano entrambe con un'identica sequenza composta da una trentina di nucleotidi allineati in ordine inverso; si ritiene che tale sequenza abbia un ruolo primario nel processo che permette ai trasposoni di separarsi dalla loro posizione originaria per inserirsi ‛a caso' in una nuova localizzazione genomica. Si sa che questo processo - tuttora ben lungi dall'essere spiegato in dettaglio - è regolato da una trasposasi, un enzima codificato dall'informazione genetica presente nelle stesse LINEs; le sequenze ripetitive di questa famiglia sono caratterizzate da un'altra connotazione esclusiva, cioè dalla presenza di un gene per la trascrittasi inversa che - come si dirà più avanti - permette loro di essere reintegrate nel DNA genomico anche come cDNA (DNA complementare) attraverso il meccanismo di trascrizione inversa dell'RNA da essi prodotto. È per questo motivo che le sequenze LINEs vengono raggruppate con i retrotrasposoni. Tra i trasposoni meglio conosciuti ci sono l'elemento Ac del mais e il fattore P di Drosophila, entrambi usati con successo per costruire i cosiddetti organismi ‛transgenici', ovvero per inserire un gene estraneo nel genoma di una cellula/organismo recettore della stessa specie o di specie diversa. Questa strategia è già stata impiegata con successo nel campo della genetica applicata al miglioramento di piante e animali domestici e - come vedremo - ha ovvie potenzialità applicative per la terapia genica delle malattie ereditarie della nostra stessa specie (v. botanica; v. genetica: Applicazioni della genetica, v. riproduzione: Tecniche di inseminazione artificiale).
Si indicano col termine ‛retrotrasposoni' quelle famiglie di elementi trasponibili capaci di creare copie di se stessi in altra parte del proprio genoma, o in genomi estranei, senza un'effettiva mobilizzazione del proprio DNA. L'informazione genetica codificata nel DNA dei retrotrasposoni viene regolarmente trascritta nell'mRNA corrispondente - come accade per qualunque gene strutturale - senza che la sequenza nucleotidica coinvolta venga necessariamente rimossa dalla sua localizzazione abituale; in un secondo momento, l'mRNA prodotto viene tradotto a ritroso per intervento dell'enzima trascrittasi inversa (v. gene, vol. VIII), codificato dagli stessi retrotrasposoni. Infine, la copia libera del DNA complementare così prodotta, identica alla sequenza originale del retrotrasposone, viene integrata nel DNA dello stesso genoma o in quello della cellula ospite, forse non proprio a caso, ma comunque in una posizione del tutto diversa da quella occupata dal retrotrasposone corrispondente. Negli ultimi anni le analogie strutturali e funzionali tra retrotrasposoni e Virus sono state intensamente studiate, specialmente da H. E. Varmus e J. M. Bishop, che nel 1986 hanno condiviso il premio Nobel per aver dimostrato l'integrazione di sequenze virali nel genoma dei Mammiferi. Come è noto, il genoma dei Retrovirus è costituito solo da RNA che viene trascritto a ritroso - a opera della stessa trascrittasi inversa codificata dai retrotrasposoni - in una sequenza nucleotidica complementare (cDNA) che finisce poi con l'essere integrata nel DNA della cellula ospite. Se si compara la struttura primaria del cDNA virale con quella del DNA dei retrotrasposoni ci si accorge che entrambi terminano alle due estremità con una sequenza ripetuta che si compone di 200-400 nucleotidi nota con l'acronimo LTR, dalle iniziali di Long Terminal Repeat (v. Singer e Berg, 1991; v. Deininger e Batzer, 1993). La differenza fondamentale fra le due strutture è la presenza, nei soli genomi virali, del gene env, e quindi del rivestimento proteico da esso codificato, che permette ai Virus di uscire nuovamente dalla cellula ospite. Senza questo rivestimento Virus e retrotrasposoni sarebbero elementi funzionalmente identici per la capacità, comune a entrambi, di spostarsi da una parte all'altra del genoma che li ospita. È per questo motivo che i Virus sono stati talora definiti come retrotrasposoni arricchiti del gene env nel corso dell'evoluzione biologica o, viceversa, i retrotrasposoni come il relitto evoluzionistico di Virus che abbiano perduto lo stesso gene env. In pratica, la distinzione tra Virus e retrotrasposoni può essere molto difficile, come si deduce dalla dimostrazione, fornita da alcuni ricercatori del laboratorio CNRS di Gif-sur-Yvette (Parigi), della capacità infettiva della sequenza nucleotidica gypsy, un retrotrasposone ben noto ai cultori della genetica degli Insetti. Un'altra analogia sorprendente tra retrotrasposoni e Virus è la capacità di entrambi di attivare gli oncogeni (v. Watson e altri, 19962 v. neoplasie: Oncologia sperimentale) inserendosi nelle sequenze nucleotidiche a essi adiacenti. D'altronde, Marie Lou Pardue, del Massachusetts Institute of Technology, ha sottolineato il ruolo stabilizzatore che gli elementi trasponibili, ma non certo i Virus, potrebbero avere nel mantenimento della struttura, della stabilità e della funzione dei cromosomi, come suggerisce l'accertato effetto protettivo esercitato dagli elementi mobili Het e Tart sui telomeri cromosomici in Drosophila. Altri autori sostengono che l'accumulo di famiglie di DNA ripetitivo in zone strategiche del genoma (come i centromeri e i telomeri) potrebbe favorire l'appaiamento regolare dei cromosomi omologhi durante la meiosi e la loro segregazione nelle cellule germinali mature. Infine, la frequente localizzazione di entrambi gli elementi SINEs e LINEs in siti cromosomici che sono anche la sede di polimorfismi multiallelici ha generato l'ipotesi di un possibile ruolo primario degli elementi trasponibili come induttori di polimorfismi da mini- e microsatelliti nelle regioni genomiche dove essi vengono integrati (v. Arcot e altri, 1995). L'ipotesi è affascinante, anche perché lo stesso tipo di associazione è stato osservato a livello delle regioni cromosomiche che sono particolarmente suscettibili a integrazioni di genomi virali e ad altri tipi di instabilità cromosomica; è chiaro, tuttavia, che - almeno per il momento - queste associazioni possono venir spiegate anche in un modo diametralmente opposto, assumendo, cioè, che esse siano conseguenze diverse della medesima circostanza, vale a dire della peculiare instabilità molecolare di alcuni siti cromosomici del nostro genoma. Quale che sia la giusta interpretazione dei fatti, si può comunque ritenere, almeno in via ipotetica, che il meccanismo che genera le minute amplificazioni e riduzioni di sequenze trinucleotidiche e i polimorfismi da mini- e microsatelliti sia lo stesso e sia riferibile a un'errata replicazione del DNA (slippage) durante il processo di divisione cellulare meiotico o mitotico. Viceversa, le duplicazioni in tandem di intere sezioni cromosomiche e la presenza di delezioni più o meno lunghe nelle stesse regioni sono da considerarsi alla stregua di occasionali errori nel corso dei due fondamentali processi biologici che sono alla base del meccanismo di produzione della variabilità genetica individuale, ovvero la ricombinazione meiotica tra cromosomi omologhi di origine parentale diversa e l'interscambio mitotico tra i cromatidi fratelli di uno stesso cromosoma (sister chromatid exchange). Secondo questa ipotesi, i due su riferiti tipi di errori occasionali delle divisioni meiotiche o mitotiche sarebbero responsabili, rispettivamente, della produzione di mutazioni ‛sezionali' del genoma associate a malattie, o semplicemente espressione della variabilità genetica ereditabile e di mutazioni non ereditabili associate al differenziamento somatico che caratterizza lo sviluppo di complessi organismi multicellulari da un singolo uovo fecondato (v. embriologia).
Per concludere, si può ragionevolmente affermare che i copiosi dati raccolti durante questi ultimi anni sulla struttura molecolare e la distribuzione intra- e interspecie degli elementi mobili non hanno forse ancora permesso di chiarire compiutamente il problema della loro origine e funzione, ma sono certamente serviti a farli rimuovere una volta per sempre dalla categoria del cosiddetto ‛DNA-spazzatura' (junk-DNA) - ammesso che una siffatta categoria esista per davvero - nella quale essi erano stati frettolosamente inseriti all'indomani della loro scoperta.
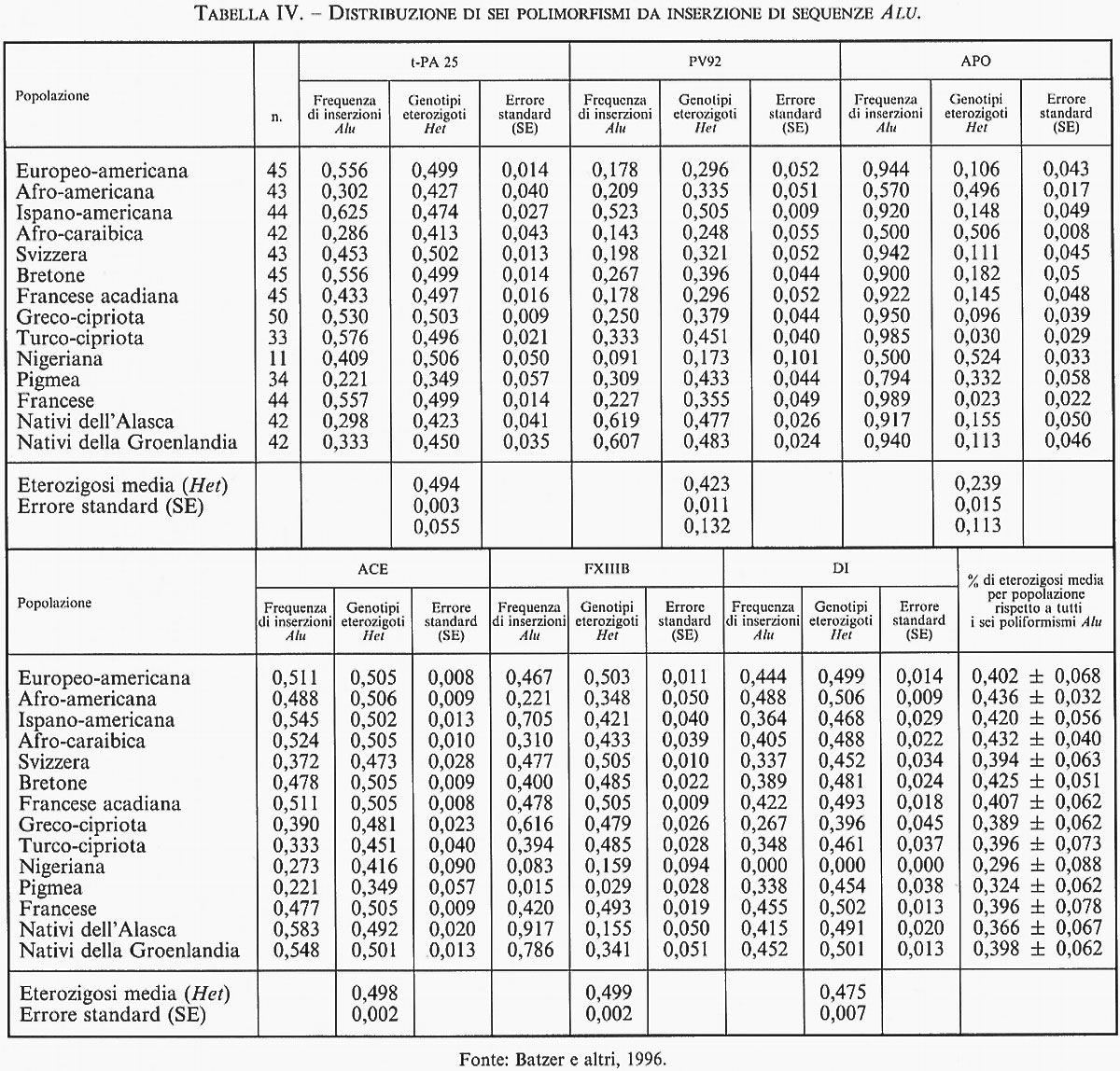
3. Elementi trasponibili e variabilità genetica. - È intuitivo che gli elementi SINEs e LINEs - per la loro caratteristica tendenza alla replicazione in tandem, alla trasponibilità e alla prevalente concentrazione in siti cromosomici che mostrano una particolare tendenza alla ricombinazione genetica in senso lato (crossing-over meiotici, sister chromatid exchanges, riarrangiamenti cromosomici e integrazioni virali) - hanno pieno diritto a essere annoverati tra i fattori responsabili della variabilità genetica, normale e patologica. La novità di questi ultimi anni, resa possibile dall'uso intensivo della PCR nella ricerca di differenze ereditabili a livello di specifici frammenti di DNA, è stata la scoperta di diversi esempi di classici polimorfismi dimorfici creati dalla presenza o assenza di un elemento Alu in determinate regioni cromosomiche, come se l'inserzione di alcune di tali sequenze fosse avvenuta una sola volta in epoca ancestrale e ciascuna di esse fosse stata diffusa nelle generazioni successive secondo i ben noti meccanismi della segregazione mendeliana nelle popolazioni. Un primo esempio di questo polimorfismo dimorfico nell'uomo è quello determinato dall'inserzione di una singola sequenza Alu di 300 nucleotidi in un introne del gene che codifica la sintesi di una proteina enzimatica di notevole importanza farmacologica. L'iter della sua scoperta è un esempio pertinente degli inattesi benefici che la ricerca di base può talvolta derivare dalle attività scientifiche rigidamente applicative del settore bioindustriale. Nel 1983, una ben nota industria californiana, interessata alla produzione commerciale dell'enzima t-PA (attivatore tessutale del plasminogeno) impiegato nella terapia degli infarti miocardici da trombosi delle coronarie, riuscì a clonare il gene responsabile della sua sintesi e a inserirlo in cellule batteriche per una massiva produzione in vitro (v. Pennica e altri, 1983). Qualche anno più tardi, un gruppo di ricercatori intenti a studiare il meccanismo di dispersione delle sequenze Alu nel genoma umano e nei Primati in genere, si accorse che una di queste sequenze, inizialmente descritta come parte integrante di un introne del gene TPA, era in effetti assente in un gran numero di individui, determinando un classico esempio di polimorfismo genetico ubiquitario in tutte le popolazioni umane studiate (v. Batzer e altri, 1996). Queste osservazioni dettero luogo a una sistematica ricerca di analoghe situazioni in altre regioni introniche del genoma, con il positivo risultato della scoperta di altri cinque casi di polimorfismi da inserzioni Alu in specifici siti dei cromosomi 1, 3, 11, 16 e 17, anch'essi ubiquitari presso tutte le popolazioni studiate, con tipiche variazioni di frequenze geniche (v. tab. IV) influenzate dal pattern migratorio e dal grado di isolamento di ciascuna di esse, e pertanto utili alla costruzione delle genealogie storiche della specie. Dal momento che il totale di elementi Alu presenti nel genoma umano è di circa mezzo milione, è possibile che il numero dei polimorfismi da inserzione Alu sia in effetti molto più elevato di quanto non appaia dai dati popolazionistici finora raccolti. D'altra parte, se la caratteristica fondamentale della grande maggioranza degli elementi Alu dovesse essere la loro mobilità intragenomica, sarebbe più verosimile attendersi che, in generale, essi non abbiano una localizzazione fissa, a eccezione della modesta aliquota che è rimasta stabilmente inclusa nella sequenze di DNA intergenico o nelle frazioni del DNA genico (introni) che non partecipano alla produzione del prodotto genico finale. La maggior parte delle altre inserzioni possibili sarebbe eliminata dalla selezione naturale, come dimostra la lunga lista di malattie gravi associate a inserzioni sporadiche di uno o più elementi Alu nelle porzioni codificanti di importanti geni strutturali del nostro genoma (v. Labuda e altri, 1995). Data l'estrema semplicità metodologica necessaria per la sua classificazione, in alcuni paesi lo studio della diversità genetica da inserzioni Alu è ormai entrato a far parte delle classi di laboratorio sperimentale delle scuole medie superiori per illustrare come si mettano in evidenza le differenze individuali a livello del DNA e sottolinearne il potenziale valore cognitivo. Non a caso, l'idea di servirsi di questa semplice strategia sperimentale per far conoscere meglio il DNA anche al grande pubblico, è partita dal DNA Learning Center, creato da James Watson (premio Nobel, insieme a Francis Crick e Maurice Wilkins, per la scoperta della struttura a doppia elica del DNA) per quella che egli chiama la sua ‟crociata contro l'analfabetismo molecolare".
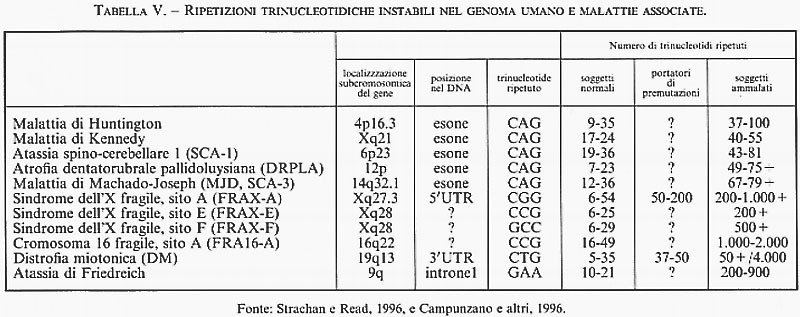
4. Amplificazioni di sequenze trinucleotidiche e malattie. - I primi due esempi di malattie ereditarie associate ad amplificazioni trinucleotidiche, descritti per la prima volta nel 1991, sono la sindrome dell'X fragile (una grave forma di ritardo mentale trasmessa con il meccanismo dell'eredità diaginica tipica delle mutazioni del cromosoma X), associata all'amplificazione della tripletta CGG nella regione cromosomica Xq27.3, e l'atrofia muscolare bulbospinale di Kennedy, associata all'amplificazione della tripletta CAG nella regione Xq21. In entrambi i casi un polimorfismo genetico normale, consistente nel numero variabile di tali triplette nucleotidiche, diventa causa di malattia quando l'amplificazione supera una determinata soglia. I pazienti con la sindrome dell'X fragile presentano diverse centinaia, e talora migliaia, di triplette CGG, mentre gli individui normali ne hanno in media qualche decina; analogamente, nelle atrofie muscolari di origine bulbospinale sono state osservate da 38 a 66 triplette CAG, invece delle 20 presenti in media negli individui normali. Nel corso degli ultimi anni la lista di malattie ereditarie legate all'espansione abnorme di specifiche triplette nucleotidiche è andata rapidamente crescendo (v. tab. V). Tra queste vi è l'espansione della tripletta CAG, responsabile della corea di Huntington e di altre malattie degenerative del sistema nervoso, quella della tripletta CTG, associata alla distrofia miotonica di tipo autosomico, e quella della tripletta GAA, propria dell'atassia di Friedreich. È interessante notare che - a eccezione di quest'ultima - tutte le malattie legate a un'espansione trinucleotidica vengono trasmesse come mutazioni dominanti con manifestazione tardiva dello stato morboso conclamato (intorno ai 45-60 anni di età) qualora si tratti di un paziente nato da genitori normali, manifestazione che diviene progressivamente più precoce nelle generazioni successive. Indagini molecolari hanno dimostrato che questo fenomeno di ‛anticipazione' dell'età di manifestazione della malattia non è soltanto l'effetto di un vizio statistico - come ci si aspetterebbe a causa della maggiore difficoltà di studiare famiglie con figli in età matura - ma corrisponde alla progressiva espansione della tripletta nucleotidica man mano che la mutazione viene trasmessa di genitore in figlio. Inoltre, la localizzazione delle espansioni trinucleotidiche CGG e CAG in siti codificanti di specifici geni (esoni) aveva fatto pensare che gli stati morbosi in oggetto fossero l'espressione diretta di alterazioni qualitative della funzione genica primaria determinate dalla crescente amplificazione trinucleotidica, e che il meccanismo di formazione di quest'ultima fosse in diretta connessione con l'eccesso di nucleotidi CG presenti in entrambe le suddette triplette. Tuttavia, queste interpretazioni sono state ovviamente messe in dubbio - almeno come spiegazioni unitarie per tutte le mutazioni di questo tipo - dal caso dell'atassia di Friedreich, che rappresenta un'eccezione rispetto a ciascuna delle caratteristiche su menzionate, in quanto si tratta di malattia a trasmissione di tipo recessivo, senza fenomeno di anticipazione e associata a espansione trinucleotidica GAA localizzata in un introne.
d) Nuovi strumenti a disposizione del ‛cacciatore di geni'.
A giudizio di chi scrive, i contributi più importanti finora recati dal Progetto Genoma (v. cap. 3; v. anche genoma, vol. X) sono stati essenzialmente due: 1) lo straordinario stimolo intellettuale che esso ha fornito all'ideazione degli strumenti necessari alla sua realizzazione; 2) l'interesse e la partecipazione diretta del settore industriale, che vi ha profuso massicci investimenti (anche se tutt'altro che filantropici), assolutamente insoliti nell'ambito del mondo scientifico. Per sottolineare l'entità di quest'ultimo contributo basta rinviare i lettori interessati agli Atti della conferenza intitolata Commercial implications of genomic research tenutasi tra il 12 e il 15 maggio 1996 presso il prestigioso, e ormai centenario, laboratorio di Cold Spring Harbor che, dall'inizio degli anni cinquanta, è riconosciuto come la più importante sede di incontri di genetica e biologia molecolare. Quanto al primo contributo, è opportuno descrivere con qualche dettaglio le più significative metodologie messe a punto nell'ultimo decennio.
1. Minicromosomi artificiali in lieviti (YAC) e batteri (BAC). - Come è intuitivo, il primo passo verso la mappatura molecolare di un genoma, a qualunque livello nella scala dell'organizzazione biologica, è stato finora la scomposizione della lunga molecola del DNA della specie in frammenti di piccole dimensioni in modo da poterli inserire, e quindi replicare indefinitamente, in vettori plasmidici e fagici (v. biotecnologie) opportunamente manipolati con tecniche di ingegneria genetica per accogliere inserti nucleotidici di lunghezza variabile da poche centinaia a qualche decina di migliaia di nucleotidi. Come è noto le ‛forbici' biologiche per operare questa frammentazione del DNA genomico sono le endonucleasi batteriche, note come ‛enzimi di restrizione' per la loro capacità di tagliare la lunga molecola del DNA all'altezza di specifiche sequenze nucleotidiche, a loro volta denominate ‛siti di restrizione'. Questa metodologia è stata ed è tuttora utilizzata con estremo successo per ottenere le cosiddette ‛mappe di restrizione' di piccole porzioni di genomi complessi, attraverso cui isolare e caratterizzare singoli geni coinvolti nella manifestazione delle varianti fenotipiche normali e patologiche di ciascuna specie di organismi viventi. È chiaro, d'altronde, che la mappa di restrizione di un intero genoma complesso - come quello umano, composto di tre miliardi di nucleotidi che ciascun individuo eredita alla nascita in doppia dose - sarebbe stata una meta difficilmente raggiungibile per questa strada. Per questo motivo, all'indomani del lancio del Progetto Genoma lo sforzo maggiore è stato concentrato sulla messa a punto di metodologie che consentissero la frammentazione genomica in frammenti di dimensioni di gran lunga maggiori di quelle su menzionate, e sulla ricerca di vettori capaci di includerli stabilmente e di permetterne la replicazione a oltranza. Era ovvio, infatti, come ciò rappresentasse la condizione indispensabile per realizzare il non facile obiettivo della mappatura subcromosomica e del sequenziamento nucleotidico dei frammenti. Lo sforzo è tutt'altro che concluso, ma si può ragionevolmente affermare che i recenti successi nella frammentazione del DNA in frammenti dell'ordine delle centinaia di migliaia di nucleotidi e il loro clonaggio in lieviti e batteri (rispettivamente noti come minicromosomi YAC, Yeast Artificial Chromosomes, e BAC, Bacterial Artificial Chromosome; v. anche biotecnologie, vol. X; v. genoma, vol. X) hanno ormai definitivamente portato il Progetto Genoma dalla fase di programmazione teorica a quella operativa. La differenza essenziale tra minicromosomi YAC e BAC è nella dimensione degli inserti: per i primi varia da poche centinaia di migliaia a oltre un milione di nucleotidi e, per i secondi, da un minimo di 50-60.000 a un massimo di 500-600.000. Pertanto, i cloni BAC - che sono anche gli ultimi arrivati - riempiono il vuoto metodologico che esisteva in precedenza relativamente alla possibilità di ottenere librerie totali del DNA umano in frammenti di dimensione gradualmente crescente, dalle poche centinaia (librerie plasmidiche), alle migliaia (librerie fagiche), alle decine di migliaia fino a circa un centinaio di migliaia (librerie P1), ai milioni di nucleotidi (librerie YAC). Le dimensioni dei cloni BAC occupano una posizione intermedia tra quelle dei P1 e quelle degli YAC, ma il vantaggio più importante dei minicromosomi BAC rispetto agli YAC è la loro maggiore stabilità e facilità di replicazione, essendo praticamente esenti dal chimerismo (ovvero l'accidentale inclusione nello stesso clone di frammenti cromosomici di origine diversa), frequentemente osservato nei cloni YAC, e richiedendo sistemi di propagazione essenzialmente simili alle semplici e rapide colture batteriche, mentre le procedure di propagazione degli YAC sono di fatto più difficili e lente delle colture di lieviti. Comunque, questi due tipi di minicromosomi (l'appellativo è giustificato dalla circostanza che la lunghezza dei cromosomi umani varia dai 50 ai 200 milioni di nucleotidi), impiegati come sonde molecolari con la tecnica dell'ibridazione in situ, hanno permesso di costruire mappe molecolari del genoma umano e di altri organismi complessi a un livello di risoluzione dell'ordine delle centinaia di migliaia di nucleotidi, contro le decine o le centinaia di milioni di nucleotidi che costituivano il livello di risoluzione medio delle mappe cromosomiche precedentemente disponibili. La strategia per la costruzione delle mappe di microcromosomi YAC e BAC (denominate genericamente contigs, in quanto descrivono i rapporti di contiguità reciproca dei vari cloni entro un gene, una regione subcromosomica, un cromosoma o un intero genoma) comporta l'uso di metodologie diverse, che vanno dall'ibridazione molecolare diretta delle sonde sui cromosomi metafasici della specie studiata, all'uso della PCR per identificare i cloni contenenti geni o frammenti di DNA di localizzazione submicroscopica già nota, all'impiego di qualunque altro dato disponibile sulla distanza (misurata in termini di ricombinazione meiotica) tra marcatori dello stesso minicromosoma o di minicromosomi contigui. La mappatura subcromosomica in termini di contigs YAC e BAC è stata già applicata all'analisi di genomi diversi, dai più semplici, quelli di Batteri e lieviti, a quelli più complessi, dei Vertebrati superiori come uomo e topo.
2. Progressi della citogenetica molecolare umana. - Col termine citogenetica - diversamente da quanto verrebbe fatto di dedurre dal significato etimologico della parola - non si indica lo studio della genetica a livello delle singole cellule, ma quello delle caratteristiche strutturali e funzionali dei cromosomi, i vettori fisici dell'eredità biologica che normalmente ciascun individuo eredita per metà da ciascun genitore e che vengono replicati nei milioni di miliardi di cellule nucleate che compongono l'organismo completamente sviluppato. Per diversi decenni dopo l'enunciazione delle leggi di Mendel, la citogenetica umana era rimasta la ‛cenerentola' del settore per la difficoltà tecnica di propagare in coltura le cellule da analizzare, a eccezione di quelle tumorali che presentano cromosomi fortemente condensati e spesso alterati nel numero, nella morfologia e nella struttura. Nella prima metà degli anni cinquanta, una fortuita scoperta effettuata nel laboratorio di Hsu dell'Università di Austin, Texas (l'erronea sospensione delle cellule in divisione in una soluzione salina ipotonica) e i successi delle ricerche pianificate, realizzate in vari laboratori, sui metodi di coltura a breve termine dei linfociti del sangue periferico e dei fibroblasti cutanei, resero possibile una serie ininterrotta di progressi tecnologici che, in poco più che un decennio, permisero di definire una precisa classificazione morfologica (il cosiddetto banding pattern, come dire ‛l'impronta digitale') di ogni singolo cromosoma umano. Il livello di precisione raggiunto fu tale da permettere il riconoscimento diagnostico di un gran numero di varianti cromosomiche normali e delle gravi alterazioni cromosomiche associate a malattie. Alla fine degli anni sessanta lo sviluppo delle nuove strategie di mappatura genica attraverso gli ibridi cellulari somatici (v. genetica) fece balzare in primissimo piano il ruolo della citogenetica come mezzo d'indagine elettivo per la mappatura del genoma umano. Questo primato ha continuato a sussistere dopo l'avvio del Progetto Genoma che, avendo portato a una dimensione industriale il problema del mappaggio fisico di innumerevoli frammenti genomici, ha senza dubbio svolto un ruolo determinante per la trasformazione della citogenetica classica, essenzialmente morfologica, nella odierna citogenetica molecolare, una disciplina ormai autonoma che annovera tra i suoi cultori numerosi fisici, chimico-fisici, biologi molecolari e computer-analysts. L'ultima straordinaria acquisizione di questa disciplina è lo sviluppo della cosiddetta FISH (Fluorescence In Situ Hybridization), una metodologia che permette di determinare la fine localizzazione subcromosomica di frammenti genomici delle più varie dimensioni (da qualche migliaio di nucleotidi dei frammenti plasmidici, alle centinaia di migliaia e ai milioni dei minicromosomi e delle preparazioni di DNA totale derivate da singoli cromosomi) attraverso l'ibridazione molecolare in situ sui cromosomi metafasici di cellule sia normali sia patologiche, a seconda del quesito specifico posto dalla ricerca in oggetto. Le prime applicazioni di questa strategia metodologica ebbero inizio - circa quarant'anni or sono, usando come sonda molecolare l'RNA ribosomiale marcato con isotopi radioattivi - con la mappatura dei geni ribosomiali, che si prestavano particolarmente agli esperimenti di ibridazione molecolare per la loro caratteristica distribuzione in clusters di numerose copie dello stesso gene in siti cromosomici ben definiti in tutte le specie esaminate, dai lieviti ai vermi e alla drosofila, ai Mammiferi e al mais (v. Singer e Berg, 1991).
I progressi tecnologici realizzati nell'ultimo decennio riguardano essenzialmente i seguenti tre aspetti fondamentali: 1) la sostituzione della marcatura radioattiva delle sonde con quella a fluorescenza ottenuta con fluorocromi di colore diverso; ciò permette la mappatura contemporanea con sonde diverse, senza peraltro incorrere negli inevitabili problemi di inquinamento dell'ambiente associati all'uso di reagenti radioattivi; 2) l'applicabilità della tecnologia alla mappatura diretta di sequenze nucleotidiche uniche di dimensione modesta che, nelle mani più esperte, può essere persino di poche centinaia di nucleotidi; 3) l'automazione computerizzata nel rilievo dei segnali fluorescenti emessi ai siti di ibridazione molecolare, attraverso la combinazione di sofisticate procedure di ottica fisica, basate sulla spettroscopia di Fourier, e l'analisi digitale degli impulsi ottici rilevabili con la CCD-camera, l'ultimo degli accessori tecnologici della microscopia a fluorenscenza.
Il livello di efficienza raggiunto dalla citogenetica molecolare non potrebbe essere meglio descritto che dalle spettacolari immagini della tav. III, A, che illustra l'analisi cromosomica di una cellula umana normale e quella di una cellula tumorale con le 24 sonde molecolari specifiche per ciascuno dei 22 cromosomi autosomici e dei due cromosomi sessuali (X e Y) propri della nostra specie. Lo scenario suggerito da queste immagini per l'immediato futuro è quello dell'automazione robotizzata dello screening citogenetico di routine per la diagnosi delle alterazioni cromosomiche nella pratica medica e per le innumerevoli applicazioni citogenetiche alla ricerca scientifica che - come si può rilevare dalla tab. VI - vanno molto al di là di quelle strettamente pertinenti al Progetto Genoma Umano. È certamente molto probabile che questi obiettivi divengano appannaggio dell'industria biotecnologica in misura sempre maggiore, ma questa prospettiva - diversamente da quanto si sente dire nei circoli meno informati - sembra a chi scrive tutt'altro che negativa, in quanto le circostanze descritte potranno liberare lo scienziato del futuro dalla frustrante schiavitù di un iterativo lavoro di routine, lasciandogli molto più tempo da dedicare all'attività di ricerca (‛dionisiaca' o ‛apollinea' che sia).

3. Librerie di ibridi cellulari con minuscoli frammenti di DNA umano. - Degli ibridi cellulari somatici e dello straordinario contributo dato alla genetica umana dagli ibridi instabili topo/uomo, ratto/uomo e criceto/uomo, si è già parlato a lungo in precedenza (v. genetica, vol. VIII). È a questa fortuita scoperta degli anni sessanta che la genetica umana deve il suo ingresso tra le scienze sperimentali: essa, infatti, permise per la prima volta di determinare sperimentalmente la localizzazione cromosomica di specifici geni umani attraverso la costruzione di ibridi cellulari opportunamente pianificati per rispondere agli interrogativi di scelta. Il procedimento è del tutto simile a quello adottato in genetica animale o vegetale per costruire le ‛mappe genetiche', ovvero per scoprire i geni localizzati sullo stesso cromosoma (gruppi di linkage) attraverso l'analisi della segregazione delle caratteristiche ereditarie da essi controllate nella discendenza di incroci opportunamente scelti: infatti, per individuare i gruppi di linkage in Homo sapiens viene utilizzata la concordanza nella conservazione o nella perdita di due o più marcatori cellulari umani in cellule ibride roditore-uomo che - andando incontro alla perdita progressiva dei soli cromosomi umani - tendono a conservare o a perdere simultaneamente i gruppi di geni con la medesima localizzazione cromosomica. Ovviamente, questa strategia sperimentale ha potuto essere applicata alla costruzione delle mappe genetiche dei cromosomi umani in misura decrescente via via che scemava il numero delle ‛differenze specie-specifiche' che restavano da mappare tra quelle espresse nelle cellule ibride e chiaramente distinguibili nei due tipi di cellule parentali. La scoperta dei polimorfismi di restrizione (RFLP) aveva nuovamente riportato in primo piano l'uso degli ibridi cellulari somatici instabili per il mappaggio del genoma umano, in quanto i frammenti di restrizione relativi a specifici RFLP umani possono essere sempre classificati senza ambiguità con l'analisi diretta del DNA delle cellule ibride coltivate in vitro. La scoperta della reazione di polimerizzazione a catena ha enormente aumentato la potenzialità degli ibridi cellulari per la mappatura del genoma umano e ne ha semplificato significativamente il protocollo di lavoro. Di fatto, l'applicazione di quest'ultima tecnica per la mappatura cromosomica dell'elevato numero dei nuovi marcatori richiede oggi minime quantità di DNA per ogni clone di cellule ibride da analizzare e può essere realizzata rapidamente con attrezzature robotiche di costo limitato e senza l'uso di nucleotidi radioattivi, com'era invece finora indispensabile per la costruzione di mappe genetiche per mezzo degli RFLP. Ma in realtà, la più efficiente applicazione odierna degli ibridi cellulari per la mappatura del genoma umano è quella condotta con librerie di ‛ibridi cellulari altamente ridotti', ovvero quelli che hanno conservato un residuo di genoma umano composto esclusivamente di minuscoli frammenti di DNA umano stabilmente fusi con i cromosomi murini o di altri roditori. Queste librerie sono in genere derivate sperimentalmente dalla fusione di cellule di Roditori (quelle di criceto danno solitamente i migliori risultati) e cellule ibride altamente ridotte (per esempio con un solo cromosoma umano) irradiate con elevate dosi di raggi X (radiation hybrids).
Un altro sistema per produrre questo tipo di ibridi ridotti (che ha il vantaggio di evitare il rischio di riarrangiamenti cromosomici indotti dalle radiazioni ionizzanti) è quello utilizzato dal gruppo di ricerca di M. Rocchi, dell'Università di Bari: cloni di cellule ibride vengono propagate in coltura per il tempo necessario alla perdita totale dei cromosomi umani intatti, con l'eccezione di minuscole porzioni di essi accidentalmente traslocate sui cromosomi murini. Una paziente ricerca permette una prima approssimativa classificazione della dimensione e dell'origine cromosomica dei frammenti residui di genoma umano negli ibridi ridotti mediante il protocollo del FISH-painting, esponendo cioè le metafasi di ciascun ibrido cellulare ridotto a sonde fluorescenti contenenti preparazioni di DNA totale derivate da singoli cromosomi isolati. Per stabilire poi la precisa localizzazione cromosomica di ciascun frammento, singole preparazioni di DNA totale - derivate dai cloni ridotti contenenti frammenti della stessa origine cromosomica - vengono a loro volta utilizzate come distinte sonde fluorescenti in esperimenti di ibridazione molecolare in situ su cromosomi metafasici di cellule umane normali. Infine, l'analisi del contenuto di ciascun frammento viene effettuata con la PCR, usando coppie di primers capaci di amplificare porzioni di geni o di sequenze nucleotidiche a localizzazione subcromosomica già nota (v. Antonacci e altri, 1995). Alla conclusione di questo paziente lavoro (che offre il vantaggio di essere necessario una sola volta), i cloni ibridi ridotti più rappresentativi vengono assemblati in ‛librerie di frammenti' che - nel loro insieme - coprono per quanto è possibile l'intero genoma.
La disponibilità di queste librerie cellulari ibride semplifica enormemente il mappaggio di nuovi geni o di sequenze nucleotidiche anonime, che infatti può essere realizzato nel giro di poche ore quando si abbia a disposizione una minuscola aliquota del DNA di ciascun clone della libreria e una serie di coppie di primers per determinare la presenza/assenza di specifici prodotti di amplificazione in ciascuno dei cloni della libreria in esame. Ovviamente, la potenzialità di siffatte librerie aumenta progressivamente con la loro utilizzazione da parte della comunità scientifica; non stupisce, pertanto, che la loro produzione e distribuzione siano divenute argomento di primario interesse sia per il settore accademico che per alcune delle industrie biotecnologiche sorte a supporto del Progetto Genoma. Di fatto, una buona parte delle librerie di radiation hybrids è ora gestita da industrie biotecnologiche cui i ricercatori interessati inviano la coppia (o le coppie) di primers-PCR necessari per determinare la localizzazione subcromosomica dei geni e/o delle sequenze nucleotidiche di loro interesse nel breve tempo richiesto per effettuare con metodologie robotiche lo screening PCR dei singoli cloni di ibridi ridotti. Alternativamente, gli interessati possono acquistare dei kits di preparazioni nucleotidiche (ciascuna contenente il DNA totale di un singolo clone ibrido ridotto) relative ai frammenti di singoli cromosomi o dell'intera collezione di frammenti genomici e procedere per proprio conto al mappaggio desiderato.
4. Librerie di frammenti di sequenze espresse (EST): un limbo di geni da scoprire. - La costruzione di librerie di DNA complementare (cDNA) partendo da miscele di RNA messaggero (mRNA) è una procedura utilizzata con successo da almeno un decennio per l'isolamento di geni che controllano la sintesi di proteine dalle spiccate qualità immunogene, cioè capaci di indurre la formazione di anticorpi specifici se iniettate in organismi di specie diversa. La strategia di elezione è quella di usare l'anticorpo specifico contro la proteina in oggetto per identificare immunologicamente il clone (o i cloni) di cDNA che codifica(no) per le sequenze amminoacidiche dotate delle corrispondenti proprietà immunogene. Com'è noto, questa tecnologia ha permesso di clonare geni che controllano importanti funzioni biologiche anche in assenza totale di informazioni sulla struttura primaria delle proteine coinvolte e delle corrispondenti sequenze nucleotidiche codificanti (v. biologia molecolare, vol. VIII). Nel corso dell'ultimo quinquennio un numeroso gruppo di ricercatori americani guidati da J. Craig Venter - fondatore, presidente e direttore dell'Istituto di Ricerca Genomica (TIGR) di Rockville, Maryland - ha preso a servirsi di questa metodologia sistematicamente e su vasta scala per ottenere in breve tempo la sequenza di quella parte di genoma umano che codifica per geni strutturali. Poiché la porzione di DNA codificante del genoma umano rappresenta soltanto il 3% dei tre miliardi di nucleotidi che lo compongono, Venter sostiene che insistere nello sforzo globale tuttora in corso per una sequenza completa del genoma significherebbe ritardare di mezzo secolo l'identificazione della maggioranza dei geni che ci interessano. Convinto della realtà di questa sua pessimistica previsione, nel 1991 Venter, quando lavorava ancora presso l'NIH (National Institutes of Health) di Bethesda, si preoccupò innanzi tutto di mettere a punto un protocollo sperimentale che permettesse di accelerare di circa mille volte la scoperta di sequenze nucleotidiche codificanti (i geni propriamente detti) rispetto alla strategia alternativa di sequenziamento del DNA genomico. Questo metodo - che combina le tecniche di sequenziamento automatizzate con l'uso di algoritmi bioinformatici - consiste: 1) nell'estrazione dell'RNA messaggero prodotto da tipi cellulari diversi dell'organismo umano; 2) nella produzione delle corrispondenti molecole di cDNA per mezzo della trascrittasi inversa; 3) nel rapido sequenziamento automatizzato e parziale delle rispettive sequenze codificanti che Venter designò con l'acronimo EST (Expressed Sequence Tags, ovvero ‛frammenti di sequenze espresse'), dimostrando che esse erano sufficienti per identificare e quantificare l'espressione di singoli geni in praticamente ogni tipo di cellula e tessuto. Usando questo elegante approccio, i ricercatori del TIGR hanno finora prodotto più di 10.000 EST capaci di riconoscere oltre la metà dei circa 100.000 geni strutturali presumibilmente presenti nel genoma umano. Per dare un'idea concreta dell'efficienza del metodo, basterà ricordare che fino al 1991 (la data di nascita ufficiale dei primi EST) il totale dei geni strutturali identificati raggiungeva a stento le 3.000 unità. L'elenco completo dei geni corrispondenti agli EST finora prodotti è stato pubblicato col titolo Genome directory in uno speciale supplemento pubblicato dal settimanale scientifico ‟Nature" il 28 settembre 1995 (v. tav. IV); esso fornisce precisi riferimenti al tessuto di origine di ciascun EST, ai tessuti con la loro maggiore espressione e ai codici necessari per l'identificazione delle sequenze nucleotidiche corrispondenti nelle banche dati finora disponibili. È intuibile, anche per il lettore non specializzato, quanto questa monumentale dovizia di dati sulle sequenze parziali di geni strutturali sia preziosa ai fini della loro rapida localizzazione cromosomica e per il loro diretto isolamento senza dover ricorrere alle frustranti esperienze del cosiddetto ‛clonaggio posizionale', che ha spesso richiesto anni di estenuante lavoro anche quando la mappatura del gene ricercato era molto accurata, nei limiti di precisione possibili in base alla sola informazione citogenetica.
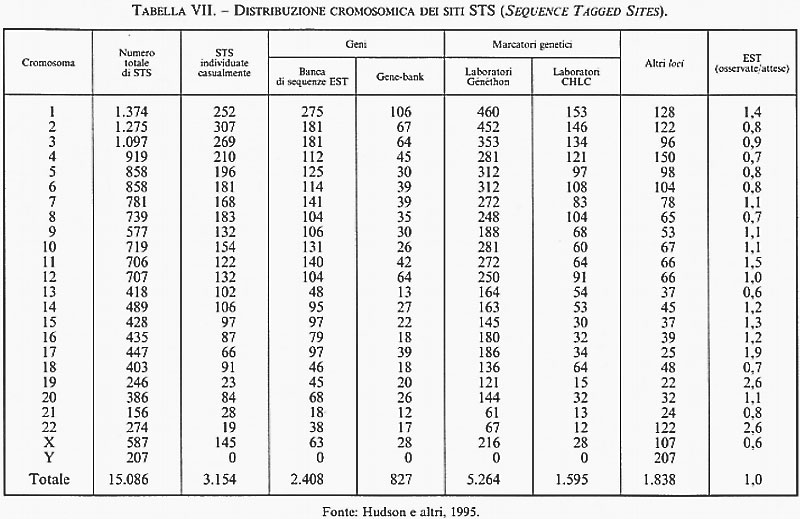
5. Sequenze di riferimento genomico (STS). - Le STS (Sequence Tagged Sites) costituiscono una collezione di ‛sequenze di riferimento genomico' che si è venuta formando nel tempo in vari laboratori partecipanti al Progetto Genoma; tali sequenze si distinguono per le seguenti favorevoli caratteristiche: 1) sono derivate dalla cosiddetta estremità 3′UT di qualunque gene o cDNA (dove UT sta per regione untraslated, ovvero una sequenza codificante un RNA messaggero che non viene tradotto nella corrispondente catena polipeptidica); 2) presentano un'elevata variabilità nei diversi geni della stessa specie e sono praticamente prive di omologia con i geni di altri mammiferi (in particolare con le specie di roditori usate come partners cellulari negli ibridi somatici; 3) sono sempre libere da introni e quindi possono essere ricostruite nella loro reale lunghezza per amplificazione tramite PCR, effettuata utilizzando primers 3′UT ottenuti dalle librerie di cloni cDNA precedentemente descritte. Per via di queste utili connotazioni, il Progetto Genoma americano ha deciso di concentrare i suoi sforzi per identificare e distribuire 30.000 sequenze STS, separate l'una dall'altra dalla distanza fisica di 100.000 nucleotidi, come pietre miliari di riferimento lungo ciascuno dei 24 cromosomi della specie (22 autosomi + X e Y). Alla fine del 1995, le sequenze STS mappate erano già più di 15.000 (v. tab. VII) e si sono rivelate di grande utilità nell'integrazione dei dati sulla mappatura fisica dei cromosomi umani. Le STS sono inoltre utilizzate come preziosi marcatori di riferimento per determinare l'allineamento reciproco (contig maps) dei singoli cloni di librerie genomiche parziali o totali, com'è il caso dei minicromosomi YAC, BAC, ecc. Per quanto le prime serie di mappe genomiche STS di uomo e di topo siano già state pubblicate e siano accessibili alla consultazione generale attraverso Internet, l'obiettivo finale di una mappa completa dei 30.000 marcatori STS di riferimento per il genoma umano è ancora lungi dall'essere raggiunto e potrebbe non esserlo mai se - come è già accaduto in precedenti occasioni (per esempio la totale perdita d'interesse della comunità scientifica verso gli RFLP che furono i primi utili marcatori per lo studio della variabilità genetica a livello molecolare) - lo sviluppo di nuove metodologie, ad esempio le mappe ottiche di restrizione e i microchips elettronici, dovesse rivoluzionare nuovamente le strategie di scelta per il mappaggio di genomi complessi.
6. Mappe ottiche di restrizione e microchips elettronici. - Nonostante i notevoli e rapidi progressi che la comunità scientifica internazionale è riuscita a ottenere nel tentativo di arrivare a una mappa fisica totale del genoma umano, non c'è dubbio che il successo dell'impresa è tuttora condizionato dalla necessità di approcci concettuali e sperimentali del tutto nuovi, in quanto le metodologie molecolari attualmente disponibili sono state sviluppate per la caratterizzazione di singoli geni, non di interi genomi. La necessità di nuove strategie metodologiche è specialmente sentita per lo studio degli aspetti popolazionistici della variabilità genomica. Le mappe ottiche di restrizione e i microchips elettronici intendono soddisfare questa particolare esigenza e rendere possibile una ricerca genomica popolazionistica. Vediamone brevemente le caratteristiche fondamentali e alcune possibili applicazioni nell'immediato futuro.
Le mappe ottiche di restrizione rappresentano il primo fortunato tentativo di mappaggio fisico di sequenze nucleotidiche di cospicue dimensioni senza ricorrere alla caratterizzazione elettroforetica dei minuti frammenti che le compongono. Il procedimento consiste nell'immobilizzazione della molecola del DNA da esaminare - di lunghezza variabile da poche centinaia a diverse migliaia di nucleotidi - in un gel di agarosio addizionato di enzimi di restrizione distribuito nell'intercapedine di una coppia di vetrini porta- e coprioggetto, in modo tale da assicurare l'estensione completa della molecola da esaminare lungo il suo asse maggiore. A questo punto l'analisi della struttura primaria del frammento viene effettuata determinando la distribuzione delle sequenze nucleotidiche specifiche per un gran numero di enzimi di restrizione, visualizzandone l'effetto attraverso l'uso di fluorocromi che rendono fluorescenti le porzioni indigerite della molecola, inframezzate a intervalli incolori corrispondenti ai siti di restrizione (v. tav. III, B). Questa metodologia è già stata applicata con successo alla costruzione di mappe di restrizione di minicromosomi YAC e BAC relativi agli autosomi umani 11 e 22. Nella sua più recente versione, la metodologia per le mappe ottiche di restrizione fa uso della PCR per la produzione di molecole di DNA umano fino a 20-30 kb di lunghezza; ciò permette di studiare la variabilità individuale a specifici siti di restrizione a livello popolazionistico senza dover ricorrere alle laboriose metodologie dell'analisi di restrizione tradizionale.
Lo straordinario potenziale dei microchips elettronici per l'analisi della variabilità genetica individuale a livello molecolare è stato il tema di un avveniristico reportage apparso nella sezione scientifica di ‟Newsweek" del 27 gennaio 1997. Quest'ultimo ritrovato della biotecnologia molecolare (prodotto da una piccola bioindustria californiana creata a bella posta per commercializzarne l'uso con l'indovinato appellativo di gene-chip) consentirà di concretizzare in un futuro molto prossimo il depistaggio simultaneo di mutazioni sfavorevoli per decine di migliaia di geni a composizione nucleotidica già nota. La strategia usata per raggiungere un così straordinario risultato si basa su una variante della tecnica FISH per il mappaggio fisico di geni attraverso l'ibridazione molecolare in situ su cromosomi metafasici. Nel caso dei gene-chips, il DNA-testo è quello dell'individuo che si vuol classificare rispetto alla presenza/assenza di specifiche sequenze nucleotidiche che vengono fissate in gran numero (dell'ordine di milioni di elementi identici) in ciascuno dei 60.000 microsettori del microchip, in modo che nell'insieme la lettura delle ibridazioni positive, visualizzate per fluorescenza mediante scansione laser, possa permettere di descrivere la diversità genetica di un gran numero di individui nel breve tempo necessario per la lettura di un signolo microchip per individuo. Naturalmente, l'intento primario è di generalizzare l'uso del gene-chip nel campo della medicina preventiva, come ad esempio nella diagnostica prenatale o nella diagnosi precoce della predisposizione ai tumori (perdita di geni oncosoppressori). D'altronde, non è necessaria una conoscenza specialistica per rendersi conto dell'enorme potenziale informativo che i microchips elettronici possono avere nello studio popolazionistico della diversità genomica a livello molecolare. La loro massiccia applicazione, insieme all'uso sistematico delle mappe ottiche di restrizione, è dunque soltanto una questione di investimenti finanziari che non dovrebbe essere difficile reperire, specialmente dal settore bioindustriale, data la sempre più palese evidenza che l'identificazione dei fattori genetici e ambientali associati alle cosiddette malattie multifattoriali comuni (ovvero quelle che maggiormente interessano l'opinione pubblica e il mondo della bioindustria) è funzione diretta del progredire delle nostre conoscenze sulle intricate interazioni tra diversità genetica e malattie.
3. Il Progetto Genoma
a) Lo stato dell'arte: progressi e problemi
Del Progetto Genoma e del ruolo primario ch'esso ha svolto nel corso dell'ultimo decennio nell'evoluzione delle nostre conoscenze genetiche, si è detto ripetutamente fin dalla premessa del presente articolo. Questo capitolo conclusivo intende riassumerne gli intenti cercando di chiarire, anche al lettore non specialista, i vari interrogativi che l'argomento pone. Fin dai suoi primi passi, il Progetto Genoma ha suscitato nell'opinione pubblica le reazioni più diverse; c'è chi ne attende l'impatto positivo sulla prevenzione e la cura delle malattie ereditarie, chi ne paventa i risvolti inquisitivi nella vita privata individuale, e chi resta del tutto indifferente, quasi per legittima difesa verso quanto è difficile capire immediatamente. Alla base di reazioni così diverse c'è lo stesso denominatore comune: la mancanza di informazione adeguata. Sarebbe vera arroganza pretendere di poter colmare questa lacuna in questa occasione, ma sarebbe già molto se questo capitolo conclusivo riuscisse a rettificare alcune inesattezze che spesso ricorrono nei mezzi di informazione. Come si può facilmente desumere da quanto si è già detto a proposito del preminente ruolo dell'industria biotecnologica in questo settore, la mappatura del genoma è diventata argomento da ‛ordine del giorno' nei consigli di amministrazione dei maggiori complessi farmaceutici e la motivazione specifica per diverse iniziative a carattere internazionale, come quella patrocinata dall'organizzazione HUGO (Human Genome Organization) che coordina le attività delle migliaia di scienziati oggi impegnati nella colossale impresa di decifrare la sequenza totale dei 3 miliardi di nucleotidi allineati in duplice copia lungo le 23 coppie di cromosomi che costituiscono il genoma di Homo sapiens .
A questo punto va precisato che, se c'è una privacy che rischia di venire infranta da questo tipo di informazione, essa è quella della specie e non del singolo individuo, come si sente ripetere talvolta anche nei circoli scientifici. Infatti, la determinazione, effettuata una sola volta, della sequenza dei 3 miliardi di nucleotidi di Homo sapiens (un'impresa di per se stessa colossale) non rivelerebbe assolutamente nulla sulle innumerevoli differenze che si potrebbero reperire allo stesso livello d'indagine in individui diversi della stessa specie, popolazione, famiglia e persino coppia di gemelli uniovulari. Per contro, l'ingente massa di dati raccolti, confrontata con quelle analoghe, raccolte con lo stesso approccio sperimentale, relative ai genomi di altre specie animali e vegetali, servirebbe meglio d'ogni altro tipo di ricerca per ricostruire le genealogie dell'evoluzione biologica e interpretare i meccanismi molecolari che le determinano. Tuttavia, se questo dovesse essere l'unico risultato di questo megaprogetto, potrebbero aver ragione gli scettici a contestarne la priorità a scapito di tante altre problematiche scientifiche non meno importanti seppur di minore effetto (poco importa se positivo o negativo) sull'immaginazione del pubblico e dei suoi rappresentanti politici e amministrativi. Ma se anche fosse vero che la motivazione primaria del Progetto è sostanzialmente quella stessa curiosità che ha spinto l'uomo del XX secolo a investire fortune immense per andare a curiosare direttamente i segreti della Luna e di Marte, chi si sentirebbe di definirla curiosità inutile? Come si può escludere la possibilità che da una siffatta lettura completa del bagaglio ereditario della nostra specie derivi la ‛stele di Rosetta' genetica indispensabile per decifrare gli innumerevoli e persistenti segreti dell'evoluzione biologica? Di fatto, si deve ammettere che il lancio del Progetto è riuscito a focalizzare l'attenzione di enti pubblici e privati di tutto il mondo sulla necessità di investimenti speciali per finanziarne la realizzazione. Questi investimenti hanno consentito il succedersi degli straordinari progressi, descritti nelle pagine precedenti, sui metodi di scoperta e di analisi della diversità genetica, ma - a circa un decennio dalla nascita del Progetto - il traguardo principale del sequenziamento dell'intero genoma è stato a malapena scalfito, in quanto la parte di genoma umano di cui si è completata la sequenza (peraltro in frammenti non necessariamente contigui) è appena del 5%. Pertanto, d'ora in avanti il vero dilemma non sarà più quello di decidere se completare o meno la sequenza totale del genoma, ma piuttosto se l'impresa merita quella priorità di investimenti economici e intellettuali che taluni vorrebbero accordarle a tutti i costi. A questo riguardo, il parere dei genetisti differisce da quello dei biologi molecolari. Questi ultimi, citando ad esempio l'universalità del codice genetico e dei meccanismi che regolano la funzione dei geni, insistono sulle ricadute che la conoscenza completa del genoma di organismi superiori (nella fattispecie, di quello che meglio conosciamo) potrà avere sulle applicazioni mediche e sulla ricerca biologica in generale; i genetisti sostengono invece l'opportunità prioritaria di sequenziare piccole parti del genoma in individui diversi, poiché da Mendel ai nostri giorni lo studio dell'eredità biologica (a qualsiasi livello esso venga condotto) significa analisi della variabilità individuale nelle popolazioni e nella discendenza di particolari tipi d'incrocio. In pratica, quanto sta succedendo presso grandi e piccoli centri di ricerca genomica, sia accademici che industriali, è qualcosa d'intermedio tra queste due estreme posizioni, come lo sforzo di mappare innanzi tutto la posizione relativa dei vari tipi di minicromosomi artificiali di varia lunghezza e contenuto genomico (cloni YAC, BAC, P1, fagi, plasmidi, ibridi cellulari ridotti) e quella delle innumerevoli sequenze di riferimento genomico (STS) ricavate dal limbo dei geni da scoprire. Come si è già detto, le prime mappe fisiche di minicromosomi YAC per l'intero genoma e minicromosomi BAC per il cromosoma 22 sono state pubblicate entro il traguardo dei dieci anni dall'avvio del Progetto Genoma, come i più ottimisti tra i suoi sostenitori avevano previsto. Tuttavia, considerato che si tratta solo di un modesto traguardo di tappa - come si è detto -, il 95% del genoma resta ancora da sequenziare. Ma il significato strategico del lavoro fatto sinora è notevole, perché la scomposizione dell'intero genoma in frammenti dall'ordine noto e di lunghezza compatibile con le metodologie di sequenziamento era la premessa essenziale per raggiungere il traguardo finale della sequenza completa dei tre miliardi di nucleotidi che costituiscono il bagaglio ereditario della nostra specie. La meta è dunque ancora terribilmente lontana, ma la strategia messa a punto per raggiungerla (ovvero il sequenziamento separato dei vari tipi di mini- e microcromosomi artificiali in cui il genoma è stato parcellizzato) ha ormai ridotto l'impresa a una semplice questione di investimento di tempo e di mezzi. Sembra ovvio, ha scritto argutamente un corrispondente scientifico del quotidiano londinese ‟The independent", che il vero problema del Progetto Genoma sia ormai non la vastità del lavoro che resta da fare e il suo costo, ma piuttosto la noia che esso rischia di generare nelle migliaia di giovani ricercatori reclutati per realizzarlo. Walter Gilbert, premio Nobel nel 1980 per aver messo a punto uno dei metodi più efficienti per sequenziare nucleotidi e difensore tra i più convinti del Progetto, ha affermato senza mezzi termini che il sequenziamento genomico è un problema di produzione, non di ricerca. Di conseguenza, egli ha sostenuto fin dall'inizio l'opportunità di affidare all'industria un ruolo predominante, se non addirittura esclusivo, nella realizzazione dell'impresa e, per coerenza, ha fondato lui stesso una delle prime industrie biotecnologiche private per produrre e vendere informazioni sulla sequenza del genoma umano. Come si è già detto, il numero delle iniziative industriali nella ricerca genomica è cresciuto rapidamente negli ultimissimi anni, non solo per fare del lavoro di sequenziamento a pagamento, ma per ogni tipo di servizio biotecnologico di grande richiesta che si presti all'automazione robotica. Qualcuno ritiene pericoloso il diffondersi di troppo stretti legami tra accademia e industria, ma non è esagerato affermare che senza gli investimenti notevoli che quest'ultima ha stanziato negli ultimi anni (prevalentemente attraverso il reclutamento di eccellenti menti ed esperte braccia e con l'allestimento ex novo di numerosi centri di ricerca specificamente progettati per la ricerca genomica) il Progetto Genoma Umano avrebbe rischiato di abortire sul nascere.
A chi scrive sembra giusto e desiderabile che il mondo produttivo privato partecipi direttamente - con i rischi relativi - a una così grossa spesa collettiva e non trova scandaloso che il movente primario di questa partecipazione industriale non sia l'impulso altruistico del mecenate, ma la calcolata aspettativa di una futura produzione industriale di sonde diagnostiche e di geni clonati su vasta scala che diventeranno i farmaci del prossimo millennio. Ma, com'è ovvio, di un tale scenario vengono additati più spesso i rischi che i benefici. Per esempio, c'è il timore del diffondersi dei brevetti industriali sulle scoperte scientifiche di ovvio potenziale economico e, in particolare, delle sequenze nucleotidiche delle regioni più importanti del nostro bagaglio ereditario.
A dimostrare la realtà di questi timori va ricordata la lunga querelle esplosa tra scienziati, industrie e organi di ricerca accademici a proposito della proposta (paradossalmente avanzata negli Stati Uniti proprio dai National Institutes of Health) di brevettare tutti i cloni delle librerie di cDNA allo scopo di imperdirne lo sfruttamento a titolo commerciale. La risposta unanime dell'opinione pubblica, del mondo degli addetti ai lavori e degli organi di controllo coinvolti nella materia è stata quella di negare la liceità di applicare un brevetto ai geni ancora ‛nel limbo', pur consentendo che ciò venisse fatto nel caso di sequenze specifiche dalle già provate potenzialità per applicazioni mediche, anche se per un periodo limitato e secondo le regole vigenti nella legislazione dei brevetti. Questa salomonica conclusione ha tenuto ovviamente conto dell'incentivo che può fornire ai singoli ricercatori e alle industrie, così che si dedichino sempre più intensamente alla realizzazione del traguardo finale del Progetto Genoma e allo sviluppo delle sue tanto attese applicazioni mediche. Ma a spazzar via le perplessità generate dalla sempre più incalzante interazione tra ricerca accademica e industriale, basterà sottolineare che molte delle più interessanti acquisizioni nel settore della medicina molecolare dell'ultimo decennio sono state realizzate (e interamente finanziate) dalle industrie biotecnologiche: è questo il caso dei primi clonaggi di geni d'interesse farmacologico (interferone, fattore VIII e IX, attivatore tessutale del plasminogeno, coinvolti rispettivamente nella terapia di malattie virali, dei tipi più comuni di emofilia e dell'infarto miocardico), e della messa a punto della reazione polimerasica a catena (PCR), che ha non solo enormemente semplificato la produzione industriale dei kits diagnostici, ma ha anche fornito un mezzo tanto semplice quanto prezioso per l'insegnamento sperimentale della biologia molecolare. È opportuno a questo proposito sottolineare che il segreto di questi successi - realizzati prevalentemente da ricercatori americani - non è questione di mezzi e di uomini che, come si usa dire, abbondano soltanto al di là dell'Atlantico; i mezzi dell'industria sono ingenti dovunque e quanto ai protagonisti delle ricerche più avanzate va sottolineato che una buona parte di essi è nata e ha studiato al di fuori degli Stati Uniti. La differenza essenziale sta nel coraggio che il privato americano mette negli investimenti a rischio, non per generosità sociale ma per la convinzione diffusa che i progressi tecnici (prevalentemente ‛apollinei') sono molto spesso il risultato di scoperte del tutto impreviste (‛dionisiache') e che pertanto il modo più efficace per alimentare la ricerca finalizzata - per paradossale che possa sembrare - resta sempre quello di sovvenzionare generosamente la ricerca di base.
b) Considerazioni etiche
Si è già precisato che dalla realizzazione dell'obiettivo fondamentale del Progetto Genoma, cioè dalla conoscenza della sequenza totale dei tre miliardi di nucleotidi che compongono il bagaglio ereditario della specie, non potrebbe derivare alcuna violazione della privacy individuale.
Ma la questione si pone in termini diversi se ci si riferisce allo studio delle innumerevoli differenze ereditabili, e oggi riconoscibili a livello molecolare, tra individui o popolazioni di individui appartenenti alla stessa specie. Alla descrizione globale di queste differenze in termini molecolari si è dato di recente il nome di ‛diversità genomica', un aspetto della ricerca genetica che di nuovo ha soltanto il livello di osservazione sulla base del quale vengono catalogati individui della stessa specie. Per restare in tema di Homo sapiens, è nozione comune che questa aspirazione a catalogare i singoli individui in base alla loro apparenza è vecchia quanto il tentativo umano di descrivere la natura: ne è testimone la teoria aristotelica dei quattro umori, di cui si trova ancora traccia nel vocabolario odierno di tutti i giorni (il ‛melanconico', il ‛bilioso', il ‛flemmatico', il ‛sanguigno'). Questa innata propensione a catalogare è ora giunta a un grado di perfezione tale da potersi concretamente affermare che al livello del DNA ciascun individuo è ‛unico' e questa sua individualità può essere oggi descritta e documentata attraverso l'esame della sequenza anche solo di poche migliaia dei suoi nucleotidi. La descrizione più completa possibile di queste ‛differenze' in popolazioni di individui della nostra specie (sulla base dei numerosi esempi di varianti molecolari) è l'ambito traguardo del progetto HGD (Human Genome Diversity), che si potrebbe definire come l'approccio popolazionistico alla sequenza del genoma umano, in quanto consiste nello sceglierne minuscole frazioni di preminente interesse biologico per studiarne la variabilità individuale nelle popolazioni. La scelta delle frazioni genomiche da studiare si ispira a due criteri fondamentali: il loro potenziale informativo (per esempio, quelle già note per essere la sede di polimorfismi genetici molto comuni e di facile classificazione) e una distribuzione cromosomica tale da avere a disposizione, per quanto possibile, un congruo numero di marcatori genetici altamente informativi per ciascuna delle 23 paia di cromosomi della specie.
La scelta delle popolazioni da studiare è un problema tutt'altro che facile da risolvere: c'è chi sottolinea l'urgenza di studiare popolazioni sopravvissute a millenni di condizioni ambientali stressanti (e attualmente a rischio di estinzione per graduale assimilazione da parte di popolazioni limitrofe) per raccogliervi preziose informazioni sul possibile valore adattativo della loro presente costituzione genetica; c'è chi preferisce focalizzare l'attenzione prevalente del progetto sull'analisi, quanto più completa possibile, della costituzione genetica di individui appartenenti a popolazioni diverse allo scopo di ricostruirne la genealogia storica sulla base delle differenze genetiche che oggi le separano (v. genetica: Genetica delle popolazioni, vol. X); c'è infine chi s'interessa all'analisi della diversità genetica in generale e di quella relativa a popolazioni isolate soltanto perché queste ultime rappresentano il materiale di studio ideale per l'identificazione dei fattori genetici e ambientali coinvolti nella manifestazione delle cosiddette malattie multifattoriali comuni.
Da quanto si è detto traspare un futuro carico di promesse per i potenziali benefici che si possono ottenere dall'insieme delle conoscenze accumulate finora sulle interazioni tra diversità genetica ed eventi normali e patologici della vita. D'altronde, appare anche evidente che è proprio l'approccio popolazionistico allo studio del genoma - e non la descrizione una tantum della sua composizione e sequenza nucletodica - che potrebbe mettere a rischio la privacy individuale in nome della conoscenza scientifica. Riassumiamo brevemente gli interrogativi etici più seri che vengono associati al Progetto Genoma insieme alle più comuni risposte degli addetti ai lavori, pienamente condivise da chi scrive.
La prima questione riguarda la possibilità che le caratteristiche genetiche dei singoli individui siano incluse negli schedari di uno Stato leviatano o, nella migliore delle ipotesi, in quelli delle compagnie di assicurazione. Una simile evenienza sarebbe ovviamente da imputare al comportamento errato dell'uomo, non della sua scienza. E l'uomo, come sappiamo, potrebbe fare di peggio, come premere i pulsanti che scatenano l'arsenale nucleare. Il rimedio non può che essere quello di una adeguata legislazione che eviti e punisca gli abusi sull'uso dell'informazione in oggetto senza sopprimerne gli innegabili vantaggi. Tra questi ultimi vale la pena di citare l'esempio degli studi di genetica molecolare popolazionistica condotti in Finlandia, che hanno recentemente portato alla scoperta delle mutazioni responsabili di quattro gravi malattie ereditarie grazie alla disponibilità (e facile accessibilità) dei censimenti delle malattie ereditarie raccolti dai servizi di statistica sanitaria del paese.
Un secondo interrogativo si pone riguardo all'utilità di predire al malcapitato portatore di una mutazione dominante come la corea di Huntington, che intorno al quarantesimo anno di età egli/ella svilupperà una così devastante malattia e che metà dei suoi figli possono essere portatori della stessa malattia. Il problema non è granché diverso da quello del rivelare la prognosi infausta al paziente affetto da un tumore maligno a lenta evoluzione. In generale - e tanto vale per qualunque diagnosi genetica precoce di malattia grave a insorgenza nell'età adulta - si potrebbe discutere all'infinito sul dilemma di informare o meno il paziente a rischio in assenza di terapie risolutive. Quando queste ultime esistono, il dilemma scompare. I progressi delle conoscenze sul genoma umano e sulle funzioni specifiche di singoli geni nello sviluppo normale degli organismi sono la premessa (e la promessa) della biologia molecolare per la creazione di dette terapie risolutive.
Un terzo e più difficile interrogativo riguarda l'opportunità di classificare, e secondo quali criteri, le caratteristiche ereditarie in favorevoli o sfavorevoli, con l'inevitabile ‛allusione' eugenica che una tale pratica comporta. La genetica molecolare descrive delle differenze a un livello di osservazione che di per sé non può essere sufficiente a prevedere l'adattamento dell'individuo al suo ambiente. A sottolineare la difficoltà di tali previsioni basti l'esempio delle mutazioni per la talassemia e la falcemia che sono letali negli individui omozigoti, ma che negli eterozigoti conferiscono protezione contro la malaria maligna.
Un dilemma ricorrente, infine, è relativo alla decisione se lasciar procedere o meno una gravidanza che porterebbe alla nascita di un individuo con gravi anomalie. L'opinione condivisa dalla maggioranza è di lasciare questa decisione ai genitori, ma il problema resterà di difficile soluzione fino a quando non si sarà scoperto un efficiente sistema per il trattamento terapeutico dell'embrione malato.
La drammaticità degli interrogativi su riportati, e dei numerosi altri che potrebbero esser posti sullo stesso tema, è funzione della persistente difficoltà a trovare un rimedio efficace per la maggior parte delle malattie che si possono identificare in epoca prenatale (e persino in un embrione di pochi blastomeri a 24 ore dalla fecondazione artificiale in vitro). È chiaro che se si potesse intervenire con il trattamento adeguato per ogni problema genetico diagnosticato nelle prime fasi dello sviluppo embrionale, non vi sarebbero né riserve etiche, né timori di violazione della privacy individuale.
La diagnosi prenatale, e persino il mantenimento in vitro del prodotto del concepimento, potrebbero essere accettati da tutti se queste pratiche dovessero costituire un inevitabile prerequisito per la terapia definitiva e quindi per la salvezza di un embrione, di una madre, di entrambi, della nostra stessa specie. È possibile che le tanto temute biotecnologie genetico-molecolari riescano a realizzare un tale scenario entro la fine di questo millennio.
Non è certo necessario essere degli esperti per capire quanto la possibilità di disporre di misure terapeutiche molecolari a favore dell'embrione e dell'individuo che da esso deriva sia chiaramente subordinata alla legittimità della ricerca embriofetale. Ma c'è chi contesta con veemenza una tale opportunità, c'è chi vede nei medici molecolari dei ‛Frankenstein' piuttosto che degli ‛Schweitzer' e ne teme ragionevolmente gli errori, sia pure involontari, o, peggio, gli abusi.
Sarebbe irresponsabile ridicolizzare queste preoccupazioni. Purtroppo, come qualcuno ha giustamente sottolineato, ‟le scoperte scientifiche arrivano senza le istruzioni per l'uso". È pertanto quanto mai urgente che queste vengano approntate, ed è naturalmente essenziale che a questo processo partecipi la società nel suo insieme, che deve esser chiamata a esprimere un voto a favore o contro le normative che vengono proposte dalle competenti commissioni governative, ispirandosi ai fatti già noti piuttosto che alle dichiarazioni di principio. È con questo scenario in mente che James Watson ha ritenuto necessario lanciare la sua crociata contro l'analfabetismo molecolare di cui si è detto a proposito del DNA Learning Center di Cold Spring Harbor, una scuola per un pubblico non specializzato dove si impara che cos'è il DNA.
BIBLIOGRAFIA
Adams, M. D., Kerlavage, A. R., Fleischmann, R. D. e altri, Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence, in ‟Nature", 1995, CCCLXXVII, suppl., pp. 3-173.
Antonacci, R., Marzella, R., Finelli, P., Lonoce, A., Forabosco, A., Archidacono, N., Rocchi, M., A panel of subchromosomal painting libraries representing over 300 regions of the humane genome, in ‟Cytogenetics and cell genetics", 1995, LXVIII, pp. 25-32.
Arcot, S. S., Wang, Z., Weber, J. L., Deininger, P. L., Batzer, M., Alu repeats. A source for the genesis of Primate microsatellites, in ‟Genomics", 1995, XXIX, pp. 136-144.
Batzer, M. A., Arcot, S. S., Phinney, J. W. e altri, Genetic variation of recent ALU insertions in human populations, in ‟Journal of molecular evolution", 1996, XLII, pp. 22-29.
Biemont, C., Brookfield, J. F., Les gènes sauteurs: patrimoine sous influence, in ‟La recherche", 1996, CCLXXXVII, pp. 50-55.
Bloom, M. V., Freyer, G. A., Micklos, D. A., Laboratory DNA science: an introduction to recombinant DNA techniques and methods of genome analysis, Menlo Park, Cal., 1996.
Bowcock, A. M., Ruiz-Linares, A., Tomfohrde, J., Minch, E., Kidd, J. R., Cavalli-Sforza, L. L., High resolution of human evolutionary trees with polymorphic microsatellites, in ‟Nature", 1994, CCCLXVIII, pp. 455-457.
Cai, W., Housman, D. E., Wang, Y., Schwartz, D. C., Ordered restriction endonuclease maps of yeast artificial chromosomes created by optical mapping on surfaces, in ‟Proceedings of the National Academy of Sciences", 1995, XCII, pp. 5164-5169.
Campunzano, V., Montermini, L., Moltò, M. D. e altri, Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion, in ‟Science", 1996, CCLXXI, pp. 1423-1427.
Cann, R. L., Stoneking, M., Wilson, A. C., Mitochondrial DNA and human evolution, in ‟Nature", 1987, CCCXXV, pp. 31-36.
Chumakov, I. M., Rigault, P., Le Gall, I. e altri, A YAC contig map of the human genome, in ‟Nature", 1995, CCCLXXVII, suppl., pp. 175-297.
Collins, J. E., Cole, C. G., Smink, L. J. e altri, A high density YAC contig map of humane chromosome 22, in ‟Nature", 1995, CCCLXXVII, suppl., pp. 367-379.
Deininger, P. L., Batzer, M., Evolution of retroposons, in Evolutionary biology (a cura di M. K. Hecht e altri), vol. XXVII, New York 1993.
Deininger, P. L., Batzer, M., SINE master genes and population biology, in The impact of Short Interspersed Elements (SINEs) on the host genome (a cura di R. J. Maraia), Georgetown, Tex., 1995, pp. 43-60.
Dib, C., Fauré, S., Fizames, C. e altri, A comprehensive genetic map of the human genome based on 5,264 microsatellites, in ‟Nature", 1996, CCCLXXX, pp. 152-154.
Drummond-Borg, M., Deeb, S. S., Motulsky, A. G., Molecular patterns of X chromosome-linked color vision genes among 134 men of European ancestry, in ‟Proceedings of the National Academy of Sciences", 1989, LXXXVI, pp. 983-987.
Gemmili, R. M., Chumakov, I., Scott, P. e altri, A second generation map of human chromosome 3, in ‟Nature", 1995, CCCLXXVII, suppl., pp. 299-320.
Hudson, T. J. e altri, An STS-based map of the human genome, in ‟Science", 1995, CCLXX, pp. 1945-1954.
Kim, U., Shizuya, H., Kang, H. e altri, A bacterial artificial chromosome-based framework contig map of humane chromosome 22q, in ‟Proceedings of the National Academy of Sciences", 1996, XCIII, pp. 6297-6301.
Krauter, K., Montgomery, K., Yoon, S., A second- generation YAC contig map of human chromosome 12, in ‟Nature", 1995, CCCLXXVII, suppl., pp. 321-334.
Labuda, D., Zietkiewicz, E., Mitchell, G. A., Alu elements as a source of genomic variation: deleterious effects and evolutionary novelties, in The impact of Short Interspersed Elements (SINEs) on the host genome (a cura di R. J. Maraia), Georgetown, Tex., 1995, pp. 1-24.
Le Beau, M. M., One FISH, two FISH, red FISH, blue FISH, in ‟Nature genetics", 1996, XII, 4, pp. 341-344.
Macdonald, F., Ford, C. H. J., Molecular biology of cancer, Oxford 1997.
McKusick, V. A., Mendelian inheritance in man: a catalog of human genes and genetic disorders, 2 voll., Baltimore, Md., 199411.
Meng, X., Benson, K., Chada, K., Huff, E. J., Schwartz, D. C., Optical mapping of lambda bacteriophage using restriction endonucleases, in ‟Nature genetics", 1995, IX, pp. 432-438.
Micklos, D. A., Freyer, G. A., DNA science, Cold Spring Harbor, N. Y., 1990.
Mullis, K. B., The unusual origin of the Polymerase Chain Reaction, in ‟Scientific American", 1990, CCLXII, 4, pp. 36-43 (tr. it.: La scoperta della reazione a catena della polimerasi, in ‟Le scienze", 1990, XLIV, 262, pp. 32-39).
Passarge, E., Taschenatlas der Genetik/Color atlas of genetics, Stuttgart-New York 1995.
Pennica, D., Holnes, W. E., Kohr, W. J. e altri, Cloning and expression of human tissue-type plasminogen activator cDNA in E. coli, in ‟Nature", 1983, CCCI, pp. 214-221.
Samad, A., Huff, E. J., Cai, W., Schwartz, D. C., Optical mapping: a novel, single molecule approach to genomics analysis, in ‟Genome research", 1995, V, 1, pp. 1-4.
Silver, L. M., Mouse genetics. Concepts and applications, Oxford 1995.
Singer, M., Berg, P., Genes and genomes, Mill Valley, Cal., 1991 (tr. it.: Geni e genomi, Bologna 1993).
Speicher, M. R., Gwyn Ballard, S., Ward, D. C., Karyotyping human chromosomes by combinatorial multi-fluor FISH, in ‟Nature genetics", 1996, XII, 4, pp. 368-375.
Strachan, T., Read, A. P., Human molecular genetics, Oxford-New York 1996 (tr. it.: Genetica umana molecolare, Roma 1997).
Watson, J. D., Gilman, M., Witkowski, J., Zoller M., Recombinant DNA, New York 19962 (tr. it.: DNA ricombinante, Bologna 1994).
Applicazioni della genetica di Jonathan Beckwith
Sommario: 1. Introduzione: a) i recenti progressi della genetica; b) la rivoluzione continua. 2. Vantaggi medici e sociali della nuova genetica: a) mappatura e identificazione di geni umani; b) vaglio genetico (genetic screening); c) test genetici prima delle nuove tecnologie del DNA; d) la nuova era e il Progetto Genoma Umano; e) trattamento delle malattie genetiche; f) la terapia genica; g) le impronte del DNA (DNA fingerprinting). 3. L'industria delle biotecnologie: a) vaccini; b) test diagnostici; c) prodotti genici; d) ingegneria genetica delle piante; e) problemi. 4. Problemi sociali ed etici dopo il 1975: a) terapia genica; b) errori d'opinione sulla genetica; c) ogni essere umano è portatore di geni responsabili di malattie genetiche recessive; d) conseguenze negative degli screenings genetici; e) la genetica del comportamento umano e il libero arbitrio; f) il rapporto tra la genetica e la legge. 5. L'eugenetica: a) l'eugenetica oggi; b) terapia genica; c) il rapporto tra la genetica e le priorità sociali. □ Bibliografia.
1. Introduzione
a) I recenti progressi della genetica
Verso la metà degli anni settanta si è verificata nel campo della genetica una profonda rivoluzione, dovuta principalmente a due fattori: in primo luogo, sono state messe a punto tecniche per la determinazione della sequenza in basi della molecola dell'acido desossiribonucleico (DNA), secondo le metodologie schematizzate in fig. 1; ciò ha permesso ai ricercatori di determinare la sequenza delle basi azotate nei geni e da questa di risalire alla sequenza amminoacidica del prodotto genico. In secondo luogo, l'uso degli enzimi di restrizione ha consentito la frammentazione del DNA, permettendo la ricongiunzione di frammenti provenienti da organismi diversi. In alcuni casi, è stato possibile prelevare i frammenti di DNA - e l'informazione in essi contenuta - e reintrodurli nell'organismo dal quale erano stati ottenuti o in altri organismi, riuscendo così a trasferire i geni dall'organismo di provenienza a un organismo estraneo. Questa tecnica, denominata del ‛DNA ricombinante' o del ‛montaggio genico' (gene splicing), è stata usata per trasferire geni fra una quantità di organismi, dai più semplici batteri agli esseri umani (v. figg. 2 e 3; v. biotecnologie, vol.VIII; v. genetica).
Gli straordinari progressi tecnici avvenuti nel rivoluzionario periodo degli anni settanta determinarono una trasformazione concettuale della genetica. Fino a quel momento i geni erano considerati relativamente inaccessibili a livello fisico, ma, allorché si compresero pienamente le potenzialità delle tecnologie del DNA, divenne rapidamente chiaro che erano finalmente disponibili molti strumenti fondamentali che permettevano di affrontare problemi scientifici sino ad allora ritenuti intrattabili. Si verificò un rapido progresso di settori della ricerca come l'immunologia, la biologia dello sviluppo, la ricerca sul cancro e la genetica umana. La possibilità di isolare geni codificanti per un anticorpo, per fattori di controllo importanti nello sviluppo embrionale, per un componente cellulare coinvolto nella genesi del cancro o per il prodotto tossico di batteri o virus patogeni, rappresentava la possibilità di studiare in dettaglio ognuno di questi processi, la qual cosa avrebbe potuto determinare numerose conseguenze pratiche.
b) La rivoluzione continua
Il sorgere dell'era del DNA ricombinante ha avuto un effetto catalitico sullo sviluppo di tecnologie ancor più avanzate per l'analisi dei geni. Prima che questa trasformazione avesse luogo, i biologi si erano concentrati, per la maggior parte, sulla soluzione di problemi biologici. La messa a punto di nuove tecnologie era stata talvolta il prodotto collaterale di tali sforzi, ma raramente aveva costituito la finalità primaria dei ricercatori in scienze biologiche.
I vantaggi, in termini sia di riconoscimenti che di possibilità di lavoro, ottenuti dai creatori di nuove tecniche non erano comparabili con quelli conseguiti da coloro che scoprivano nuovi principî biologici. Comunque, il progredire della ricerca di base e applicata, dovuto agli straordinari successi delle tecnologie del DNA ricombinante, ha trasformato lo sviluppo di nuovi approcci genetici in un modo considerevolmente più proficuo per conseguire un avanzamento nella carriera accademica. Di conseguenza, molti biologi hanno abbandonato l'analisi dettagliata dei sistemi biologici attraverso tecniche non molecolari, rivelatasi poco dinamica, per diventare tecnologi, usando la loro creatività in una direzione estremamente diversa. Gran parte dell'inventiva scientifica del periodo successivo è stata dedicata alla creazione di nuove tecniche per lo studio di problemi biologici e al tentativo di renderle sempre più efficienti. Ciò ha portato, nei decenni successivi, a una serie di raffinamenti di quelle tecniche e un grande numero di genetisti molecolari si è dedicato allo sviluppo di strategie sempre più ingegnose per studiare i geni e la loro funzione. Inoltre, le potenziali applicazioni pratiche della nuova genetica hanno portato i genetisti molecolari a creare una nuova attività industriale, quella della biotecnologia. Questa attività coinvolge oggi migliaia di imprese in tutto il mondo e l'intensa competizione fra tali industrie ha fatto sì che esse concentrassero molti dei loro sforzi sugli sviluppi tecnologici, aumentando in tal modo le possibilità di creare nuovi prodotti. Molte di queste imprese hanno commercializzato equipaggiamenti (kits) e materiali per gli scienziati che hanno agevolato la ricerca, sia nell'ambito accademico che nell'attività biotecnologica più in generale.
Due applicazioni illustrano bene i progressi fatti negli ultimi venti anni. La prima è costituita da un approccio per mappare la collocazione e l'ordine reciproco di geni sui cromosomi umani, nota come tecnica dei polimorfismi della lunghezza dei frammenti di restrizione (Restriction Fragment Length Polymorphism, RFLP). Eccetto un piccolo numero di casi di individui che posseggono cromosomi supplementari o mancano di un cromosoma, tutti i membri della specie umana possiedono 46 cromosomi nelle loro cellule (con l'eccezione delle cellule germinali che ne possiedono solo 23, cioè metà del corredo cromosomico); in generale la stessa serie di geni è localizzata nella stessa posizione sui nostri cromosomi. Pertanto, in quest'ottica macroscopica, la composizione genetica della stragrande maggioranza della popolazione umana è la stessa.
Tuttavia, è noto già da diverso tempo che, qualora si esamini la composizione genetica degli individui in un'ottica microscopica, per esempio a livello di proteine specifiche, emerge un certo numero di variazioni fra individui, non correlabili a stati patologici o a malfunzionamento dei processi dell'organismo: queste variazioni sono denominate ‛polimorfismi'. Con l'avvento delle nuove tecnologie del DNA, la capacità di individuare polimorfismi è aumentata enormemente. In primo luogo, si possono prendere in considerazione tratti confrontabili di DNA di due individui e verificare la loro risposta a enzimi di restrizione, cioè a enzimi che riconoscono particolari sequenze nel DNA e producono una scissione della molecola in quei siti. Differenze nel profilo di frammentazione di due tratti di DNA indicano differenze nella sequenza di DNA di quei frammenti; diversità nella localizzazione dei siti di riconoscimento sono messe in evidenza dalle variazioni di lunghezza dei frammenti di DNA prodotti dal trattamento con l'enzima di restrizione. La RFLP (e le sue varianti più recenti e perfezionate) è stata usata per costruire una mappa dettagliata dei cromosomi umani e di altri organismi; tale mappa, unitamente alla conoscenza dello spettro di polimorfismi lungo la mappa stessa, permette la localizzazione di geni associati a vari caratteri umani. La posizione di un gene sulla mappa è determinata dalla sua vicinanza a un marcatore polimorfico noto. La costruzione di una mappa dettagliata ha così permesso di localizzare numerosi geni associati a malattie umane (v. Botstein e altri, 1980).
Un secondo esempio di progresso tecnologico che ha avuto un profondo impatto su biologia e biotecnologia è costituito dalla reazione di polimerizzazione a catena (Polymerase Chain Reaction, PCR), illustrata in fig. 3B. Questa tecnica permette ai ricercatori di ottenere grandi quantità di DNA corrispondenti a una regione specifica del cromosoma una volta che sia nota la sequenza del DNA di quella regione, che può comprendere molti geni o un singolo gene: la PCR prevede che l'informazione relativa alla sequenza di DNA che comprende la regione di interesse sia utilizzata per sintetizzare piccoli frammenti di DNA (oligonucleotidi) corrispondenti alle estremità di tale regione. Questi oligonucleotidi vengono poi ibridati mediante le loro sequenze complementari al campione di DNA che deve essere analizzato. Un enzima replicherà il DNA usando come siti di partenza (priming sites) le due posizioni al confine fra la regione nella quale gli oligonucleotidi sono ibridati e il DNA a singolo filamento. Ripetendo questo processo molte volte, la quantità di copie di DNA della regione d'interesse può essere amplificata enormemente, producendo quantità di materiale sufficiente a essere sottoposto a manipolazione genica e ad altri tipi di analisi del DNA. Questo significa, per esempio, che singoli geni possono essere estratti in forma pura dall'intero corredo delle migliaia di geni presenti nei cromosomi di un organismo. La tecnica è talmente sensibile che può essere usata per ottenere DNA amplificato da campioni contenenti quantità minime di materiale cellulare. Un singolo follicolo pilifero umano ha fornito DNA sufficiente da permetterne l'amplificazione tramite PCR.
2. Vantaggi medici e sociali della nuova genetica
a) Mappatura e identificazione di geni umani.
A partire dal 1980, la RFLP ha permesso un rapido progresso di nuovi approcci finalizzati alla localizzazione di geni umani correlati con varie malattie; il loro impiego, insieme ad altri metodi di analisi del DNA come la PCR, ha reso infinitamente più semplice il compito di mappare e caratterizzare geni responsabili di svariate malattie umane e codificanti per numerosi tratti somatici. In particolare, tratti - sia dominanti che recessivi - che mostrino un semplice modello ereditario sono un bersaglio ideale per questo genere di analisi.
b) Vaglio genetico (genetic screening)
Ancor prima dell'avvento delle nuove tecnologie del DNA, erano state già identificate numerose malattie genetiche e, in alcuni casi, erano stati messi a punto test di screening per verificare la presenza di alterazioni genetiche tali da dar luogo a malattia in un individuo o in qualche componente della sua progenie. Si possono distinguere diversi tipi di alterazioni genetiche: in primo luogo, l'alterazione della struttura o del numero dei cromosomi può causare seri problemi di sviluppo; per esempio, la sindrome di Down, che conduce a un grave ritardo mentale e a numerosi problemi fisici, è dovuta alla presenza di una copia supplementare del cromosoma 21 in tutte le cellule dell'organismo. Le malattie genetiche più comuni, come la corea di Huntington, la fibrosi cistica e l'anemia falciforme, non sono però dovute a vistose modificazioni dei cromosomi, ma sono per lo più il risultato di alterazioni di singoli geni. Tali disordini della funzione genica possono essere a loro volta suddivisi in due classi: la prima comprende quelli, come la corea di Huntington, causati da mutazioni dominanti; la malattia si manifesta in individui che possiedono anche solo una copia del gene alterato. La seconda classe è costituita dalle malattie genetiche recessive in cui, finché l'individuo possiede almeno una copia funzionale del gene, i sintomi non sono manifesti; affinché ciò accada, l'individuo deve possedere il gene mutato in entrambe le copie del cromosoma nel quale è localizzato. L'individuazione di mutazioni direttamente sul DNA è oggi possibile grazie a diverse tecniche, quali la PCR e l'ibridazione molecolare.
c) Test genetici prima delle nuove tecnologie del DNA
Un test genetico individua le alterazioni genetiche che rendono l'individuo predisposto all'insorgenza di una malattia o all'eventualità di manifestare problemi durante lo sviluppo. Prima dell'avvento delle nuove tecnologie del DNA, era possibile individuare un numero limitato di alterazioni genetiche mediante vari approcci; le tecniche che permettevano la visualizzazione di aberrazioni cromosomiche vistose erano le sole in condizione di esaminare direttamente il DNA. Visualizzando, per esempio, l'intero corredo dei cromosomi umani con il microscopio, fu possibile individuare il cromosoma supplementare presente nelle cellule di individui affetti dalla sindrome di Down. Un certo numero di malattie genetiche causate da specifiche anomalie biochimiche poté essere individuato eseguendo test chimici, per esempio, su campioni di urina; la fenilchetonuria, una malattia genetica recessiva, viene diagnosticata in base alla presenza di alti livelli di fenilalanina e dei suoi metaboliti nell'urina; l'anemia a cellule falciformi, altra malattia recessiva, può essere riconosciuta per la presenza nel sangue di globuli rossi dalla morfologia inusuale (a forma di falce). Quando si scoprì che l'anemia a cellule falciformi e altre malattie dei globuli rossi erano causate da specifiche alterazioni dell'emoglobina, divenne possibile individuare l'alterazione genetica più direttamente, esaminando l'emoglobina dei pazienti. Tuttavia, per molte malattie genetiche, la diagnosi poteva essere fatta esclusivamente sulla base dei sintomi e dell'anamnesi familiare e non c'era altro metodo per predire con certezza quali individui ne avrebbero sofferto.
Nondimeno, la disponibilità di un crescente numero di test ha portato alla messa a punto di una varietà di programmi di screening, eseguibili a vari stadi della vita e con varie finalità. Una coppia che desidera avere un bambino può sottoporsi a screening per verificare se entrambi i genitori siano portatori di un gene recessivo, condizione che fisserebbe in 1 su 4 la probabilità di generare un bambino con entrambe le copie del gene alterato; tali informazioni possono essere utili per la pianificazione familiare. Supponiamo che i due potenziali genitori apprendano di essere entrambi portatori del gene responsabile di una malattia recessiva: potrebbero scegliere di sottoporre il feto allo screening prenatale con tecniche come l'amniocentesi. Se il feto fosse omozigote per l'alterazione genetica, i genitori potrebbero in ultima analisi scegliere di interrompere la gravidanza o, in alternativa, prepararsi a qualsiasi cura si rivelasse necessaria a un bambino con un quadro genetico alterato; infine, la coppia potrebbe scegliere di usare la contraccezione e di non generare bambini. Lo screening neonatale è particolarmente utile per le malattie che si possono curare; negli Stati Uniti, per esempio, viene eseguito diffusamente lo screening per la fenilchetonuria - malattia che può essere trattata limitando l'assunzione di fenilalanina nella dieta del bambino - che consente di prevenire il severo ritardo mentale e i problemi fisici che si manifestano in assenza di questa dieta particolare; similmente, lo screening neonatale per l'anemia a cellule falciformi è eseguito al fine di prevenire, mediante un'adeguata terapia antibiotica, le patologie infettive debilitanti che spesso accompagnano tale malattia.
Lo screening dei neonati o degli adulti fornisce, agli individui sottoposti al test, informazioni tali da consentire loro di cambiare stile di vita o mantenersi sotto sorveglianza medica, al fine di evitare eventuali problemi di salute. Per esempio, individui le cui cellule non sono in grado di produrre la proteina α1-antitripsina possono evitare di fumare o di frequentare ambienti lavorativi inquinati da alti livelli di materiale particolato, riducendo la possibilità di andare incontro ai gravi problemi polmonari cui sono particolarmente suscettibili.
d) La nuova era e il Progetto Genoma Umano
Malgrado l'esistenza, ancor prima della nuova era della genetica, di metodologie in grado di rivelare la presenza di quadri genetici alterati, la grande maggioranza delle malattie genetiche non poteva essere individuata in anticipo, ma soltanto - e limitatamente ad alcune malattie - all'apparire dei sintomi. L'anamnesi familiare di una malattia genetica era di grande aiuto nella diagnosi.
Con i progressi conseguiti nell'isolamento e nella caratterizzazione dei geni e con lo sviluppo di metodi semplici per la scoperta di alterazioni genetiche si sono verificati enormi cambiamenti, anche grazie all'attuazione del ‛Progetto Genoma Umano', che ha accelerato i tempi di ricerca (v. Guyer e Collins, 1993). Una delle principali finalità del Progetto Genoma Umano è quella di definire una mappa dettagliata del genoma stesso: questo comporta l'identificazione di polimorfismi genetici marcatori in ciascun cromosoma, situati così vicini fra loro che mappare geni responsabili di molte variazioni genetiche umane risulti abbastanza semplice. Anche se gran parte dell'attività di ricerca che ha condotto all'identificazione di geni umani era ben avviata già prima del lancio del Progetto, il supporto economico e il livello di coordinazione che esso ha fornito hanno enormemente facilitato il progresso in quest'area. Come risultato delle nuove tecnologie e del concertato impegno nella mappatura e nell'identificazione di geni umani, si è verificata una straordinaria crescita del numero di scoperte di geni associati a malattie e variazioni del genotipo umano; ogni anno, le scoperte di nuovi geni legati a disfunzioni umane aumentano a un ritmo esponenziale. In quasi tutti i casi, i geni caratterizzati rappresentano esempi di singole alterazioni geniche correlate con specifici disordini fisiologici umani: è il caso della corea di Huntington, della distrofia muscolare, della predisposizione al cancro della mammella e del colon. Negli Stati Uniti, studi sulla genetica del cancro della mammella, campo nel quale si sono ottenuti i maggiori successi, hanno accertato che tra il 5 e il 10% dei casi il cancro si riscontra in donne che hanno avuto o hanno familiari con lo stesso tipo di patologia. I ricercatori, grazie alle nuove tecnologie di mappatura genica, hanno individuato i geni le cui mutazioni predispongono i membri femminili di alcune di queste famiglie a sviluppare il cancro della mammella e quello dell'utero: osservazioni statistiche hanno dimostrato che una donna che possieda anche solo uno di questi geni mutati ha l'80% di probabilità di sviluppare il cancro della mammella entro i settant'anni. Molte donne contraggono la malattia in età molto più precoce e, nel caso di uno di questi geni (il gene BRCA1), oltre ottanta mutazioni che predispongono al cancro della mammella sono state individuate esaminando le famiglie con una storia della malattia alle spalle (v. Miki e altri, 1994). Affinché sia possibile identificare i portatori del gene mutante, è necessario studiarne la natura specifica in ogni componente della famiglia, in modo che ognuno sappia se è geneticamente predisposto alla malattia oppure no. Ciò consente a chi risultasse positivo al test di prendere misure precauzionali e, se necessario, di intervenire chirurgicamente in maniera radicale (mastectomia bilaterale) nella speranza di ridurre le probabilità di contrarre il cancro. È importante sottolineare che questi geni della predisposizione sono responsabili solo di una piccola parte dei cancri della mammella, mentre il rimanente 90% è causato da fattori ambientali, processi stocastici all'interno delle cellule somatiche, altri fattori genetici meno chiari o una combinazione di tutti questi elementi (v. Biesecker e altri, 1993).
La mappatura dei geni associati a condizioni determinate da singoli loci è per lo più abbastanza semplice, ma vi sono alcune malattie poligeniche - cioè risultanti dalla combinazione di molteplici alterazioni geniche - che presentano maggiori difficoltà di studio: è il caso di alcune forme di diabete. Le nuove tecnologie aiuteranno a svelare le combinazioni dei geni che sono coinvolti in tali complesse malattie, come già dimostrano i primi risultati ottenuti.
Secondo il parere di molti studiosi, un'altra importante frontiera nella caratterizzazione della base genetica della variazione umana è rappresentata dallo studio dei tratti comportamentali. Nel XX secolo la questione dell'influenza della genetica e dell'ambiente sul comportamento umano è stata oggetto di una vivace disputa tra gli scienziati. Attualmente, molti scienziati si stanno impegnando nel tentativo d'identificare geni ritenuti responsabili di esercitare un'influenza sul comportamento umano, specie coloro che ritengono molto probabile non solo che tale influenza esista, ma anche che sia molto forte. Gli studi svolti vanno dall'analisi di malattie mentali, come la sindrome maniaco-depressiva e la schizofrenia, fino all'approfondimento di tratti che molti considerano parte integrante del quadro normale del comportamento umano, come l'intelligenza e l'orientamento sessuale. Per diversi motivi, non si sono ancora avuti degli esiti soddisfacenti, non risultando ancora chiaro se le difficoltà nel localizzare i geni correlati con questi comportamenti siano dovute a forti influenze ambientali, alla natura poligenica dei tratti stessi o a una combinazione di entrambi i fattori. Volendo essere ottimisti sul futuro della genetica comportamentale, i ricercatori del campo hanno definito questi tratti umani come ‛oligogenici', termine usato al posto di ‛poligenici' per identificare la tesi secondo cui tali tratti sono influenzati da pochi anziché da molti geni.
Quale che sia il successo di quest'ultima impresa scientifica, appare chiaro che la quantità di informazione genetica disponibile sta comunque crescendo a un passo estremamente rapido. Charles Cantor, uno dei leaders del Progetto Genoma Umano, ha affermato che se il susseguirsi delle scoperte, il ritmo del perfezionamento della tecnica e la riduzione del suo costo continueranno alla velocità attuale, sarà possibile ottenere nel XXI secolo la sequenza completa del DNA di ogni singolo individuo nel mondo. Walter Gilbert, uno dei primi sostenitori del Progetto, ha previsto che, entro un non lontano futuro, ognuno potrà portare con sé la propria sequenza completa del DNA, inclusa l'interpretazione dell'informazione, in un compact disc. Queste proiezioni potrebbero sembrare troppo ottimistiche, ma l'entusiasmo degli scienziati del genoma riflette gli straordinari progressi già realizzati in questo campo.
e) Trattamento delle malattie genetiche
Individuare e localizzare un gene responsabile di una particolare malattia può avere come conseguenza un gran numero di benefici; conoscere la sua funzione e la struttura del suo prodotto può infatti condurre alla messa a punto di un trattamento curativo.
I genetisti confidano che, col crescente perfezionamento delle tecniche biologiche - che vanno dalla genetica all'analisi e alla modificazione della struttura proteica - le informazioni sui prodotti genici associati a stati patologici finiranno per portare in molti casi a trattamenti e cure definitive. Nel passato, la caratterizzazione biochimica delle malattie genetiche ha portato a vari trattamenti, da cambiamenti nella dieta (fenilchetonuria e galattosemia) a trasfusioni di sangue (β-talassemia) e salasso (emocromatosi). Per esempio, la malattia genetica recessiva fenilchetonuria (PKU) porta a un severo ritardo mentale e ad altri problemi di salute; la scoperta che le mutazioni responsabili della malattia conducono a una deficienza nel metabolismo dell'amminoacido fenilalanina ha suggerito che una dieta priva dell'amminoacido potesse alleviare i sintomi. In realtà, il numero di malattie genetiche assoggettabili a trattamenti curativi semplici è molto ristretto. Per esempio, è noto da tempo che la malattia di Tay-Sachs, che influenza il feto durante il suo sviluppo, è causata da un difetto nel metabolismo dei mucopolisaccaridi. Fino a ora, questa importante informazione non ha fornito alcun metodo per il trattamento della malattia; d'altra parte, potrebbero verificarsi molti casi in cui l'identificazione di un gene responsabile della malattia e la conseguente comprensione della funzione del suo prodotto genico conducano all'immediato sviluppo di un'efficace strategia di trattamento.
La logica alla base della ricerca procede come di seguito descritto: la determinazione della sequenza del DNA di un gene associato a una malattia può fornire informazioni sulla sua funzione; successivamente, dalla sequenza del DNA del gene si può dedurre la sequenza amminoacidica del suo prodotto. Negli ultimi anni sono stati compiuti grandi passi in avanti nella capacità di predire le proprietà di una proteina semplicemente esaminando la sua sequenza amminoacidica. La localizzazione della proteina nella cellula, la sua interazione con piccoli metaboliti cellulari e anche la sua reale funzione possono spesso essere postulate su questa base. Per esempio, la sequenza del DNA del gene responsabile della fibrosi cistica è stata determinata quando, dopo aver rilevato la presenza di amminoacidi idrofobici lungo la sequenza proteica, è stato possibile predire la struttura secondaria del prodotto genico, e ciò ha fatto supporre che si trattasse di una proteina di membrana (v. Riordan e altri, 1989). Confrontando la sequenza della proteina con altre presenti in un'aggiornata banca dati, si è giunti alla scoperta che la proteina mostrava un'estesa omologia di sequenza con un gruppo di proteine di funzione nota. Queste proteine sono localizzate sulle membrane cellulari e sono responsabili del trasporto di diversi tipi di molecole attraverso la barriera costituita dalla membrana stessa. Tali scoperte hanno suggerito che la proteina CFTR (Cystic Fibrosis Transmembrane conductance Regulator) fosse responsabile del trasporto dello ione cloruro attraverso la membrana, un processo notoriamente difettivo negli individui affetti da fibrosi cistica. Poiché questa informazione è stata acquisita di recente, non è stata ancora impiegata con successo nella cura della malattia ed è impossibile predire quali difficoltà si incontreranno nel conseguire dei buoni risultati. D'altra parte si presume che tale informazione risulterà alla fine utile a mettere a punto un trattamento contro la malattia.
La conoscenza della normale funzione della proteina di per sé non porta necessariamente a una soluzione atta a compensare il difetto rinvenuto in individui che ne posseggano forme alterate; è stato già citato l'esempio della malattia di Tay-Sachs. Tuttavia, un'estesa conoscenza dei processi cellulari può generare nuovi approcci per aggirare o compensare il processo difettivo. Un esempio è dato dai recenti progressi nel trattamento dell'anemia a cellule falciformi, malattia riscontrata in individui che presentano due copie di una forma alterata del gene codificante per una delle due subunità proteiche dell'emoglobina (v. sangue: Anemie emolitiche). Il difetto specifico alla base di questa malattia è noto già dal 1957, ma il fatto che questa informazione non abbia portato a un trattamento efficace per oltre 30 anni evidenzia il difficile percorso che va dalla conoscenza della causa che provoca la malattia alla sua soluzione medica. In ogni caso, la comprensione più approfondita dell'espressione e della regolazione dei geni codificanti per varie forme di emoglobina ha portato a un trattamento in qualche modo efficace della malattia. In particolare, si è trovato che l'alimentazione di individui adulti con idrossiurea riattivava l'espressione di un'emoglobina riscontrata solitamente solo nel feto e che questa emoglobina fetale può sostituire quella adulta difettiva nei pazienti affetti da anemia a cellule falciformi e ridurre il numero di crisi dolorose. La strategia fu messa a punto solo dopo aver scoperto che alcuni individui, in cui era presente la mutazione alla base dell'anemia a cellule falciformi, non ne soffrivano in maniera grave, in quanto erano portatori anche di mutazioni compensatorie del difetto, che consentivano l'espressione dei geni dell'emoglobina fetale negli adulti.
Un altro approccio al trattamento di quelle malattie genetiche in cui è coinvolto un prodotto genico difettivo consiste nell'intervenire per ripristinare il funzionamento della proteina difettiva. Per ora i presupposti per progettare strategie terapeutiche di questo genere sono scarsi. Tuttavia, col progredire delle conoscenze sulle strutture proteiche e sul modo per alterarle, può divenire possibile in qualche caso introdurre nell'organismo un agente che, legandosi alla proteina e alterandone la struttura in modo appropriato, ne ripristini la corretta attività.
f) La terapia genica
Un approccio più generale al trattamento delle malattie genetiche è costituito dall'uso della terapia genica, applicata secondo le modalità sperimentali descritte nella fig. 5. Nelle malattie che sono dovute a un prodotto genico difettivo o assente, l'introduzione di una copia del gene normalmente funzionante nelle cellule appropriate dell'organismo può, in linea di principio, essere risolutiva. Per esempio, come già descritto, la fibrosi cistica è dovuta al malfunzionamento di un importante canale dello ione cloruro nelle membrane cellulari. Questa deficienza diviene particolarmente critica nei polmoni, dove il muco si accumula a tal punto da rendere difficile la respirazione e provocare l'insorgenza di frequenti infezioni. Se si riuscisse a introdurre una copia funzionante del gene codificante per la proteina CFTR nelle cellule degli alveoli polmonari, queste sarebbero in grado di produrre un canale per lo ione cloruro perfettamente funzionante e i sintomi della malattia verrebbero sostanzialmente alleviati.
Molti dei passi necessari per applicare la terapia genica sono stati compiuti: una volta identificato un gene, produrlo in forma purificata è spesso semplice e, mediante particolari tecniche, è possibile introdurre geni così purificati in cellule umane. Questi procedimenti vengono eseguiti incorporando i geni in versioni modificate di virus che normalmente infettano cellule umane, oppure associando a liposomi (piccole vescicole, ricche in acidi grassi, in grado di penetrare nelle cellule) la molecola di DNA che comprende il gene. Anche l'iniezione di DNA purificato nelle cellule può, come riportato nella letteratura scientifica, condurre in molti casi al ripristino della funzione genica. Tali metodi sono stati applicati in primis su pazienti affetti da fibrosi cistica; secondo resoconti iniziali, tale trattamento avrebbe avuto successo anche nel caso di due pazienti affetti da una rara malattia genetica, la deficienza di adenosin-deaminasi, che porta a gravi difetti del sistema immunitario dell'individuo. Peraltro, successivi tentativi di ottenere gli stessi risultati con altri individui affetti dalla stessa malattia sono falliti.
A tutt'oggi ci sono ancora alcuni limiti nell'applicazione della terapia genica in quanto, nella maggior parte dei casi, il gene - una volta introdotto nelle cellule - non diviene parte integrante del materiale ereditario cellulare ed è in seguito perso o la sua espressione molto attenuata. Il trattamento necessita così di un'applicazione continua in modo da soddisfare il continuo fabbisogno del paziente di prodotto genico funzionante. Esistono metodologie il cui perfezionamento potrebbe superare queste limitazioni, consentendo al gene di essere integrato stabilmente nei cromosomi delle cellule: il gene sarebbe così espresso in permanenza e trasferito alle cellule figlie a ogni divisione cellulare come parte integrante del materiale ereditario. Comunque, si deve fare attenzione che tale integrazione non alteri i cromosomi causando essa stessa un danno al paziente. Le preoccupazioni sono particolarmente gravi nel caso della terapia genica applicata con l'uso di vettori virali ‛inabilitati'. Nonostante l'alterazione del virus, esiste la preoccupante possibilità che i geni virali ancora attivi abbiano effetti negativi sul paziente e, inoltre, che l'espressione del gene sfugga alla sua usuale regolazione cellulare comportando altri problemi.
La procedura attualmente è limitata anche dalla ridotta capacità di indirizzare con precisione la terapia genica verso le cellule dell'organismo che la richiedono; questa limitazione in molti casi può essere superata usando vettori virali che hanno come bersaglio specifiche cellule o specifici organi; in altri casi, si dovrebbe riuscire a iniettare direttamente il vettore per terapia genica nelle cellule appropriate. Sebbene le strade aperte dalla terapia genica si siano rivelate fino a oggi deludenti, il progresso in quest'area è così rapido da suggerire che, in molti casi, questi problemi saranno superati (v. Watson, 1993).
Infine, il tipo di terapia genica finora descritto è limitato a quelle malattie genetiche dovute all'assenza dell'attività di un determinato prodotto genico. Alcune malattie genetiche (spesso le malattie con genotipo dominante) sono causate da proteine alterate che distruggono attivamente la funzione cellulare; in questi casi, l'introduzione di un gene funzionante normalmente nelle cellule non è in grado di contrastare l'effetto della copia distruttiva del gene. In altre malattie genetiche, vistose alterazioni della struttura del cromosoma sono alla base dei sintomi; in questi casi non ci si può attendere che l'introduzione di un singolo gene riesca a compensare un'alterazione genetica di tale estensione.
La terapia genica può anche essere usata per il trattamento di malattie che non sono necessariamente genetiche. Se un paziente ha una deficienza causata da un trauma, dallo stile di vita, dall'esposizione ad agenti tossici o ad altri fattori ambientali, l'inserimento di un gene in grado di esprimere un prodotto che compensi quella deficienza potrebbe migliorare il suo stato di salute. Un esempio di applicazione della terapia genica a una malattia di tipo non ereditario è dato dai tentativi di trattamento del cancro. L'approccio è basato sulla scoperta che la trasformazione neoplastica di una cellula sopprime la sua capacità di esprimere proteine MHC (Major Histocompatibility Complex) che servono alla cellula per presentare gli antigeni al sistema immunitario. Se le proteine MHC fossero funzionanti, le cellule tumorali sarebbero probabilmente riconosciute e distrutte dal sistema immunitario. Per risolvere questo problema, i ricercatori stanno introducendo nelle cellule tumorali geni codificanti per composti come l'interferone gamma, che esalta l'espressione delle proteine MHC: la speranza è che l'organismo venga così stimolato a distruggere le cellule cancerose.
La terapia genica è in realtà ancora allo stadio embrionale: non si può ancora predire con certezza il grado di successo che si raggiungerà in quest'area.
g) Le impronte del DNA (DNA fingerprinting)
La tecnica RFLP, come già detto, è usata per determinare il profilo dei polimorfismi del DNA sui cromosomi di una persona; la possibilità di combinazione dei polimorfismi è così vasta che non esistono due individui con lo stesso complesso di variazioni del DNA. Pertanto, l'analisi dei polimorfismi produce l'equivalente dell'impronta digitale di un individuo, un insieme di informazioni che lo identificano senza ambiguità. La RFLP è stata ampiamente usata per la messa a punto del DNA fingerprinting; esiste però un altro metodo per distinguere tra frammenti di DNA di due individui ed è quello di determinare la lunghezza di particolari sequenze che sono ripetute molte volte (sequenze ipervariabili o in tandem). Il numero di tali ripetizioni può variare da individuo a individuo e appare chiaro che il numero delle differenze nei polimorfismi tra una qualunque coppia di individui è così grande che, se si potesse definirne un numero sufficiente, si dimostrerebbe che non esistono sulla terra due individui (eccetto forse i gemelli omozigoti) con lo stesso profilo di DNA.
Come le impronte digitali sono usate in numerosi casi per stabilire senza ambiguità l'identità di persone che hanno commesso atti criminali, lo stesso risultato si può ottenere confrontando il DNA di un individuo con il DNA ottenuto da campioni di sangue, ciocche di capelli, tracce di sperma, frammenti di pelle e ogni altro tipo di materiale cellulare rinvenibile sulla scena del delitto. L'analisi del campione di DNA ha inizio con l'amplificazione di alcune sequenze usando la PCR e l'analisi dei prodotti mediante RFLP; i polimorfismi sono poi posti a confronto con un campione di DNA ottenuto da un individuo sospettato di un crimine e viene quindi determinata la presenza o l'assenza di una completa identità. Se fosse possibile caratterizzare un numero sufficiente di polimorfismi, si potrebbe raggiungere una conclusione senza alcun tipo di ambiguità ma, in pratica, solo un limitato numero di polimorfismi può essere analizzato, a causa dei costi e del tempo a disposizione. La validità delle metodiche attualmente in uso è stata messa in discussione, in quanto, come già detto, la tecnica è basata sull'uso di rari polimorfismi, che non sono cioè diffusi a tal punto che un particolare corredo (set) possa essere comunemente rinvenuto in molti individui: è necessario quindi che sia disponibile il quadro completo della frequenza di specifici polimorfismi nella popolazione analizzata. Ma, al di là di questo, si può assumere che individui appartenenti a un particolare gruppo etnico abbiano un set di polimorfismi diverso da quello caratteristico di altri gruppi etnici, ma che questi polimorfismi siano abbastanza comuni all'interno del gruppo. Se l'individuo non è posto a confronto con il gruppo di controllo corretto, si potrebbe arrivare a una conclusione errata. Tali problemi dovrebbero essere superati con l'uso di un sufficiente numero di polimorfismi e grazie alla disponibilità di ampie conoscenze dello spettro dei polimorfismi stessi nell'ambito di differenti gruppi etnici (v. Balding e Donnelly, 1994; v. Lander e Budowie, 1994).
Questo genere di analisi trova applicazione anche in un progetto, denominato Progetto Diversità del Genoma Umano, che si propone di mettere a confronto i polimorfismi presenti nel DNA di differenti gruppi etnici e di individui provenienti da diverse regioni e paesi del mondo. Questi confronti dovrebbero consentire di chiarire le origini dell'uomo e la storia delle sue migrazioni.
3. L'industria delle biotecnologie
Nonostante non sia ancora presente sul mercato un grande numero di prodotti ottenuti dall'industria delle biotecnologie, ci sono stati però alcuni successi degni di essere menzionati.
a) Vaccini
Una delle finalità primarie dell'industria biotecnologica è lo sviluppo di vaccini; bersagli di queste ricerche sono le malattie come l'AIDS e una nuova forma di tubercolosi antibiotico-resistente. Alla base di gran parte del lavoro sta la capacità di trasferire geni codificanti per importanti antigeni di agenti patogeni in organismi appropriati che possono così divenire efficaci agenti immunizzanti. Un'altra strada è rappresentata dalla manipolazione genetica di organismi patogeni così che siano vitali ed esprimano fattori immunogenici, ma senza essere virulenti, e quindi riescano a indurre una risposta immune nel corso di un'infezione. Si sta seguendo questa strada nel tentativo di ottenere vaccini anticolerici.
Un efficace vaccino contro il virus dell'epatite B è stato messo a punto introducendo un gene codificante per una delle proteine del rivestimento virale in cellule di lievito, dalle quali furono poi ottenute particelle in grado di immunizzare i pazienti. Alcuni passaggi del procedimento di clonaggio di un vaccino umano sono riassunti in fig. 9. Nel campo della zootecnia, un vaccino contro il virus della malattia di Newcastle (NDV) e il virus fowlpox (FPV), agente del vaiolo aviario, è stato ottenuto manipolando geneticamente lo stesso virus FPV. I geni che causano la malattia sono stati rimossi e i geni codificanti per gli antigeni del virus della malattia di Newcastle sono stati introdotti nel materiale genetico del virus FPV.
b) Test diagnostici
La capacità di individuare e di amplificare piccole regioni di DNA permette la produzione di kits che consentono la diagnosi di svariate malattie causate da agenti infettivi. La presenza di HIV, l'agente causale dell'AIDS e di numerose altre malattie batteriche, virali o parassitarie, può essere messa in evidenza con l'ausilio di nuove tecnologie come la PCR. In questi casi, piccole sequenze di DNA, che notoriamente sono presenti nell'organismo patogeno, vengono ibridate con il DNA ottenuto dal paziente. Se le appropriate regioni del DNA sono poi amplificate con la PCR usando queste sonde specifiche, si ottiene la prova della presenza di un particolare agente patogeno. La capacità di diagnosticare rapidamente l'agente causale di una malattia è spesso cruciale nell'applicazione di un tempestivo trattamento.
c) Prodotti genici
La capacità di purificare geni e di trasferirli da un organismo a un altro ha consentito ai ricercatori di ottenere grandi quantità di prodotti genici, introducendo i relativi geni in un organismo o in una linea cellulare appropriata. Uno dei primi prodotti commerciali a essere fabbricati in questo modo è stata l'insulina umana: una volta isolato il suo gene codificante, è stato possibile introdurlo in vettori in grado di persistere e divenire parte del materiale ereditario delle cellule di alcuni batteri (plasmidi di Escherichia coli). Sono stati utilizzati grandi fermentatori per una produzione massiva, così che tali batteri sono divenuti fabbriche per la produzione di insulina. Da quel momento in poi, una grande varietà di molecole terapeutiche è stata prodotta con questa tecnica, fra cui l'attivatore tissutale del plasminogeno, usato per dissolvere i trombi nel trattamento dei pazienti colpiti da infarto del miocardio, l'ormone della crescita umano, usato per il trattamento di certi tipi di nanismo ereditario, e diversi fattori di crescita cellulari coadiuvanti nel trattamento di varie malattie. Per esempio, il fattore stimolante le colonie di granulociti (GCSF) viene usato dopo il trattamento chemioterapico del cancro, per stimolare la formazione di globuli bianchi; l'eritropoietina, che induce la proliferazione dei globuli rossi, è usata per favorire la sopravvivenza dei pazienti in emodialisi; l'enzima desossiribonucleasi allevia alcune delle difficoltà respiratorie degli individui affetti da fibrosi cistica. Tutte queste proteine sono ora prodotte mediante strategie basate sulla tecnica del DNA ricombinante. Si tratta di significativi passi in avanti in quanto nel passato era molto difficile e costoso produrre grandi quantità di queste molecole in forma pura: l'ormone della crescita umano, per esempio, era ottenuto da tessuto cerebrale di individui deceduti e, in alcuni casi, la sua somministrazione ha causato la trasmissione al paziente di una grave, finanche fatale, malattia, la malattia di Jacob-Creuzfeldt. L'ingegneria genetica è stata utilizzata anche per semplificare la produzione di prodotti alimentari: il caglio del vitello, contenente l'enzima (rennina) che coagula il latte, è usato nella preparazione di molti formaggi. Il clonaggio del gene codificante per questo enzima ne ha permesso una produzione economicamente vantaggiosa in grandi quantità; inoltre, l'aggiunta di somatotropina bovina (ormone della crescita, prodotto con la tecnica del DNA ricombinante) alla dieta delle mucche ha incrementato la produzione del latte di oltre il 10%. In molti paesi si sono verificate resistenze all'introduzione di questo additivo: i timori dei piccoli allevatori di essere esclusi dal mercato e anche la possibile insorgenza di effetti collaterali sulla salute delle mucche, così come le ansie dei consumatori, hanno fatto sì che negli Stati Uniti e in altri paesi si arrivasse alla limitazione del suo uso. L'alimentazione dei maiali con somatotropina suina ha portato alla produzione di carne con meno contenuto di grassi. I reali benefici di questi prodotti zootecnici e la loro accettazione o meno da parte della pubblica opinione non sono ancora chiari.
La gamma dei prodotti, medici e non, provenienti dall'uso di queste nuove tecnologie genetiche potrà aumentare; ciò avverrà in funzione della scoperta - in seguito a studi genetici e biologici - di nuove molecole, come gli ormoni, in grado di influenzare vari processi fisiologici. Peraltro, le tecniche produttive stanno costantemente migliorando e alcune aziende stanno manipolando geneticamente ovini e bovini affinché producano latte arricchito con sostanze che ne aumentino il valore commerciale. Questa impresa è resa possibile dalla capacità di creare animali transgenici, portatori di geni estranei come parte integrante del loro materiale ereditario (v. farmacologia e sperimentazione animale, volx).
d) Ingegneria genetica delle piante
Una fiorente componente dell'industria biotecnologica si dedica al raffinamento dei prodotti agricoli e, in un ristretto numero di casi, alcuni prodotti sono già stati lanciati sul mercato.
Il pomodoro Flavr-Savr è stato geneticamente manipolato così da essere meno soggetto alla marcescenza (v. botanica, vol. X); nel materiale ereditario del pomodoro è stato introdotto un tratto di DNA che disattiva l'espressione dell'enzima poligalatturonasi, che causa la marcescenza. Ciò permette la raccolta del pomodoro maturo e il suo trasporto anche per lunghe distanze senza rischio di deperimento rilevante. L'espediente consente di aggirare la pratica di raccogliere il pomodoro quando è ancora verde, ma meno saporito, e poi farlo maturare artificialmente. Negli Stati Uniti si pensa che questi nuovi pomodori siano particolarmente utili nei mesi invernali, quando in ampie aree del paese, troppo fredde per la coltivazione di questi ortaggi, devono essere importati da regioni distanti.
Un'altra attiva area di ricerca è lo sviluppo di piante resistenti agli erbicidi o agli insetti nocivi: gli erbicidi potrebbero essere usati per distruggere le malerbe senza timore di uccidere la pianta stessa. Esistono in realtà timori che l'esistenza di tali piante incoraggi l'abuso di erbicidi con conseguenze negative sull'ecosfera. Piante resistenti agli insetti nocivi sono state ottenute introducendovi geni del batterio Bacillus thuringiensis, le cui tossine agiscono da pesticidi naturali. Le piante così modificate, essendo esse stesse resistenti agli insetti nocivi, non richiedono trattamento o irrorazione con pesticidi.
Oltre ai successi commerciali delle industrie biotecnologiche finora descritti, i prodotti ottenuti da queste imprese sono stati utili in molte delle applicazioni descritte nei paragrafi precedenti, come nella messa a punto dei kits per il DNA fingerprinting e dei test per varie malattie genetiche.
e) Problemi
Le controversie sono sopraggiunte negli anni settanta, con la nuova era della genetica molecolare. I primi resoconti sulla manipolazione genetica hanno destato la preoccupazione che il trasferimento di geni da una specie all'altra potesse creare nuovi, pericolosi organismi. Studi iniziali in quest'area riguardavano la manipolazione genetica in batteri come Escherichia coli, un normale ospite dell'intestino umano, e in virus come SV40, che causa il cancro nelle scimmie. Un certo numero di scienziati temeva che organismi e virus risultanti da esperimenti di ingegneria genetica potessero avere nuove proprietà in grado di renderli agenti infettivi virulenti e che il rilascio di organismi geneticamente manipolati nell'ambiente potesse causare seri danni ecologici.
Nel 1975, un gruppo di autorevoli biologi molecolari, molti dei quali impegnati in prima persona in questo tipo di ricerca, proposero una sospensione degli studi di ingegneria genetica e una conferenza per valutare il rischio potenziale di questa nuova tecnologia e anche per individuare delle possibili linee guida da seguire nella sperimentazione. La conferenza, che si tenne ad Asilomar, in California, definì gli argomenti che dovevano essere esaminati e stabilì una temporanea sospensione sulla ricerca, fino a quando non si fosse definito un codice deontologico.
Mentre gli scienziati speravano di controllare in qualche modo la messa a punto del codice e di poter garantire progressi continui della scienza una volta prese adeguate precauzioni, il pubblico divenne sempre più sensibile ai rischi di questa nuova tecnologia e sempre più timoroso delle sue conseguenze. Ciò determinò un attivismo politico sul tema della manipolazione genetica negli Stati Uniti e in Europa e l'imposizione in alcune comunità e paesi di nuove linee guida per la ricerca. In alcuni casi, come in Germania, queste erano considerevolmente più restrittive di quanto non fosse nelle intenzioni degli scienziati.
Negli anni successivi al 1975, le preoccupazioni circa i possibili rischi ambientali e sanitari della ricerca sul DNA ricombinante sono sensibilmente diminuite. Nondimeno, un certo numero di restrizioni è rimasto in vigore in quelle aree di ricerca che ancora destano timori per l'ambiente o la salute. È il caso degli studi che coinvolgono geni provenienti da organismi patogeni come batteri, virus e parassiti. Il declino dell'interesse nei confronti del dibattito è dovuto a molti fattori: l'aumentata esperienza di lavoro con le nuove tecnologie, gli studi effettuati per verificare la verosimiglianza di alcuni degli scenari pericolosi ipotizzati e le analisi più approfondite su alcune questioni di biologia delle popolazioni convinsero la maggioranza degli scienziati che i timori originari fossero, per la maggior parte, infondati. La ripresa degli studi e l'allentamento delle restrizioni poste alla ricerca possono anche essere attribuiti al crescente entusiasmo per la nuova tecnologia e al suo straordinario potere nell'analisi di quei problemi biologici che in precedenza erano sembrati intrattabili. A causa della reazione dell'opinione pubblica e delle conseguenti restrizioni alla ricerca, alcuni scienziati si pentono oggi di aver dato inizio a questo processo di autocritica. Altri, al contrario, ritengono che si sia trattato di una lodevole manifestazione di responsabilità sociale nell'ambito della scienza.
4. Problemi sociali ed etici dopo il 1975
a) La terapia genica
Negli anni settanta, la principale preoccupazione di coloro che si mostravano cauti verso la nuova era della genetica era costituita dagli effetti sulla salute e l'ambiente. Comunque, per alcuni un ulteriore motivo di discussione era l'applicazione dell'ingegneria genetica alla specie umana. Se si potevano manipolare geneticamente le piante (e in seguito gli animali), cosa sarebbe accaduto con gli esseri umani? Per quelli che erano interessati alle applicazioni mediche della manipolazione genetica, la possibilità di curare le malattie genetiche usando la terapia genica costituiva un forte richiamo.
L'inizio della sperimentazione nell'area della terapia genica portò al timore generalizzato che questa potesse essere applicata in misura più estesa per manipolare geneticamente le persone, secondo gli specifici interessi di qualcuno. Lo spettro di una ripresa dell'eugenetica nazista fu costantemente agitato, ma in realtà la tecnologia alla base della terapia genica si è sviluppata tanto lentamente che i progressi in un'altra area, la mappatura dei geni, hanno allontanato le preoccupazioni etiche e sociali dal campo dell'ingegneria genetica.
Le implicazioni sociali, etiche e legali delle tecniche di mappatura genica sono molto ampie. Per la maggior parte, le questioni che vengono poste si richiamano a quelle che sorsero con lo spettro della manipolazione genetica della specie umana. Per questo la maggior parte del resto di questo articolo sarà dedicata alle implicazioni insite nei progressi compiuti dallo screening genetico. La loro rilevanza nelle questioni riguardanti la terapia genica verrà messa in evidenza alla fine.
b) Errori d'opinione sulla genetica
La parte rimanente di questo articolo verterà sulla descrizione dei rischi potenziali, per la società e per i singoli individui, derivanti dalla crescente facilità nell'acquisizione di informazioni genetiche sulle persone. Comunque, prima di entrare nel vivo di questa discussione, desidero sottolineare alcuni errori d'opinione relativi alla genetica che influenzano il modo in cui queste informazioni sono considerate e utilizzate.
Accade spesso che il concetto di gene venga associato a concezioni deterministiche. Il ruolo dei geni e la loro importanza è perfettamente sintetizzato dalle parole di James Watson, scopritore con Francis Crick della struttura del DNA e vincitore del premio Nobel nel 1962, che, in un'intervista al popolare settimanale americano ‟Newsweek", ebbe a dire: ‟Abbiamo sempre pensato che il nostro destino risiedesse nelle stelle. Ora sappiamo che, perlopiù, esso risiede nei nostri geni".
Le conseguenze che tale prospettiva deterministica comporta sono due: in primo luogo, essa implica che molti, forse la maggior parte, degli aspetti connessi con la nostra salute e il nostro comportamento siano influenzati dai geni; in secondo luogo, volendo forzare questa interpretazione fino alle estreme conseguenze, essa postula che, essendoci sempre una base genetica dietro qualsiasi aspetto della personalità umana, sia esso una caratteristica, una qualità o una tendenza, tale aspetto sia virtualmente immutabile. Questo senso di ineluttabilità che l'immaginario collettivo associa alla genetica, è all'origine di alcuni problemi che ostacolano la realizzazione di programmi di screening, come documentano molti studi che hanno rilevato la presenza di elementi di difficoltà di natura psicologica. Inoltre, i risultati di questi test di screening possono indurre o rafforzare, nelle persone che vi si sottopongono e qualora l'esito fosse negativo, un atteggiamento pessimistico e fatalista nei confronti della vita; nel caso opposto, possono tradursi in una sensazione di sollievo con conseguenze positive dal punto di vista psicologico (v. Holtzman, 1989).
Tuttavia lo studio della genetica non giustifica affatto questi atteggiamenti fatalistici; al contrario, una valutazione più equilibrata dell'importanza della genetica non può che riconoscere l'azione che geni e ambiente esercitano gli uni sull'altro e viceversa: i geni esprimono prodotti il cui effetto non è rigidamente prestabilito, ma è influenzato in maniera significativa dall'ambiente. Il termine ‛ambiente', inteso in questo caso nella sua accezione più generale, include le condizioni di vita intrauterina, le cure ricevute durante i primi anni di vita (compreso il tipo di alimentazione e la cura delle malattie), i fattori ambientali esterni quali l'inquinamento e l'esposizione ai raggi ultravioletti, la possibilità di usufruire di servizi sociali, i condizionamenti psicologici della famiglia e altro.
La fenilchetonuria è una delle numerose malattie genetiche gravi che possono essere affrontate con cure tali da sopprimere o quanto meno ridurre gli effetti deleteri da essa provocati. Come abbiamo già detto, questa malattia consiste in una disfunzione genetica recessiva consistente nell'incapacità di metabolizzare l'amminoacido fenilalanina; ciò causa un accumulo di fenilchetoni nell'organismo che incide negativamente su alcune funzioni cerebrali, provocando, negli individui nati con questa disfunzione, un serio ritardo a livello mentale unitamente a disturbi fisici di altro tipo. Tuttavia, se un neonato affetto da questa malattia viene alimentato con una dieta estremamente povera di fenilalanina e questa precauzione viene mantenuta per tutta la durata dell'adolescenza, il bambino raggiunge un quoziente d'intelligenza con valori nella media ed è completamente sano. Quindi, una disfunzione genetica che in particolari situazioni ambientali (presenza di fenilalanina nella dieta) condanna il bambino a una grave inabilità, in situazioni ambientali differenti (basso livello di fenilalanina nella dieta) non si manifesta affatto.
Un altro caso di influenza dell'ambiente sull'espressione di un carattere è rappresentato dagli individui che manifestano una deficienza nella produzione di α1-antitripsina, un quadro genetico che, come abbiamo detto, rende gli individui portatori particolarmente sensibili al fumo delle sigarette o a particelle dell'aria spesso presenti nei luoghi di lavoro e che può essere causa di enfisema, un grave disturbo polmonare. Se questi soggetti evitano di fumare e lavorano in ambienti liberi da polveri riducono drasticamente la probabilità di manifestare la malattia. Inoltre, sebbene le conoscenze in materia siano ancora a uno stadio decisamente preliminare, è possibile che nei casi di forme ereditarie di predisposizione a particolari tipi di cancro o a disturbi cardiaci, seguendo una dieta controllata ed evitando gli altri fattori ambientali che possono provocare la malattia, si riesca a ritardarne e perfino a evitarne l'insorgenza.
Un altro punto di notevole importanza è costituito dal fatto che, per molte malattie genetiche, l'isolamento di geni associati alle malattie stesse riveste un valore diagnostico limitato, come mostrato dai numerosi casi in cui il rinvenimento di un gene con alterazioni caratteristiche non solo non permette di determinare a che età nell'individuo portatore insorgerà la malattia o la gravità degli effetti che essa comporterà, ma nemmeno consente di stabilire con certezza se l'individuo portatore ne sarà mai affetto. Abbiamo già citato il caso delle donne appartenenti a famiglie in cui è stata identificata una predisposizione genetica al tumore della mammella, che hanno, secondo questa analisi, una probabilità dell'80% di contrarre la malattia entro i settant'anni di età. In quell'80% di donne predisposte la malattia può manifestarsi in qualsiasi momento della vita; il rimanente 20% delle donne non sviluppa affatto il cancro. Un altro esempio è rappresentato dal quadro genetico alla base della neurofibromatosi, il cui sintomo principale è rappresentato dalla presenza di tumori benigni multipli che compaiono in prossimità delle terminazioni nervose, generalmente accompagnati da altri sintomi meno noti la cui espressione è estremamente variabile. Coloro che presentano il gene alterato connesso con tale condizione morbosa possono sviluppare sintomi gravi della malattia e morire in età molto precoce o non esserne mai affetti e avere una vita di durata nella media.
L'incredibile variabilità dimostrata nella manifestazione di determinate malattie genetiche può essere dovuta a un gran numero di fattori: in alcuni casi l'insieme di condizioni ambientali a cui l'individuo è esposto può influenzare l'evolversi della malattia, in altri è la particolare organizzazione dei geni dell'individuo che può modificare la natura e il decorso temporale della malattia. Infine, eventi casuali, quali per esempio gli effetti dovuti a un naturale tasso di mutazione, possono determinare se e quando insorgeranno i sintomi.
Ciò nonostante esiste un certo numero di malattie genetiche incurabili che seguono un decorso prevedibile e documentato; per esempio, lo stato di salute dei bambini nati con la malattia di Tay-Sachs peggiora progressivamente e la maggior parte di loro non supera il quarto anno di vita.
c) Ogni essere umano è portatore di geni responsabili di malattie genetiche recessive
Perché un soggetto manifesti una particolare disfunzione genetica recessiva, è necessario che entrambi i genitori siano portatori del gene associato alla disfunzione stessa nella forma mutata. In questi casi c'è una probabilità su quattro che un bambino nato da tali genitori sia portatore di entrambe le copie cromosomiche del gene alterato e quindi manifesti la malattia. Normalmente, però, i portatori sani di un carattere patologico recessivo non mostrano alcun problema di salute a esso correlabile; per esempio, i portatori sani del gene responsabile della malattia di Tay-Sachs o della fenilchetonuria non manifestano alcun sintomo di queste malattie.
In base alla frequenza con cui si manifesta un gran numero di malattie genetiche è stato calcolato che ognuno di noi è portatore sano di un numero di geni responsabili di malattie genetiche recessive variabile tra 5 e 8. Dunque è sbagliato discriminare coloro che risultino positivi a un test per le malattie genetiche, poiché ciò è parte integrante della variabilità umana (v. Billings e altri, Discriminaton as..., 1992).
d) Conseguenze negative degli screenings genetici
Se da una parte si guarda con ammirazione alla possibilità di reperire nell'immediato futuro e con estrema facilità una gran quantità di informazioni genetiche su noi stessi, dall'altra ci si interroga sulle numerose conseguenze sociali, etiche e legali che questa evoluzione comporterà. Quale sarà la nostra reazione psicologica, una volta messi al corrente degli sviluppi futuri delle nostre condizioni di salute? Per quanto tempo informazioni di questo tipo resteranno private? Cosa accadrebbe se i nostri vicini di casa, i nostri colleghi di lavoro o l'intera comunità ne venissero a conoscenza? E se fossero usate da parte di compagnie assicuratrici, datori di lavoro, scuole, tribunali o altre istituzioni sociali per discriminarci?
1. Discriminazioni in campo assicurativo e sul lavoro. - Assumendo che sia stata riscontrata la presenza di alterazioni genetiche conosciute in un individuo, pur considerando il limitato valore diagnostico delle informazioni genetiche, oggigiorno è possibile in molti casi valutare la probabilità che quell'individuo contragga una malattia genetica o riporti un'invalidità. Questa informazione può spesso rivelarsi utile per la persona che si è sottoposta al test, consentendole di affrontare in anticipo i problemi di salute e tentare di attenuarne i sintomi; in alcuni casi il controllo della dieta o degli ambienti cui si è esposti, il ricorso a un intervento chirurgico o la prescrizione di particolari cure mediche possono persino prevenire il manifestarsi della disfunzione o migliorare le condizioni della persona che ne è affetta. Ma esistono anche numerose organizzazioni che considerano vantaggiosa ai propri fini la possibilità di acquisire informazioni genetiche relative alle singole persone.
Le compagnie che stipulano polizze assicurative sulla salute hanno la necessità di assumere la maggior quantità possibile di dati relativi alle condizioni di salute del loro cliente, per stabilire il tipo di polizza da stipulare, valutarne l'importo o perfino decidere se stipularla. Spesso un'anamnesi delle condizioni di salute che riveli che il cliente o altri membri della sua famiglia abbiano contratto determinate malattie in passato rappresenta la causa della mancata stipula di una polizza assicurativa. Quanto più è accurata la stima dei rischi che la salute del cliente corre, tanto più le compagnie assicuratrici possono innalzare il prezzo da pagare per ottenere ciò che considerano l'esatto valore della loro polizza. L'esistenza di un ‛profilo genetico' di ogni cliente aumenterebbe notevolmente la loro capacità di stabilire in anticipo quali di essi avanzeranno in futuro sostanziose richieste di finanziamento per curarsi.
Sebbene non sia noto se le compagnie abbiano realmente iniziato una politica di acquisizione sistematica di informazioni genetiche sui loro clienti, è tuttavia chiara - e per la verità per nulla sorprendente - la loro intenzione di servirsene qualora fossero disponibili. Negli Stati Uniti si sono verificati numerosi episodi che indicano il modo in cui queste informazioni possono essere usate; uno di questi riguarda una donna che, dopo aver avuto un figlio malato di fibrosi cistica, rimase nuovamente incinta. Test genetici dimostrarono che il feto era portatore, in entrambe le copie cromosomali, della mutazione genica responsabile della fibrosi. Quando i genitori decisero di non interrompere la gravidanza, la compagnia assicuratrice comunicò loro che se avessero perseverato nel volerla portare a termine la loro polizza sarebbe stata revocata; soltanto la minaccia di un'azione legale, avanzata dalla famiglia, fece recedere la compagnia dalle sue intenzioni. Un altro esempio riguarda le numerose persone che si sono viste rifiutare la stipula di una assicurazione sulla salute dopo che la compagnia aveva appreso dell'esistenza, all'interno della famiglia del potenziale cliente, di casi di individui affetti dalla malattia di Huntington; a portatori sani del gene responsabile dell'anemia a cellule falciformi non è stata concessa un'assicurazione, pur non essendo omozigoti per quel gene, dunque non affetti dalla malattia; a un'intera famiglia di cui un membro aveva sofferto di emocromatosi, sebbene si fosse curato e la malattia fosse ormai sotto controllo, fu riservato lo stesso trattamento.
Questi esempi permettono di trarre conclusioni importanti: in primo luogo, si può facilmente osservare che alcune delle decisioni assunte dalle compagnie assicuratrici riflettono dei luoghi comuni relativi alla genetica; spesso, per esempio, la sola presenza di un gene in qualche modo connesso con eventuali patologie viene considerata causa del manifestarsi della malattia in modo più determinante di quanto non sia in realtà. In secondo luogo, la grande confusione presente in materia tra la gente comune si riflette sull'operato delle compagnie assicuratrici che spesso non distinguono tra la condizione di portatore sano e quella di malato; infine, va considerato che, sotto la spinta dell'interesse puramente commerciale che guida le scelte delle compagnie assicuratrici, si possono verificare pressioni sui genitori costringendoli a effettuare dei test prenatali o a sottoporsi ad aborti. Se in futuro questo tipo di pressioni fosse accettato dalla società, l'espansione di una simile politica porterebbe a uno sfruttamento dell'eugenetica da parte di un particolare settore della società esclusivamente a fini commerciali; questa strada non può essere intrapresa senza una valutazione seria e partecipata delle implicazioni future.
Le accresciute possibilità a disposizione delle società che forniscono assicurazioni sulla salute di valutare la reale predisposizione dei loro clienti a future malattie, solleva numerosi interrogativi sulla liceità del sistema su cui sono basate. Mentre l'insorgenza della maggior parte delle malattie rimarrà determinata da eventi casuali e da fattori ambientali, la conoscenza della genetica si espanderà a tal punto che larga parte della popolazione sarà classificata in base alla predisposizione che ha nei confronti di eventuali malattie o disfunzioni. Se tutto ciò verrà attuato senza alcuna forma di controllo, questo processo ci costringerà presto a interrogarci sulla reale funzione che un sistema assicurativo di questo tipo deve esercitare. Fino a quando, infatti, a causa della limitata capacità di valutare in anticipo gli eventuali problemi di salute del cliente, la percentuale della popolazione a cui è rifiutata un'assicurazione sulla salute rimarrà bassa, questo tema non susciterà l'interesse dell'opinione pubblica. Non è difficile però immaginare un'epoca, neanche troppo lontana, in cui la genetica verrà usata come una sfera di cristallo attraverso cui scrutare il futuro e in cui solo i pochi che non mostreranno alcuna predisposizione a malattie genetiche otterranno una polizza assicurativa. Allora, forse, ci si renderà conto della definitiva scomparsa del criterio della suddivisione dei rischi tra la popolazione su cui erano basati i comportamenti delle vecchie compagnie assicuratrici. Di fronte a una simile prospettiva si è arrivati a chiedere che le compagnie assicuratrici abbandonino al più presto i progetti intrapresi per giungere a una razionale valutazione delle possibili malattie future dei propri clienti (v. Murray, 1992).
Il mondo del lavoro è un altro settore in cui l'informazione genetica può essere presa in considerazione nella valutazione di una persona: in particolare, la capacità di un lavoratore di svolgere un compito, i giorni persi a causa di una eventuale malattia o, nel caso della sua insorgenza, il costo delle cure a carico del datore di lavoro, sono tutti fattori che possono influenzare l'andamento di una carriera. Se un datore di lavoro avesse accesso a informazioni genetiche relative ai dipendenti o agli aspiranti tali potrebbe usarle per ridistribuire i compiti, licenziare o rifiutare l'assunzione di candidati. Con la sempre maggiore accessibilità delle informazioni genetiche, una pratica di questo tipo finirebbe con avere effetti su larghe fasce della popolazione: molte persone non solo avrebbero difficoltà a ottenere una polizza assicurativa sulla salute a causa di reali o potenziali disfunzioni, ma potrebbero avere anche problemi nel trovare un lavoro stabile e duraturo. Perfino nell'eventualità che si trovi una cura per le loro potenziali malattie, la loro situazione non sarebbe risolta, poiché le spese mediche si rivelerebbero molto costose e le compagnie assicuratrici si rifiuterebbero di pagarle. Anche nel mondo del lavoro esistono numerosi esempi di discriminazione genetica più o meno in linea con quanto osservato a proposito delle compagnie assicuratrici.
Un altro caso in cui lo screening genetico è stato utilizzato nel mondo del lavoro riguarda l'impiego di test genetici per determinare se un individuo sia o meno particolarmente suscettibile a eventuali agenti tossici presenti nell'ambiente di lavoro, come nel caso, già menzionato, dei soggetti affetti da deficienza nella produzione di α1-antitripsina, che possono, in presenza di pulviscolo o di particelle sospese nell'aria, sviluppare particolari affezioni del polmone. Gli operatori sanitari che lavorano nel settore industriale hanno recentemente proposto l'utilizzo di test genetici a cui sottoporre i lavoratori in via di assunzione, per individuare quelli fra loro particolarmente predisposti a questo tipo di disturbi e avere così la possibilità di valutare i rischi presentati dalle differenti richieste di lavoro. Purtroppo è accaduto che molti datori di lavoro, anziché adoperarsi per diminuire o eliminare gli agenti tossici presenti nell'ambiente di lavoro, abbiano preferito, adottando la soluzione economicamente più conveniente, respingere le domande d'impiego di alcuni aspiranti lavoratori geneticamente predisposti a malattie dell'apparato respiratorio. Ma l'esclusione di questi individui non elimina i rischi per la salute degli altri lavoratori in presenza di determinati agenti tossici; gli agenti a cui un soggetto predisposto è sensibile sono normalmente dannosi in misura rilevante anche per gli altri individui. Per evitare quindi che l'uso dei test genetici risulti utile esclusivamente al datore di lavoro e per difendere i diritti dei lavoratori geneticamente predisposti a determinate malattie è stato proposto che le informazioni genetiche sul conto dei lavoratori debbano rimanere in possesso degli interessati e inaccessibili alle istituzioni non autorizzate.
2. Emarginazioni. - Le persone con particolari genotipi non solo sono soggette a discriminazioni per motivi economici da parte dei datori di lavoro e delle compagnie assicuratrici, ma spesso vengono etichettate dalla società e conseguentemente emarginate. Questo è il risultato di un'errata valutazione delle conseguenze che una particolare caratteristica genetica può indurre o di un distorto senso della società, secondo cui una persona con caratteristiche genetiche particolari sia da ritenere in qualche modo inferiore; le conseguenze dell'emarginazione possono essere molto profonde.
È noto, per esempio, il caso della β-talassemia, una malattia genetica particolarmente frequente nelle popolazioni mediterranee, fra cui quelle greche e italiane. La malattia, che colpisce i globuli rossi, è causata da mutazioni del gene dell'emoglobina ma, grazie ai notevoli progressi compiuti dalla ricerca medica in questo campo negli ultimi anni, i malati che ne sono affetti riescono ad avere una vita relativamente lunga. Ciò nonostante è assolutamente indispensabile che i malati ricevano frequenti trasfusioni di sangue e siano loro somministrati farmaci piuttosto costosi. Allo scopo di informare le coppie in attesa di figli sulla possibilità di essere portatrici sane, in modo da poter, eventualmente, ricorrere all'aborto nei casi di feti affetti dalla malattia, le popolazioni di Cipro e della Sardegna sono state sottoposte a programmi di screening genetico (v. Cao e altri, 1991). I ricercatori che effettuarono questi programmi raccontano che, durante i primi anni, gli individui riconosciuti come portatori sani della mutazione del gene dell'emoglobina venivano emarginati e indicati nelle loro comunità come non adatti al matrimonio; la condizione di portatore sano, cioè, la cui pericolosità si sarebbe manifestata solo nel caso di matrimonio con un'altra persona a sua volta portatrice sana, era considerata come un indicatore di inferiorità genetica.
3. Conseguenze psicologiche dei test genetici. - Per finire, vanno considerate le ripercussioni psicologiche, spesso gravi, indotte nelle persone messe a conoscenza di determinati aspetti del proprio quadro genetico. Ormai non solo si è in grado di conoscere la base genetica dei propri problemi di salute in atto, ma sempre più spesso si può prevedere quali saranno i rischi di contrarre malattie in futuro. Oggi esistono test che consentono di prevedere in alcune famiglie l'insorgenza in tarda età del morbo di Alzheimer o della corea di Huntington, di stimare la probabilità di sviluppare il cancro della mammella o del colon e di stabilire la predisposizione ad avere problemi alle coronarie. Informazioni genetiche di questo tipo possono rappresentare un'arma a doppio taglio: da una parte alcuni individui preferiscono essere messi a conoscenza dei risultati di questi test, poiché ciò permette loro di programmare in anticipo la propria vita in relazione alle informazioni acquisite; dall'altra, molte persone accusano invece un profondo senso di angoscia quando apprendono di essere portatrici di un gene che può predisporle alla malattia. In passato, a causa di notizie di questo tipo si sono verificati casi di suicidio, dimissioni ingiustificate dal posto di lavoro e rotture di matrimoni; il numero di coloro che mostrano reazioni di questo tipo, anche se attualmente rimane limitato, è destinato a crescere con l'aumento del numero e della frequenza dei test genetici a cui la popolazione sarà sottoposta.
È altrettanto vero che la diffusione dei test genetici dovrebbe portare a una maggiore comprensione della genetica e, conseguentemente, alla progressiva scomparsa di quei luoghi comuni a essa relativi. È auspicabile che la pratica dei test genetici venga condivisa da larga parte della popolazione, in modo che il concetto di previsione genetica entri a far parte della cultura e diventi talmente radicato negli individui da non provocare più i traumi psicologici oggi frequenti. Gli effetti che questa integrazione può provocare a livello etico e morale sono al momento tanto imprevedibili quanto i progressi scientifici che saranno compiuti nel campo medico e tecnologico per la cura delle disfunzioni genetiche. Naturalmente, la disponibilità di cure appropriate ridurrà notevolmente alcuni dei problemi qui accennati.
Abbiamo descritto alcune delle possibili conseguenze dannose dei risultati dei test genetici; esse evidenziano la necessità di una programmazione attenta e responsabile della loro esecuzione, se non di un vero e proprio regolamento che ne controlli l'applicazione. Questi test sono però spesso ideati e finanziati da società che appartengono al mondo delle biotecnologie industriali; quindi, in alcuni casi, la necessità di una loro introduzione controllata si scontra con gli interessi economici delle industrie: in vista di nuovi e ampi mercati per i loro prodotti, ne viene accelerata quanto più è possibile la commercializzazione. Per esempio, in seguito alla scoperta del gene e delle mutazioni responsabili dell'insorgenza della fibrosi cistica, molte società iniziarono negli Stati Uniti a pubblicizzare presso i medici il test che ne rivelava la presenza. Alcuni gruppi di ricercatori e di medici erano a conoscenza del carattere ancora troppo sperimentale e dell'elevato margine d'errore che questo test presentava e indicarono subito le dannose conseguenze che un utilizzo del test in forma massiva e incontrollata avrebbe avuto. Non solo, ma alcuni di questi gruppi chiesero che fossero adottate precauzioni e che fossero imposte delle restrizioni nell'uso del test fino a quando non si fosse raggiunto un maggiore livello di sicurezza nell'interpretazione dei risultati e fino a quando non fossero stati del tutto chiari gli effetti del test sulla popolazione. Si convenne di garantire alle famiglie che presentavano casi di fibrosi cistica la possibilità di sottoporsi al test, ma il suo utilizzo massivo fu giudicato prematuro e l'inizio di programmi pilota di screening esercitò un'influenza moderatrice sulla tendenza alla diffusione incontrollata del test (v. Wilfond e Fost, 1990).
Da allora numerosi altri test sono stati introdotti sul mercato dalle industrie biotecnologiche, compresi alcuni che rivelano la presenza di marcatori genetici che possono indicare l'insorgenza di gravi malattie già presenti nell'anamnesi di una famiglia. In questi casi sorgono numerosi interrogativi sui benefici e sui rischi che questa nuova pratica della medicina comporta: è giusto promuovere la diffusione di alcuni test i cui risultati non concedono di fatto alcun vantaggio al paziente e che provocano invece spesso gravi traumi psicologici? È giusto sottoporre i bambini allo screening genetico? È necessario informare i familiari più stretti della diagnosi genetica riguardante un proprio congiunto? Tutte queste domande meritano studio e ponderazione, ma fino a quando non si raggiungerà un accordo sul comportamento da adottare in questa materia, l'attuazione in maniera sistematica di programmi di screening genetico e l'utilizzo dei test rimarranno problemi di difficile soluzione.
e) La genetica del comportamento umano e il libero arbitrio
Sempre più frequentemente si ricorre all'impiego delle nuove tecnologie nel campo della genetica al fine di studiare aspetti, anormalità e tendenze del comportamento umano. Questo genere di studi trae la sua origine da programmi di ricerca precedentemente intrapresi, in cui si analizzavano le relazioni esistenti tra le componenti genetiche e il comportamento umano nelle famiglie con gemelli o figli adottati. Mentre i risultati degli studi precedenti, elaborati cioè prima della risoluzione della struttura del DNA, erano stati accolti con scetticismo dalla comunità scientifica, l'interpretazione attuale dei dati rafforza la teoria secondo cui esisterebbe un ampio coinvolgimento dei geni nella definizione del comportamento umano. Come naturale conseguenza di ciò, recentemente si è assistito alla pubblicazione di un numero sempre crescente di studi preliminari, basati sull'analisi del DNA, che associano alcune caratteristiche del comportamento umano a loci genetici ben definiti; in particolare fino ad ora si è registrata la pubblicazione di lavori che hanno come oggetto la ricerca dei geni responsabili dell'omosessualità, dell'alcolismo, dell'aggressività, della psicosi maniaco-depressiva, della schizofrenia e dei disturbi dell'apprendimento (v. Baron e altri, 1990; v. Risch, 1990; v. Alper e Natowicz, 1993).
La genetica del comportamento è un'area di ricerca estremamente complessa: è difficile definire un fenotipo per ogni caratteristica del comportamento con la stessa precisione con cui lo si può stabilire nel caso di una malattia a base genetica come il cancro o la fibrosi cistica. Questo limite riconosciuto della genetica del comportamento induce la scelta di strategie di studio del problema spesso molto complicate. Inoltre, è largamente accettato da tutti l'assioma secondo cui così come esiste una base genetica per ogni caratteristica del comportamento, allo stesso modo esisterà una componente ambientale che svolgerà un ruolo determinante. Così, non tutti coloro che ereditano un gene che predispone a un determinato comportamento necessariamente lo esprimeranno: alcuni fattori ambientali ne faciliteranno l'espressione, altri la inibiranno. Per esempio, è noto come sia più probabile che una malattia mentale basata su una componente genetica si manifesti in particolari condizioni di stress ambientale, in alcuni casi provocate dall'individuo stesso. Un'altra difficoltà nello studio della genetica del comportamento è rappresentata dall'eventualità che un gene collegato a una caratteristica comportamentale sia sotto il controllo di altri geni che possono modificarne l'azione fino al punto di annullarne gli effetti. Questo è per molti ricercatori del campo un fenomeno così comune da ritenere in pratica che ogni carattere coinvolto nella determinazione del comportamento umano possa essere influenzato da più geni.
La complessità di analisi in quest'area della ricerca rende perciò estremamente difficile stabilire delle connessioni tra specifici geni e specifici comportamenti; in alcuni casi (ad esempio le psicosi maniaco-depressive, la schizofrenia o l'alcolismo) si è assistito alla ritrattazione di dati che erano stati già pubblicati, mentre altri risultati non hanno mai ottenuto il pieno consenso della comunità scientifica. Tutte queste controversie non implicano assolutamente che le relazioni tra geni e comportamento siano da ritenere inesistenti: esse indicano semplicemente la grande complessità che presenta quest'area della ricerca.
Nonostante ciò, numerosi studi di tipo genetico in questo campo sostengono l'ipotesi secondo cui i geni hanno una grande importanza nel determinare il modo in cui ci comportiamo; inoltre, l'interpretazione che la gente comune dà dei risultati di questi lavori va decisamente in una direzione deterministica. Basti ricordare che quando uno studio suggerì che le donne erano geneticamente inferiori agli uomini nello sviluppo delle capacità d'apprendimento della matematica, il popolare settimanale ‟Newsweek", affermò in un articolo che ‟se queste [le differenze] sono genetiche, noi dobbiamo imparare ad accettarle". Come abbiamo già avuto modo di affermare, la genetica non è sinonimo di predestinazione e non può essere usata per avvalorare un punto di vista così deterministico.
Molte sono le cause di questa errata interpretazione del ruolo della genetica: in primo luogo, le responsabilità sono da attribuire in parte ad alcuni ricercatori che nelle loro affermazioni in pubblico hanno caricato di eccessiva importanza il ruolo dei fattori genetici; è questo il caso, ad esempio, di James Watson, primo direttore del Progetto Genoma Umano negli Stati Uniti, che, come dicevamo, ha divulgato la sua visione fatalistica della genetica. In alcuni casi, poi, i ricercatori, nel riferire i propri risultati, ne hanno gonfiato l'importanza al fine di renderli più interessanti. In secondo luogo, il passaggio di un articolo da una rivista scientifica a un quotidiano o a un settimanale spesso si risolve in una semplificazione dei contenuti e delle conclusioni, a tutto vantaggio delle interpretazioni di tipo deterministico che forniscono spiegazioni a un livello molto più semplice, soprattutto se si tratta di temi inerenti alla genetica del comportamento. Va inoltre considerato il fatto che le tesi su base genetica sono spesso utilizzate per fornire una spiegazione di comodo a problemi sociali, mascherandoli come difetti relativi ai singoli individui o ai singoli gruppi. In questo modo la società, avendo attribuito alla genetica la responsabilità del problema, può sentirsi esentata dalla necessità di introdurre cambiamenti a livello sociale e proporre una soluzione basata sull'applicazione di trattamenti medici. Le spiegazioni su base genetica si mostrano così come un escamotage per sollevare la società da enormi responsabilità morali ed economiche, ed esonerarla dall'obbligo di fornire soluzioni a problemi come il crimine, la disuguaglianza sociale, ecc. In terzo luogo, a causa degli straordinari successi ottenuti nel definire i geni responsabili di una serie di malattie genetiche umane meno complesse e della vasta eco che questi successi hanno avuto presso l'opinione pubblica, la consapevolezza del ruolo primario svolto dai geni nel definire i comportamenti umani è notevolmente cresciuta anche tra i non specialisti. Purtroppo, però, questa consapevolezza ha portato a una diffusione incontrollata di teorie relative alla genetica di stampo chiaramente fatalistico, spesso al di là del nostro reale livello di conoscenza (v. Alper e Beckwith, 1993).
Le tesi di genetica del comportamento di stampo propriamente deterministico vengono spesso avanzate nelle discussioni sul libero arbitrio e la responsabilità individuale, e le conclusioni possono avere notevoli effetti sulla politica sociale. In seguito saranno discusse le conseguenze dal punto di vista sociale nel campo del diritto, ma prima è necessario elencare alcuni dei risultati a cui la genetica del comportamento è realmente giunta (v. Billings e altri, The genetic analysis..., 1992).
Quello dell'alcolismo è un caso molto indicativo, soprattutto perché in quest'area la scienza si mostra particolarmente attiva e fornisce risultati controversi: allo stato attuale delle conoscenze si ritiene possibile che alcune forme di alcolismo abbiano una base genetica, mentre altre siano di chiara origine ambientale. L'idea che l'alcolismo sia da considerarsi come una sorta di malattia è rafforzata dal riconoscimento di una predisposizione genetica esistente in alcuni casi; per alcuni individui è di grande sollievo pensare che il loro stato di alcolizzati sia da ricondurre a una forma ereditaria, dunque incontrollabile, e spesso ciò ne facilita il recupero; inoltre la loro condizione è più facilmente accettata dai loro amici e familiari. Uno degli effetti socialmente scomodi che questa interpretazione offre è però rappresentato dalla tendenza della società, e degli stessi alcolizzati a cui è riconosciuta una predisposizione genetica, a condannare in maniera definitiva coloro che invece devono il loro stato di alcolismo a una esclusiva influenza ambientale. Questi ultimi vengono considerati persone alle quali richiedere un maggiore autocontrollo.
Spesso la gente comune, venendo a sapere che un determinato comportamento può esser correlato a un certo fattore genetico, reagisce pensando che quel comportamento sfugga al controllo dell'individuo: si pensa che il soggetto sia più responsabile di un comportamento non influenzato geneticamente in quanto tale comportamento potrebbe essere modificato attraverso l'esercizio del libero arbitrio. La controversia sul rapporto tra fattori genetici e libero arbitrio è stata di recente amplificata dai dibattiti sull'incidenza dei fattori biologici sull'omosessualità; due ricercatori, Michael Bailey e Richard Pillard, hanno di recente pubblicato i risultati di alcuni studi compiuti su gemelli identici, che indicano l'esistenza di una componente genetica alla base dell'omosessualità (v. Bailey e Pillard, 1991). Gli autori sostengono che se fosse possibile dimostrare l'esistenza di una base genetica per l'omosessualità, questa non sarebbe più da ritenersi il risultato di una scelta esercitata liberamente, e quindi probabilmente la società sarebbe maggiormente disposta ad accettare gli omosessuali senza discriminazioni, sapendo che il loro modo di essere è in qualche modo predeterminato. Questa idea si basa sulle seguenti premesse: a) se l'omosessualità non è di natura genetica, allora si dovrà ritenere che le persone che si rivelino omosessuali lo siano per libera scelta o a causa di una serie di fattori ambientali, come per esempio le relazioni familiari; b) se l'omosessualità è determinata da fattori genetici, allora un individuo potenzialmente omosessuale non esercita in realtà alcuna scelta su quelli che saranno i propri gusti sessuali. Se fosse vera la prima premessa, chi sostiene che l'omosessualità è una condizione innaturale potrebbe affermare che gli individui omosessuali abbiano deliberatamente e coscientemente scelto una strada sbagliata; oppure, accettando l'ipotesi dell'esistenza di condizionamenti ambientali, coloro che avversano l'omosessualità potrebbero sostenere la necessità di curarla come una delle tante malattie psichiatriche di origine non genetica. Se invece fosse vera la seconda premessa, l'omosessualità sarebbe necessariamente da considerare ‛naturale' e quindi da accettare come parte delle manifestazioni del comportamento umano.
Considerazioni analoghe possono essere proposte anche nella discussione sull'alcolismo. Le conclusioni a cui si giunge negando l'esistenza di una base genetica in assoluto sono perfettamente adattabili anche al caso dell'alcolismo, ma la premessa opposta andrebbe comunque modificata, visto che ben poche persone sarebbero disposte ad accettare l'alcolismo, seppure legato a una predisposizione genetica, come una condizione ‛naturale' o ‛normale'. Ne consegue che anche se l'omosessualità fosse geneticamente determinata e quindi ‛naturale', coloro che rifiutano il comportamento degli omosessuali non accetterebbero mai di considerarlo come una normale manifestazione del comportamento umano.
Inoltre, va considerato che non c'è motivo di ritenere che i caratteri influenzati dall'ambiente siano più flessibili di quelli controllati geneticamente; quindi non è affatto detto che noi possiamo esercitare il libero arbitrio sul comportamento influenzato dall'ambiente, e ne siamo perciò responsabili, mentre non possiamo esercitarlo sul comportamento controllato geneticamente. Per esempio, una persona che soffre di squilibri ormonali dovuti ad alterazioni del genotipo può rivelarsi in grado di controllare i propri scatti d'ira molto più efficacemente di un'altra che ha subito degli abusi durante l'infanzia.
Infine, si immagini che in futuro venga dimostrato che, nonostante l'omosessualità sia di natura genetica, ciò non sia comunque sufficiente a determinare le attitudini sessuali di una persona (cioè che un individuo con un genotipo che indica omosessualità non necessariamente diventerà omosessuale). Gli stessi studi di Bailey e Pillard (v., 1991), in effetti, dimostrano che individui con lo stesso corredo genetico (i gemelli identici) rivelano solo nel 52% dei casi un'analoga tendenza all'omosessualità. Come si spiega che nel restante 48% dei casi tale concordanza non si manifesti? Ipotizzando l'esistenza del ‛genotipo omosessuale', che si rivela nelle attitudini sessuali di uno dei due gemelli, si deve concludere che l'altro, di tendenze eterosessuali, abbia volontariamente scelto la propria condizione o che i fattori ambientali (per esempio le pressioni della società in cui vive) siano stati sufficientemente forti da soffocare le sue attitudini omosessuali. Questi dati convergono nel far ritenere che se anche fosse provata l'esistenza di una componente genetica dell'omosessualità, essa non basterebbe a determinare il comportamento omosessuale manifesto.
C'è un certo pericolo nell'utilizzare le teorie genetiche con la finalità di combattere le discriminazioni a cui gli omosessuali sono sottoposti. Cosa accadrebbe se fosse in seguito dimostrato che il cosiddetto ‛gene del gay' in realtà ha una valenza ridotta o non esiste affatto? L'aver sostenuto l'esistenza di una base genetica per indurre a un maggiore rispetto nei confronti degli omosessuali, di fronte a una scoperta di questo tipo finirebbe coll'avvalorare le tesi di coloro che osteggiano gli omosessuali. Non è detto che la ‛naturalità' di un comportamento, la flessibilità nello sviluppo di un comportamento, l'opportunità di modificare un comportamento o la responsabilità di un individuo nei confronti di un proprio comportamento, possano essere giudicati esclusivamente stabilendo se la loro origine è prevalentemente genetica o ambientale. Le ragioni che sostengono la richiesta di uguali diritti per gli omosessuali devono essere basate su presupposti etici e morali e non su considerazioni genetiche.
In alcuni casi può comunque verificarsi che la distinzione tra un comportamento determinato geneticamente e uno determinato a livello ambientale possa contribuire a valutare la reale responsabilità di un individuo. Questo potrebbe essere per esempio il caso dei pazienti affetti dalla sindrome di Gilles de la Tourette, che spesso profferiscono pesanti oscenità verbali e violenti insulti in pubblico senza riuscire a controllarsi. Ovviamente l'essere a conoscenza del fatto che un individuo affetto da questa sindrome non ha in realtà alcun controllo sul proprio comportamento cambierà completamente il modo in cui verrà giudicato.
f) Il rapporto tra la genetica e la legge
Anche nel campo del diritto è sorta la discussione sulla necessità di distinguere tra azioni compiute per libera scelta o per condizionamento genetico, e le implicazioni conseguenti hanno avuto effetti significativi soprattutto nel ramo del diritto penale. Nel 1965 Patricia Jacobs e i suoi collaboratori pubblicarono uno studio sull'esistenza in alcuni uomini di un cromosoma Y in più (maschi XYY). Essi notarono che un'alta percentuale di questi individui era in carcere e ciò li portò ad affermare che la presenza di un cromosoma Y supplementare conferiva all'individuo un'aggressività maggiore e quindi giustificava il gran numero di questo tipo di uomini tra i criminali. Ai risultati di questo studio fu dato grande risalto e in breve tempo molti avvocati negli Stati Uniti e in Australia cominciarono a sfruttare questa condizione per difendere i propri clienti, affermando, in accordo con le teorie della Jacobs, che non potevano essere ritenuti responsabili dei crimini dei quali erano accusati coloro che risultavano essere portatori del cromosoma Y in più e quindi non dovevano essere condannati. L'argomentazione addotta si basava evidentemente sul presupposto che ‛genetico' fosse come sinonimo di ‛immutabile', e che quindi le spiegazioni di un comportamento in termini genetici potessero essere usate per sostenere che i criminali non erano responsabili dei delitti commessi.
Come per molti altri studi compiuti in questo campo, successive e più approfondite ricerche dimostrarono che le conclusioni in realtà erano errate e che la maggior parte degli uomini XYY conduce una vita normale e socialmente produttiva. Oggigiorno un individuo maschio di tipo XYY non viene più classificato come portatore del ‛cromosoma criminale' dalla comunità dei genetisti.
Più recentemente, un gruppo di ricercatori olandesi ha pubblicato una serie di articoli sui risultati preliminari di una ricerca riguardante una famiglia in cui molti individui maschi mostravano un comportamento particolarmente aggressivo; in tali articoli, l'aggressività viene associata alla mutazione del gene responsabile della produzione dell'enzima monoamminossidasi (MAO) (v. Brunner e altri, Abnormal behavior..., e X-linked..., 1993). Entro un anno dalla pubblicazione, uno degli autori è stato convocato per testimoniare come perito in un processo contro un uomo accusato di omicidio, condannato alla pena di morte nello Stato della Georgia, negli Stati Uniti. Allo scienziato è stato chiesto di fornire prove dell'esistenza di una base genetica del comportamento aggressivo, poiché l'avvocato difensore riteneva che ciò sarebbe servito a mitigare l'entità della pena inflitta al suo assistito.
Numerosi sono gli esempi analoghi in cui in un processo si è fatto ricorso a teorie genetiche per smontare una tesi accusatoria. Negli Stati Uniti, in Colorado, durante il dibattito riguardante l'approvazione di leggi relative ai diritti degli omosessuali è stato chiamato a testimoniare il ricercatore che più in quel momento si dimostrava interessato alla ricerca del gene dell'omosessualità. Egli espose i suoi risultati circa l'esistenza di una base genetica per questa condizione e alcuni sperarono che le prove biologiche avrebbero aiutato la promulgazione di leggi non discriminatorie in tema di omosessualità, mostrando che tale tendenza sessuale non era in realtà il frutto di una scelta volontaria. In California il riconoscimento di una predisposizione genetica all'alcolismo è servito a contenere la pena inflitta a un individuo accusato di appropriazione indebita.
5. L'eugenetica
Concezioni deterministiche della genetica sono state usate in passato come base per l'attuazione di programmi eugenetici da parte dei governi di alcuni paesi. Il presupposto che le caratteristiche di alcuni gruppi razziali o etnici fossero geneticamente determinate e immutabili ha permesso ad alcuni governi di considerare questi gruppi come inferiori e intrinsecamente corrotti. Tali argomentazioni portarono a programmi di sterilizzazione, alle restrizioni dell'immigrazione di determinati gruppi etnici e, come estrema manifestazione di questo atteggiamento, allo sterminio di milioni di persone considerate inferiori da parte dei nazisti.
a) L'eugenetica oggi
Storicamente, uno dei più sconvolgenti effetti delle applicazioni della genetica è stato lo sviluppo, da parte di alcuni governi, di programmi eugenetici che oggi, visti i profondi cambiamenti sociali, politici e culturali che sono successivamente intervenuti nella società, ci appaiono difficilmente ripetibili in un immediato futuro. In particolare, il ricordo del più estremo dei programmi eugenetici - quello nazista - è rimasto come un monito per molti dei paesi dell'Europa occidentale e per gli Stati Uniti, dove le conseguenze di una politica simile sono state avvertite direttamente. Tuttavia, altri paesi, che non hanno subito le conseguenze dirette di quel periodo storico, sotto la spinta dei propri governi attuano ancora oggi programmi eugenetici. I governi di Singapore e della Malaysia hanno entrambi incoraggiato le unioni matrimoniali tra membri di gruppi etnici considerati geneticamente superiori e, viceversa, hanno osteggiato quelle tra membri di gruppi considerati inferiori. La Cina incoraggia la pratica degli aborti, specie nei casi di feti considerati anormali, coerentemente con una politica che mira alla riduzione del numero di bambini presenti in una famiglia.
A prescindere dalla remota possibilità di un ritorno all'attuazione di programmi eugenetici tipici del passato, esistono altri modi attraverso cui le pratiche eugenetiche potrebbero oggi essere impiegate su vasta scala (v. Horgan, 1993). L'evolversi di tecniche di screening prenatale quali l'amniocentesi e l'analisi dei villi coriali permette di identificare una vasta gamma di variazioni genetiche nel feto a uno stadio relativamente precoce dello sviluppo. I consulenti genetisti o altri medici specialisti possono informare i genitori sul significato di determinate variazioni genetiche e sulle conseguenze che esse comportano per lo sviluppo del bambino. I genitori che fanno ricorso a tali tecniche possono quindi, in base alle informazioni ricevute, decidere se portare a termine o meno la gravidanza. In alcuni paesi, si richiede ai consulenti genetisti di non condizionare la scelta dei futuri genitori. Ciò significa che il loro personale punto di vista non dovrebbe trapelare. Negli Stati Uniti, ad esempio, l'autonomia dei genitori nel prendere una decisione è considerata uno degli obiettivi primari dei consulenti genetisti. Teoricamente, quindi, le analisi di screening prenatale forniscono importanti informazioni ai genitori e consentono loro di prendere decisioni sul proprio futuro in maniera libera e razionale. Se si trascurano per un attimo le implicazioni di natura morale che l'aborto comporta, si deve convenire che queste analisi sono di indubbia utilità per le famiglie.
Tuttavia, il concetto di autonomia delle famiglie in questi casi è da considerare in qualche modo illusorio; infatti, non solo non tutti coloro che esercitano la professione di consulente genetista si prefiggono l'obiettivo di non influenzare le famiglie, ma inoltre è molto difficile che il comportamento dei consulenti riesca a non far trapelare la loro personale opinione. Così, come si evince da molti rapporti, i medici possono influenzare in maniera significativa, con il loro modo di pensare, le scelte dei futuri genitori.
Inoltre, più in generale, anche gli atteggiamenti prevalenti nel contesto sociale possono condizionare la decisione dei genitori. Oggi, in molte società, si registra una crescente riluttanza a sovvenzionare progetti in favore dei meno fortunati. I genitori che scelgono di portare avanti una gravidanza che si concluderà con la nascita di un figlio affetto da una malattia certa e che comporterà un grave sforzo economico, devono spesso fronteggiare ostilità e ostracismi per avere compiuto una scelta simile. La consapevolezza di vivere in una società sempre meno disposta a fornire cure mediche o altre forme di assistenza a bambini disabili, che ‟non era necessario mettere al mondo", può esercitare una notevole pressione sui genitori, i quali possono decidere di non portare a termine la gravidanza. È già accaduto, come detto in precedenza, che compagnie assicuratrici abbiano indotto alcuni genitori a interrompere la gravidanza, ponendoli davanti alla prospettiva, in caso non avessero accettato, della perdita della polizza per l'intera famiglia.
La complessità di questo tema è ben resa dal caso della β-talassemia, precedentemente descritto: con i miglioramenti ottenuti dalla ricerca nella cura di questa malattia, il costo del mantenimento in vita e in buona salute di individui β-talassemici è aumentato vertiginosamente. A Cipro, dove un bambino su 158 nasce β-talassemico, i medici e il governo reputarono che il sostegno e le cure per questi bambini fossero una spesa troppo elevata per il bilancio dello Stato. Si giunse quindi all'attuazione di un programma di prevenzione da parte del governo che, seppur non coercitivo, di fatto indirizzava le famiglie verso l'interruzione delle gravidanze nel caso di feti che si rivelassero β-talassemici (v. Angastiniotis e altri, 1988). L'approccio ebbe un tale successo che portò alla riduzione di più del 90% delle nascite di bambini malati, ottenendo la decisa approvazione della società e garantendosi anche il plauso della Chiesa ortodossa. Si può quindi immaginare un futuro in cui, con l'avanzare della ricerca scientifica e l'aumentare del costo delle cure mediche, le istituzioni preposte al loro sostegno economico (lo Stato o le compagnie private di assicurazione) eserciteranno pressioni sempre maggiori per impedire la nascita di bambini affetti da malattie che prevedono cure dispendiose.
Il rapido aumento delle conoscenze e il miglioramento delle tecniche per ottenere diagnosi genetiche consentiranno, in un futuro abbastanza prossimo, di analizzare un gran numero di malattie ereditarie. Inoltre, è prevedibile che anche le tecniche per il rilevamento prenatale di varianti genetiche miglioreranno sostanzialmente: presto i ricercatori saranno in grado di ottenere senza difficoltà cellule fetali da un campione di sangue della madre; queste cellule potranno essere sottoposte a test che rivelino la presenza di marcatori genetici e la tecnica, se avrà successo, consentirà di analizzare i feti in maniera molto più semplice e meno rischiosa, aprendo le porte a una serie di analisi prenatali ad ampio raggio.
Infine, esigenze commerciali spingeranno sempre più le industrie del settore biotecnologico ad allargare il mercato dei test genetici; questo aspetto, se rimarrà incontrollato, finirà con l'esercitare un'ulteriore spinta verso l'abuso dei test anche nel campo dell'eugenetica. Al fine di rendere più chiari gli effetti derivanti dalla commercializzazione di un farmaco ottenuto mediante l'ingegneria genetica, cito il caso dell'ormone umano della crescita, GH. Questo ormone è utilizzato nella cura dei bambini che manifestano deficienze nella crescita e che sarebbero dunque soggetti a gravi forme di nanismo. Il numero di individui affetti da questa deficienza è tuttavia troppo basso per garantire un mercato tale da giustificare la produzione della medicina su larga scala; quindi, per aumentarne il commercio, una delle industrie che la produceva ha finanziato un programma di screening per studenti delle scuole inferiori, allo scopo di individuare quelli che avevano un'altezza con valori al di sotto della media. ‛Valori al di sotto della media' erano considerati quelli al di sotto dei 162 cm. Alcuni medici hanno quindi consigliato ai genitori di questi bambini l'uso del GH, sottolineando la delusione che avrebbero provato nel constatare che il loro figlio avrebbe raggiunto una statura al di sotto della media.
Nella prospettiva di una rapida espansione della disponibilità di test genetici, entrerà in gioco una serie di fattori sociali che determineranno se noi entreremo o meno in una nuova era di sperimentazione eugenetica. Ci si può chiedere se, con la scoperta delle relazioni esistenti tra i geni e alcune forme di comportamento sociale, come l'alcolismo, le malattie mentali o particolari inclinazioni, si giungerà all'estensione degli screenings genetici prenatali anche a queste caratteristiche. In questa prospettiva giocheranno un ruolo determinante sia le pressioni sociali, dalla spesa sanitaria agli interessi commerciali, sia l'evoluzione dell'atteggiamento con cui tutti considereranno la diversità genetica all'interno della specie.
b) La terapia genica
L'idea di manipolare geneticamente gli esseri umani modificando la costituzione genetica delle loro cellule fa venire in mente la creazione di mostri simili a quello di Frankenstein e, se si considera l'eventualità che una tale operazione abbia conseguenze impreviste, si capisce come queste paure abbiano un fondo di verità. Tuttavia, in generale si può dire che il principio dell'uso della terapia genica per la cura di malattie genetiche gravi, come anche di altre disfunzioni che non hanno necessariamente un'origine genetica, sia da considerare valido, anche perché tale terapia, sotto molti aspetti, è analoga a quella farmacologica. È noto ad esempio che certe forme di diabete si possono curare mediante la somministrazione di insulina; un approccio basato sulla terapia genica consisterebbe nell'introduzione dei geni responsabili della sintesi dell'insulina all'interno delle cellule appropriate, per ottenere gli stessi effetti della somministrazione dell'ormone. Tuttavia, si dovrà porre grande attenzione nell'assicurarsi che i geni incorporati non siano espressi in modo tale da provocare essi stessi dei problemi o da danneggiare altre funzioni cellulari.
È necessario considerare alcune questioni generali sull'utilizzo della terapia genica. In primo luogo, va esaminato il rischio che questa pratica venga impiegata per alterare caratteristiche fisiche umane che non sono considerate malattie: con la stessa logica con cui si è promossa la campagna per la vendita e la somministrazione dell'ormone della crescita a bambini al di sotto di una certa statura, si potrebbe introdurre una ‛terapia' con il gene responsabile della sua sintesi. Tentativi simili potrebbero essere compiuti per modificare altre caratteristiche umane, sia fisiche che comportamentali.
In secondo luogo, da sempre si guarda con grande apprensione all'applicazione della terapia genica alle cellule germinali, che avrebbe l'effetto di trasmettere i cambiamenti e le manipolazioni introdotte da una generazione all'altra. Ulteriore motivo di preoccupazione in questo caso particolare è dato dalla possibilità che queste alterazioni abbiano poi degli effetti, ancora ignoti, deleteri per le cellule sottoposte a trattamento. Inoltre, l'introduzione o la modifica di una caratteristica umana in un particolare contesto storico e culturale potrebbero non essere accettate dalle generazioni successive. Un gran numero di scienziati, teologi e personalità pubbliche ha chiesto che l'impiego della terapia genica sulle cellule germinali venga proibito, adducendo tra gli argomenti citati a supporto della loro richiesta la considerazione secondo cui con una pratica simile si modificherebbero le caratteristiche genetiche di un bambino senza che egli possa esprimere il proprio parere al riguardo.
c) Il rapporto tra la genetica e le priorità sociali
È stato descritto il modo in cui l'impiego degli screenings genetici e della terapia genica può apportare benefici a persone affette da particolari patologie. Si è anche parlato dell'interesse suscitato dalla ricerca di geni associati a determinati aspetti del comportamento umano, che può, seppur sotto altri punti di vista, presentare dei lati positivi. Ma molti sostengono che vi sia una troppo marcata tendenza a ricondurre tutti i problemi di salute e sociali a cause genetiche, e che tale tendenza possa distrarre l'opinione pubblica dai fattori ambientali e sociali che concorrono in maniera determinante a creare tali problemi. Per esempio, mentre è accertato che alcune persone dimostrano una forte tendenza verso un comportamento violento a causa di una componente biologica, sembra improbabile che la stessa predisposizione genetica possa considerarsi fondamentale nello spingere una persona a compiere un crimine. Anche nei casi in cui si accertasse la presenza di una predisposizione genetica, la manifestazione di un determinato modo di agire dipenderebbe comunque da fattori ambientali e sociali. E tuttavia, i rapporti preliminari sui ‛geni dei criminali' vengono in genere intesi come se fornissero una spiegazione del comportamento antisociale. Inoltre, la relativa semplicità delle spiegazioni genetiche è particolarmente attraente, in quanto rivela una nuova via per affrontare il problema in maniera definitiva e quantitativa: l'identificazione di un gene responsabile del comportamento violento sembra essere la via più facile da seguire, soprattutto se confrontata con le complesse spiegazioni di stampo sociologico che spesso cercano di dipanare un groviglio di altri fattori, quali le condizioni di vita durante la crescita, il tenore di vita o il retroterra culturale. Le conseguenze dell'attribuire tanta importanza ai fattori genetici possono essere il calo d'attenzione verso gli elementi sociali e ambientali che contribuiscono a formare un comportamento criminale e la esclusiva ricerca di presunti difetti congeniti dei criminali. Ne deriva una forte tendenza a sollevare la società da ogni responsabilità di miglioramento delle condizioni sociali creando al contempo una distinzione tra i possessori di geni ‛buoni' e geni ‛cattivi'.
L'adozione di una soluzione di tipo medico per i problemi sociali potrebbe un giorno essere accettata così come l'impiego della terapia genica o di altre terapie analoghe basate sulla genetica. Forse un giorno arriveremo a curare i criminali e gli omosessuali con la terapia genica, o addirittura promuoveremo l'aborto dei feti che mostreranno di possedere i geni della criminalità e dell'omosessualità. La preferenza accordata a spiegazioni mediche e biologiche potrebbe determinare l'ordine di priorità delle scelte governative; probabilmente si arriverà a un aumento dei finanziamenti per stabilire la relazione esistente tra la genetica e il comportamento sociale. Ciò potrebbe portare a una conseguente diminuzione dei finanziamenti di quei programmi che considerano globalmente gli effetti delle influenze ambientali e sociali. Se ne deduce che mentre l'attenzione nei confronti dell'identificazione di geni connessi con varie malattie porterà indubbi vantaggi, un'esclusiva focalizzazione su questo aspetto della ricerca può rivelarsi un'arma a doppio taglio. Ad esempio, gli studi epidemiologici volti a stabilire i fattori ambientali responsabili dell'acuirsi o del manifestarsi di determinate malattie potrebbero essere sottovalutati e in un prossimo futuro persino abbandonati per mancanza di fondi.
Questa tendenza ad attribuire forzatamente una base genetica ai problemi sociali è sicuramente il risultato dei progressi compiuti in genetica. Analizzando i fenomeni che ruotano intorno all'essere umano secondo un punto di vista olistico, le soluzioni dovrebbero essere cercate integrando le informazioni di tipo biologico con quelle di tipo ambientale: combinando l'identificazione di nuovi geni con gli studi di tipo epidemiologico si dovrebbe in realtà riuscire ad arrivare a una nuova sintesi che ristabilisca l'approccio olistico. Lo studio compiuto sulle famiglie che presentano geni che predispongono al cancro o a disturbi cardiaci potrebbe coinvolgere una ricerca volta a stabilire se l'alimentazione o altri fattori possano o meno avere una certa influenza su persone con queste predisposizioni, determinando differenti condizioni di salute. Analogamente, negli studi sugli individui portatori di geni che predispongono alla psicosi maniaco-depressiva si potrebbe tentare di correlare i risultati ottenuti con le analisi compiute sui vari aspetti della vita del paziente.
Lo sviluppo di una visione equilibrata del ruolo della genetica e dell'ambiente nel campo della salute e del comportamento sociale non può che risultare di grande beneficio per l'intera società (v. Andrews e altri, 1994).
BIBLIOGRAFIA
Alper, J. S., Beckwith, J., Genetic fatalism and social policy. The implications of behavior genetics research, in ‟Yale journal of biology and medicine", 1993, LXVI, pp. 511-524.
Alper, J. S, Natowicz, M. R., On establishing the genetic basis of mental disease, in ‟Trends in neurosciences", 1993, X, pp. 387-389.
Andrews, L. B., Fullarton, J. E., Holtzman, N. A., Motulsky, A. G., Assessing genetic risks: implications for health and social policy, Washington 1994.
Angastiniotis, M., Kyriakidou, S., Hadjiminas, M., The Cyprus thalassemia control program, in ‟Birth defects: original article series", 1988, XXIII, pp. 417-432.
Bailey, M., Pillard, R., A genetic study of male sexual orientation, in ‟Archives of general psychiatry", 1991, XLVIII, pp. 1089-1096.
Balding, D. J., Donnelly, P., How convincing is DNA evidence?, in ‟Nature", 1994, CCCLXVIII, pp. 285-286.
Baron, M., Novel strategies in molecular genetics of mental illness, in ‟Biological psychiatry", 1994, XXXV, pp. 757-760.
Baron, M., Endicott, J., Ott, J., Genetic linkage in mental illness. Limitations and prospects, in ‟British journal of psychiatry", 1990, CLVII, pp. 645-655.
Biesecker, B. B., Boehnke, M., Calzone, K., Markel, D. S., Garber, J. E., Collins, F. S., Weber, B. L., Genetic counseling for families with inherited susceptibility to breast and ovarian cancer, in ‟Journal of the American Medical Association", 1993, CCLXIX, pp. 1970-1974.
Billings, P. R., Beckwith, J., Alper, J. S., The genetic analysis of human behavior: a new era?, in ‟Social science and medicine", 1992, XXXV, pp. 227-238.
Billings, P. R., Kohn, M. A., deCuevas, M., Beckwith, J., Alper, J. S., Natowicz, M. R., Discrimination as a consequence of genetic testing, in ‟American journal of human genetics", 1992, L, pp. 476-482.
Botstein, D. L., White, M. H., Skolnick, M. H., Davis, R. W., Construction of a genetic linkage map in man using restriction fragment length polymorphisms, in ‟American journal of human genetics", 1980, XXXII, pp. 314-331.
Brunner, H. G., Nelen, M., Breakefield, X. O., Ropers, H. H., Oost, B. A. van, Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A, in ‟Science", 1993, CCLXII, pp. 578-583.
Brunner, H. G., Nelen, M. R., Zandvoort, P. van, Abeling, N. G. G. M., Gennip, A. H. van, Wolters, E. C., Kuiper, M. A., Ropers, H. H., Oost, B. A. van, X-linked borderline mental retardation with prominent behavioral disturbance: phenotype, genetic localization and evidence for disturbed monoamine metabolism, in ‟American journal of human genetics", 1993, LII, pp. 1032-1039.
Cao, A., Rosatelli, C., Pirastu, M., Galanello, R., Thalassemias in Sardinia: molecular pathology, phenotype-genotype correlation, and prevention, in ‟The american journal of pediatric hematology-oncology", 1991, XIII, pp. 179-188.
Cook-Deegan, R., The gene wars: science, politics, ad the human genome, New York 1994.
Guyer, M. S., Collins, F. S., The human genome project and the future of medicine, in ‟American journal of diseased children", 1993, CXLVII, pp. 1145-1152.
Holtzman, N. A., Proceed with caution: predicting genetic risks in the recombinant DNA era, Baltimore, Md., 1989.
Horgan, J., Eugenics revisited, in ‟Scientific American", 1993, CCLXVIII, 6, pp. 92-100.
Krimsky, S., Genetic alchemy: the social history of the recombinant DNA controversy, Cambridge, Mass., 1982.
Lander, E. S., Budowie, B., DNA fingerprinting dispute laid to rest, in ‟Nature", 1994, CCCLXXI, pp. 735-738.
Lewontin, R, C., The fallacy of biological determinism, in ‟Sciences", 1976, XVI, 2, pp. 6-10.
Lewontin, R. C., Rose, S., Kamin, L. J., Not in our genes: biology, ideology and human nature, New York 1984.
Miki, Y., Swensen, J., Shattuck-Eidens, D., Futreal, P. A., Harshman, K., Tavtigian, S. e altri, A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1, in ‟Science", 1994, CCLXVI, pp. 66-71.
Murray, T. H., Genetics and the moral mission of health insurance, in ‟Hastings Center report", 1992, XXII, 6, pp. 12-17.
Riordan, J. R., Rommens, J. M., Kerem, B.-S., Alon, N., Rozmahel, R., Grzelczak, Z. e altri, Identification of the cystic fibrosis gene: cloning and characterization of the complementary DNA, in ‟Science", 1989, CCXLV, pp. 1066-1073.
Risch, N., Genetic linkage and complex diseases, with special reference to psychiatric disorders, in ‟Genetic epidemiology", 1990, VII, pp. 3-16.
Strohman, R., Epigenesis: the missing beat in biotechnology, in ‟Bio-technology", 1994, XII, pp. 157-164.
The Ad Hoc Committee on Genetic Testing-Insurance Issues, Background statement: genetic testing and insurance, in ‟American journal of human genetics", 1995, LVI, pp. 327-331.
Watson, F., Human gene therapy. Progress on all fronts, in ‟Trends in biotechnology", 1993, XI, pp. 114-117.
Wilfond, B. S., Fost, N., The cystic fibrosis gene: medical and social implications for heterozygote detection, in ‟Journal of the American Medical Association", 1990, CCLXIII, pp. 2777-2783.
Wolf, U., The genetic contribution to the phenotype, in ‟Human genetics", 1995, XCV, pp. 127-148.
Genetica delle popolazioni di Alberto Piazza
sommario: 1. Definizioni e concetti generali. 2. Evoluzione nel tempo e distribuzione nello spazio delle frequenze geniche. 3. Genetica delle popolazioni europee: a) l'effetto di un cambiamento tecnologico: rivoluzione agricola ed evoluzione biologica; b) variabilità genetica e cambiamento linguistico. 4. La struttura genetica delle popolazioni umane: a) l'evoluzione biologica della nostra specie è punteggiata da espansioni demiche; b) espansioni nel Paleolitico; c) espansioni neolitiche e post-neolitiche. 5. Genetica delle popolazioni umane e coevoluzione genetico-culturale. 6. Il concetto di razza umana è scientificamente inconsistente. □ Bibliografia.
1. Definizioni e concetti generali
Se il termine ‛genetica', seguendo la definizione usata originariamente nel 1905 dal biologo inglese William Bateson, indica ‟la scienza che studia l'eredità e la variazione cercando di scoprire le leggi che governano le somiglianze e le differenze negli individui che sono in rapporto di discendenza", la ‛genetica delle popolazioni' è quella parte della genetica che studia i meccanismi che regolano l'evoluzione delle popolazioni. In questo articolo si tratterà esclusivamente di popolazioni della nostra specie, Homo sapiens sapiens, non solo perché a queste siamo più interessati, ma anche per il fatto che, avendo lasciato molte più tracce di sé delle popolazioni animali (almeno dall'epoca in cui sono state documentate le prime manifestazioni culturali), lo studio quantitativo della loro evoluzione nel tempo è risultato controllabile anche attraverso riferimenti di natura non strettamente biologica. Sebbene, in linea di principio, l'evoluzione delle popolazioni umane possa essere studiata a livello del singolo individuo - e le moderne tecniche di analisi del DNA rendono già possibile questo approccio - fino a oggi tali studi si sono basati sui caratteri ereditari di cui sia stato possibile determinare le regole di trasmissione da una generazione all'altra; di questi vengono calcolate le frequenze percentuali in campioni costituiti da un numero variabile di individui, e perciò variamente rappresentativi di popolazioni più estese, ma comunemente collocate nella stessa area geografica da cui tali caratteri sono tratti. La definizione dell'oggetto di studio, la ‛popolazione', è perciò più funzionale che rigorosa, ma malgrado ciò, se vengono usate alcune precauzioni, i risultati che si ottengono sono piuttosto attendibili e riproducibili. Poiché l'errore di campionamento è inversamente proporzionale alla radice quadrata delle dimensioni del campione, più numeroso è il campione più precisa è la stima delle frequenze che si vogliono calcolare: in pratica, i campioni con meno di 100 individui dovrebbero essere analizzati con molta cautela, quelli con meno di 50 non considerati affatto. Il riferimento più usato per collegare il campione alla popolazione di appartenenza è quello geografico (villaggio, città, regione, Stato, continente), ma spesso - e quasi sempre fuori dall'Europa - è considerato più risolutivo il riferimento linguistico: si parla, per esempio, di popolazione basca, e non di popolazione francese o spagnola, anche se i Baschi vivono sia in Francia sia in Spagna.
La ricostruzione dell'evoluzione dell'uomo dalle sue origini è, tra i temi analizzati in genetica delle popolazioni, quello più affascinante, certo il più ambizioso. A tal fine sono stati messi a punto alcuni modelli, ai quali si accennerà più avanti; tuttavia, affinché tali modelli possano essere ragionevolmente utilizzati, occorre che le popolazioni dalle quali provengono i campioni siano: a) ‛aborigene', intendendo con questo termine che il loro insediamento nelle regioni geografiche in cui oggi esse vengono esaminate abbia preceduto almeno le grandi scoperte geografiche e le colonizzazioni (convenzionalmente, quindi, prima del 1492), così da evitare di interpretare una variabilità genetica come dovuta all'evoluzione della stessa popolazione nel corso del tempo invece che alla combinazione relativamente recente di due o più popolazioni geneticamente distanti; b) ‛mendeliane', cioè popolazioni i cui individui si sposino a caso, senza che la selezione del coniuge avvenga in base a caratteri ereditari: se tale condizione non è soddisfatta, non è più valida la legge più importante della genetica di popolazioni, la legge dell'equilibrio di Hardy-Weinberg (1908), su cui sono basate molte predizioni sull'evoluzione delle frequenze geniche di generazione in generazione.
L'informazione genetica presente nel nucleo di ciascuna cellula di un organismo è contenuta in strutture chiamate cromosomi, di cui nella nostra specie, Homo sapiens sapiens, esistono 23 coppie per cellula; ogni coppia è costituita da un cromosoma trasmesso dal padre e da uno trasmesso dalla madre, che sono morfologicamente indistinguibili. I cromosomi sono fondamentalmente costituiti da acido desossiribonucleico (DNA), e i geni si possono operativamente definire come segmenti di DNA deputati alla sintesi di prodotti genici. Oggi possiamo studiare direttamente il DNA con metodi chimici, ma fino a poco tempo fa dovevamo limitare il nostro studio al fenotipo, cioè a un prodotto genico che si poteva manifestare nell'aspetto esterno dell'organismo (come il colore azzurro degli occhi o la positività del fattore Rh del sangue, ecc.). La determinazione genetica del carattere fenotipico esaminato, cioè il controllo che esso sia effettivamente ereditario, si accerta esaminandone la trasmissione famigliare dai genitori ai figli. La nostra specie ha circa 100.000 geni, ma quelli identificati (al 1997), cioè di cui è stata individuata e registrata la posizione nel cromosoma, sono poco meno di 7.000, di cui circa 1.500 non ancora assegnati a un cromosoma specifico: tutti questi dati sono conservati in una apposita banca dati, la Genome Data Base, che può essere consultata sul sito Internet http://gdbwww.gdb.org.
A ogni divisione cellulare il DNA viene replicato, così che le due cellule figlie risultanti contengono l'identica catena di DNA della cellula madre. Con una probabilità molto piccola (dell'ordine di 10-8) questo processo di replicazione del DNA avviene con qualche errore, per esempio un nucleotide viene sostituito da un altro dei quattro possibili, oppure vengono aggiunte o cancellate sequenze più o meno estese di nucleotidi. Si parla in questi casi di ‛mutazione'. L'effetto di una mutazione sull'organismo dipende dall'alterazione della funzione del gene specifico cui è associato il DNA alterato. Le mutazioni a carico delle cellule non sessuali (somatiche) possono indurre conseguenze gravi (per esempio tumori) nell'organismo in cui si manifestano, ma non vengono trasmesse ai figli. Le mutazioni a carico delle cellule sessuali, quale che sia il loro effetto, vengono invece trasmesse ai figli, e sono quelle che interessano la genetica. L'effetto della mutazione è la creazione di un nuovo gene, che è in genere leggermente diverso dal vecchio gene mutato; i due tipi vengono chiamati ‛alleli' di quel gene. Se l'individuo in cui è avvenuta la mutazione raggiunge lo stadio adulto e ha dei figli, può avvenire che il nuovo allele generato per mutazione venga trasmesso a uno dei figli, e così via di generazione in generazione. Tuttavia, può anche avvenire che il gene mutato venga ‛perso', oppure ancora che, per influenza di meccanismi evolutivi che saranno descritti in seguito, aumenti la sua frequenza sino a sostituire completamente l'allele vecchio. Questa sostituzione di un allele ‛vecchio' con un allele ‛nuovo' costituisce il processo elementare dell'evoluzione: l'integrazione di questi processi elementari estesa a tutti i geni da una generazione all'altra determina un cambiamento graduale, molto lento nel tempo, della struttura genetica di una popolazione. La maggior parte delle mutazioni avviene in geni differenti, ma nel tempo lo stesso gene può mutare più volte. In questo modo possono coesistere nella stessa popolazione molti alleli dello stesso gene: si dice allora che il gene è polimorfico, oppure che è presente un polimorfismo. I geni che controllano le malattie ereditarie sono polimorfici, perché esistono comunemente due alleli dello stesso gene: quello che genera la malattia, molto poco frequente, e quello comune o ‛normale', presente nell'individuo che non manifesta la malattia. Esistono tuttavia anche polimorfismi non associati alla manifestazione di una malattia, di alcuni dei quali è ignota la funzione: questi vengono chiamati operativamente (fino a prova del contrario) polimorfismi neutri, e sono quelli più comunemente usati per ricostruire la variabilità genetica tra individui e/o tra popolazioni. In questo senso i prodotti di tali geni, o le sequenze nucleotidiche dei geni stessi, vengono anche denominati ‛marcatori genetici', i più importanti dei quali possono classificarsi come segue.
1) Gruppi sanguigni e antigeni di istocompatibilità identificati mediante tecniche immunologiche: sono sostanze che si trovano sulla superficie dei globuli rossi o bianchi (eritrociti o leucociti) del sangue (per es., AB0, Rh, Kell, Duffy, HLA; v. sangue: Genetica del sangue, vol. VI).
2) Proteine ed enzimi, identificati mediante tecniche elettroforetiche: le proteine prodotte dai geni vengono fatte muovere in un campo elettrico (elettroforesi) e dallo studio della loro mobilità, determinata dalla loro carica elettrica a sua volta dipendente dalla loro struttura chimica, si deducono gli alleli dei geni che ne controllano la sintesi. I polimorfismi più studiati sono quelli relativi a proteine o enzimi che si trovano nel plasma o nel siero del sangue o sulla superficie degli eritrociti (per esempio, emoglobina, aptoglobina, fosfatasi acida).
3) Immunoglobuline, identificate mediante tecniche immunologiche: sono i cosiddetti anticorpi del sistema immunitario, per esempio le gammaglobuline.
4) Polimorfismi del DNA, identificati non più dal loro prodotto ma dall'analisi diretta di segmenti di DNA, resa possibile dalle nuove tecnologie dell'ingegneria genetica. Una tra queste, chiamata ‛analisi dei polimorfismi dei siti di restrizione' (RFLP, Restriction Fragments Lenght Polymorphism), consiste nell'impiego di enzimi batterici (enzimi di restrizione) che hanno la proprietà di tagliare le sequenze nucleotidiche del DNA in siti specifici (siti di restrizione) costituiti da 4, 6 o, raramente, più nucleotidi. Una mutazione in queste sequenze specifiche può impedire il taglio o può introdurne uno nuovo, dando così luogo a frammenti di DNA di diverse lunghezze, identificabili mediante elettroforesi. Attualmente si usa in prevalenza un altro metodo per identificare polimorfismi del DNA: l'esistenza all'interno del genoma di segmenti di DNA ripetuti (‛microsatelliti' se l'unità di ripetizione è costituita da pochi nucleotidi, comunemente da 2 a 4; ‛minisatelliti' se l'unità di ripetizione è costituita da più nucleotidi) e il carattere ereditario del numero delle ripetizioni hanno reso possibile l'identificazione di migliaia di polimorfismi, mappati più o meno regolarmente su tutti i cromosomi. Il metodo più diretto e definitivo di identificazione dei polimorfismi del DNA consiste naturalmente nell'analisi di sequenze di DNA, in prospettiva della sequenza di tutto il DNA del nostro genoma, e poiché il numero di nucleotidi nel DNA umano è stimato attorno a circa 3 × 109, esso è anche l'approccio più ambizioso sul quale sono impegnati molti progetti e risorse finanziarie a livello internazionale (v. genoma, vol. X). Si noti che l'utilità di sequenziare il DNA dei geni umani è indiscutibile se lo scopo è l'identificazione di polimorfismi. Molto più complesso è tuttavia il problema di associare a ciascun polimorfismo, che per definizione può esser diverso da individuo a individuo, la sua funzione biologica. Sfruttando la metafora del linguaggio, del DNA conosciamo l'alfabeto, potremo presto identificare l'intera sequenza delle lettere, ma non sarà facile distinguere una parola dall'altra e soprattutto decifrarne il significato.
2. Evoluzione nel tempo e distribuzione nello spazio delle frequenze geniche
Si consideri per esempio il gene che determina la positività o la negatività del fattore sanguigno Rh. Ogni individuo può essere positivo o negativo, com'è facilmente dimostrabile con un test immunologico: la distribuzione percentuale di individui positivi e individui negativi in un campione in esame è, come si è già detto, tanto più precisa quanto più esteso è il campione, e in genere differisce da campione a campione. Se, per esempio, si considera un campione sufficientemente numeroso degli abitanti della Sardegna, si trova che la frequenza degli individui Rh negativi è pari al 4%; ma lo stesso tipo di indagine compiuto sulla popolazione basca, che vive nella regione dei Pirenei tra Francia e Spagna, rivelerebbe che fino a 30 Baschi su 100 hanno il gruppo sanguigno Rh negativo. Questi sono i due valori estremi nella variazione delle frequenze del gene Rh in Europa. I geni che esistono in almeno due forme riconoscibilmente diverse (come la positività e la negatività per l'Rh) mostrano per lo più variazioni della frequenza dell'una o dell'altra forma in popolazioni diverse: anche popolazioni vicine sono raramente identiche fra loro.
La legge dell'equilibrio di Hardy-Weinberg semplifica l'analisi della distribuzione dei geni in una popolazione, prevedendo che se in una popolazione gli individui sono sufficientemente numerosi, si sposano a caso (condizioni di panmixia), e sono assenti mutazioni e altri meccanismi evolutivi che verranno spiegati in questo capitolo, le frequenze dei geni non cambiano di generazione in generazione. Questa legge è del tutto analoga alle leggi di conservazione del moto o dell'energia della fisica classica: così come la materia, la quantità di moto o l'energia non si creano né si distruggono, ma si conservano in assenza di fattori di perturbazione, così i geni (e perciò le loro frequenze percentuali) si conservano di generazione in generazione in assenza di meccanismi che perturbino tale situazione di equilibrio.
Il valore assoluto della differenza di frequenza dello stesso gene in due popolazioni misura una ‛distanza genetica' fra di esse; di solito si usano formule più elaborate che la semplice differenza tra le frequenze, ma è soprattutto importante prendere in esame geni diversi e calcolarne una distanza genetica media. La variabilità genetica tra popolazioni, che si può ricavare dalle distanze genetiche fra le varie coppie di popolazioni, costituisce il ‛documento' di base sul quale lavora chi studia l'evoluzione biologica delle popolazioni umane.
Rispetto al documento storico o archeologico, quello genetico è caratterizzato da un difetto e da un pregio. Il difetto è quello di riferirsi alle popolazioni oggi esistenti, e perciò di rappresentare il risultato sincronico di un processo evolutivo la cui sequenza diacronica va ricostruita a ritroso, senza possibilità di verifica sperimentale delle varie fasi del processo stesso (oggi abbiamo la possibilità di studiare il DNA di alcuni geni di cadaveri antichi, ma permangono problemi tecnici da risolvere). Il pregio consiste in una proprietà importante del cambiamento biologico: la sua estrema lentezza. Il processo fondamentale del cambiamento biologico di tutti gli organismi viventi è la mutazione, che consiste, come si è detto, in un cambiamento casuale del messaggio chimico costituito dal DNA che forma un gene. La mutazione avviene in un individuo ed è trasmissibile ai suoi discendenti, ma siccome ogni individuo ha un numero limitato di discendenti immediati, devono passare molte generazioni prima che quella mutazione si diffonda a un numero di individui sufficiente da poter essere facilmente osservabile in una popolazione. I cambiamenti genetici possono quindi fornirci informazioni di eventi più antichi di cui non esiste memoria storica.
Le frequenze geniche variano nello spazio e nel tempo. Per un meccanismo di natura statistica, che verrà specificato in seguito e che prende il nome di ‛deriva genetica', le proporzioni dei geni in una popolazione reale variano nel tempo - in altre parole la popolazione ‛evolve' dal punto di vista biologico - in modo tanto più significativo quanto più piccolo è il numero di individui che la compongono. Poiché, soprattutto in passato, le popolazioni erano di piccole dimensioni e si distribuivano in gruppi più o meno isolati tra loro (l'isolamento dipende soprattutto dalla caratterizzazione geografica dell'insediamento, cioè da distanze e barriere geografiche, ma anche da ragioni culturali, per esempio la lingua o l'usanza di sposarsi tra consanguinei), e poiché, inoltre, ciascun gruppo evolve più o meno indipendentemente dagli altri, dalla differenziazione nel tempo di ognuno di essi discenderà anche una differenziazione nello spazio: storia e distribuzione geografica dei geni sono strettamente correlate.
Si è già parlato della mutazione: essa costituisce il meccanismo fondamentale del cambiamento genetico che genera nuovi alleli e, nel caso in cui venga duplicato per caso un intero gene, anche nuovi geni. Senza mutazione non vi sarebbe evoluzione. Tuttavia, si tratta di un evento molto raro: nell'uomo, a seconda dei geni, capita in una su 10.000-1.000.000 cellule germinali (spermatozoi e uova) per gene per generazione, il che significa che la sua possibilità di diffondersi in una popolazione è minima se non vi sono altri meccanismi in grado di aumentarne la frequenza.
Oltre alla mutazione, i meccanismi evolutivi che possono cambiare le frequenze alleliche sono la migrazione, la selezione naturale e la deriva genetica casuale. La migrazione ha un effetto analogo a quello della mutazione, in quanto l'introduzione di nuovi individui immigrati può introdurre geni nuovi, o comunque alterare la distribuzione di frequenze degli alleli nella popolazione che li riceve; gli alleli appena introdotti potranno anche essere trasmessi alle generazioni successive. In questo senso, la migrazione può comportarsi in modo simile alla mutazione. Comunemente la migrazione tende a far diminuire le differenze genetiche tra popolazioni: se due o più popolazioni hanno frequenze geniche diverse, lo scambio migratorio reciproco tende a rendere le loro frequenze geniche più simili, e se lo scambio continua per molte generazioni, tende a renderle eguali. L'effetto genetico della migrazione dipende allora in modo critico dal rapporto fra il numero di individui delle due popolazioni, un dato demografico che purtroppo si può ricavare dalle fonti archeologiche e storiche solo raramente e indirettamente, per soli ordini di grandezza. Conviene comunque distinguere qualitativamente il caso in cui tale rapporto sia molto piccolo e quello in cui sia molto grande: il primo caso si verifica quando lo spostamento riguarda individui, famiglie o piccole comunità, si distribuisce per lo più su piccole distanze e causa una continua mescolanza all'interno delle popolazioni; il secondo riguarda invece spostamenti di grandi masse di popolazione e avviene per lo più su grandi distanze, con episodi di espansione e successivi insediamenti in terre spesso disabitate. È stato questo secondo tipo di migrazione umana, che si può più propriamente definire ‛colonizzazione', il maggiore responsabile dei grandi cambiamenti genetici che hanno caratterizzato singoli popoli o addirittura interi continenti.
La selezione naturale è il meccanismo per il quale i fenotipi aumentano o diminuiscono la loro frequenza a seconda della maggiore o minore capacità di adattamento all'ambiente naturale degli individui che li portano. Fu intuizione rivoluzionaria di Charles Darwin l'aver posto a fondamento dell'evoluzione delle specie il meccanismo dell'adattamento biologico per selezione naturale: un maggiore o un minore adattamento (fitness darwiniana) significa capacità di generare figli che possano raggiungere l'età fertile in numero maggiore o minore rispetto alla media degli individui che vivono nello stesso ambiente. Si tratta di un processo per tentativi ed errori, di cui si può prevedere l'andamento sulla base esclusiva di variabili demografiche. L'ambiente seleziona i tipi più ‛adatti', cioè quegli individui che manifestano un'aumentata fertilità o una probabilità maggiore di sopravvivenza fino all'età fertile; in questo modo aumenterà la frequenza degli alleli che determinano i tipi più adatti a quel particolare ambiente, e il processo di selezione naturale continuerà una generazione dopo l'altra fino a quando altre condizioni ambientali selezioneranno altri tipi genetici.
Per meglio illustrare questo meccanismo che sta alla base del nostro processo evolutivo conviene fare un esempio. L'incapacità di digerire il latte costituisce un disturbo abbastanza comune nelle popolazioni europee: di natura ereditaria, è dovuto alla mancanza dell'enzima lattasi, che permette di metabolizzare il lattosio, uno zucchero contenuto nel latte. Comunemente la lattasi è presente nel neonato fino allo svezzamento, ma nell'individuo adulto se ne trovano quantità minime: la tolleranza al lattosio (dovuta a un gene denominato ‛persistenza della lattasi') si riscontra tuttavia con una frequenza tra il 50 e il 100% nelle popolazioni europee per le quali il latte rappresenta un alimento di primaria importanza. Si suppone che la mutazione che permette alla lattasi di persistere nell'individuo adulto abbia avuto modo di affermarsi e diffondersi con l'introduzione in Europa dell'agricoltura, quando l'addomesticamento di bovini e ovini ha reso disponibile e, forse, per qualche popolazione, necessario il latte quale risorsa alimentare. In determinate condizioni ambientali, gli individui tolleranti al lattosio sono stati avvantaggiati rispetto a quelli intolleranti, e il gene della persistenza della lattasi si è così diffuso fino a raggiungere le frequenze che si osservano oggi. Si noti che tale gene è inutile in popolazioni la cui dieta è priva di latte, nelle quali in effetti la sua frequenza è pressoché nulla. Se è lecito datare l'origine della mutazione che ha determinato la persistenza della lattasi all'inizio dell'agricoltura nel Neolitico, 10.000 anni fa, è possibile avere un'idea quantitativa del vantaggio biologico, o coefficiente di fitness, che devono avere gli individui con quel fenotipo rispetto a coloro che ne sono privi: esso è dell'ordine dell'1%, ovvero, se si paragona un gruppo di genitori senza quel fenotipo con un gruppo di uguale numero di genitori con quel fenotipo, nell'arco di una generazione il primo avrà generato 100 figli, e il secondo 101. Questa differenza, che sembra piccola, accumulata di generazione in generazione può modificare la struttura genetica di una intera popolazione, e in effetti la selezione naturale è un meccanismo evolutivo che, a differenza della mutazione, è in grado di trasformare la nostra specie in tempi relativamente brevi, dello stesso ordine di grandezza dell'arco di tempo in cui è svolta l'esistenza di Homo sapiens. Poiché la selezione è specifica rispetto a un determinato ambiente, essa influenzerà non solo l'evoluzione temporale, ma anche la distribuzione geografica delle frequenze geniche.
Esistono molte forme di selezione naturale. Alla mutazione della persistenza della lattasi si associa l'espressione ‛selettivamente vantaggiosa' per far notare che l'allele che conferisce un vantaggio a chi lo porta in un determinato ambiente tende ad aumentare la sua frequenza nel tempo (in quell'ambiente) fino a raggiungere il 100%. Analogamente, esistono mutazioni selettivamente svantaggiose (chiamate anche ‛deleterie'): tutte le patologie genetiche sono, in genere, selettivamente svantaggiose, anche se dal punto di vista della genetica il vantaggio o lo svantaggio si misura dalla possibilità o meno di avere figli che raggiungano l'età fertile, per cui le patologie che insorgono dopo tale età hanno poca o nessuna rilevanza.
Di molti geni non si conosce il ruolo selettivo; numerosi sono selettivamente neutri, nel senso che, almeno per quel che è dato sapere al momento, non conferiscono a chi li porta né vantaggi né svantaggi. Ovviamente, un gene selettivamente neutro in un dato ambiente e in un certo periodo di tempo può diventare vantaggioso o svantaggioso al mutare delle condizioni ambientali. Tale relatività storica e geografica rende la selezione naturale un meccanismo estremamente elusivo e poco misurabile: esso può far aumentare, ma anche diminuire, la variabilità genetica entro o tra popolazioni, e l'unico modo qualitativo per caratterizzarlo rispetto agli altri meccanismi è il fatto che è gene-specifico, cioè capace di modificare la frequenza dei vari geni in un modo differenziato che dipende dal contesto storico (evoluzione), geografico (ambiente) e fenotipico degli individui sui quali agisce.
Un caso di polimorfismo genetico che spiega il permanere di frequenze geniche elevate correlate a patologie ereditarie letali (causate cioè da mutazioni deleterie che la selezione naturale tenderebbe a far scomparire) prende il nome di polimorfismo ‛bilanciato': ne sono esempi tipici l'anemia falciforme e la talassemia, malattie dei paesi dell'area mediterranea consistenti rispettivamente nella sintesi di emoglobina difettosa e nella sintesi insufficiente di determinate catene emoglobiniche (v. sangue: Anemie emolitiche, vol. VI). Entrambe le malattie sono probabilmente mantenute nel tempo dal fatto che gli individui eterozigoti (con un solo gene difettoso dei due trasmessi dai genitori) sopravvivono meglio degli individui normali in ambienti dove è presente la malaria: quindi, anche se gli individui omozigoti (con tutti e due i geni difettosi trasmessi dai genitori) muoiono per effetto della malattia, il gene che la causa non scompare perché conferisce agli eterozigoti una sopravvivenza maggiore in aree malariche (la malaria è stata in passato una malattia molto diffusa).
La ‛deriva genetica', o deriva genetica casuale, è un meccanismo evolutivo che dipende dalle dimensioni finite delle popolazioni e consiste in una fluttuazione, tanto più ampia quanto più piccole sono le dimensioni della popolazione, delle frequenze geniche da una generazione all'altra. Si considerino in una popolazione due alleli dello stesso gene, A e B, con frequenza entrambi del 50%, e si immagini per semplicità che ogni individuo porti un solo allele di quel gene invece di due (uno di origine paterna e uno di origine materna). Ora, per rendere l'esempio più chiaro anche se poco realistico, si immagini che in quella popolazione i possibili genitori abbiano potuto generare solo tre figli; vi sono quattro possibilità: 1) i tre figli sono tutti A; 2) i tre figli sono tutti B; 3) due figli sono A e uno è B; 4) un figlio è A e due sono B. Le ultime due combinazioni sono le più probabili, perché sono le più vicine alla distribuzione delle frequenze geniche nella popolazione di origine dei genitori. Tuttavia, le frequenze di A e B cambiano dalla generazione dei genitori a quella dei figli, perché da una distribuzione di A e B del 50% nella popolazione dei genitori, nel campione dei figli la distribuzione di A e B può essere di 2/3 e di 1/3 o viceversa. Inoltre, se i figli invece di essere tre fossero stati solo uno (riduzione estrema della popolazione), questo avrebbe potuto essere solo A o B, e il cambiamento sarebbe stato ancora più drastico: la frequenza dell'allele A dal 50% sarebbe aumentata al 100% o diminuita allo 0% (e viceversa per B).
Un effetto di queste oscillazioni delle frequenze geniche è quello di eliminare (in un numero di generazioni che si calcola approssimativamente pari al doppio del numero degli individui che compongono la popolazione) tutti gli alleli di una popolazione tranne uno. Un allele viene eliminato quando raggiunge, per caso, la frequenza dello 0% e solo una mutazione (o la migrazione da un'altra popolazione) può nuovamente reintrodurlo nella popolazione. L'effetto della deriva genetica è tanto più rilevante quanto più piccole sono le dimensioni della popolazione considerata. È lecito allora supporre che questo meccanismo evolutivo abbia avuto molta importanza nei primi stadi dell'evoluzione umana, quando la distribuzione della nostra specie, sia in Europa sia negli altri continenti, era frammentata in molti gruppi di dimensioni ridotte. Se nelle grandi città di oggi l'effetto della deriva genetica deve essere minimo, negli agglomerati con pochi abitanti può risultare ancora notevole. Un valido esempio è offerto dai comuni di montagna che per molti mesi all'anno rimangono praticamente isolati, nei quali è comune riscontrare un numero estremamente limitato di cognomi: ora il cognome, pur non essendo il prodotto di un gene, ne segue le regole di trasmissione lungo la linea maschile, poiché viene trasmesso di padre in figlio esattamente come un gene. Quando le dimensioni della popolazione sono piccole o si riducono per cause di forza maggiore (guerre, migrazioni, malattie, ecc.) il numero dei cognomi, che si possono formalmente paragonare a degli alleli, si riduce per deriva. Da una generazione all'altra alcuni cognomi scompaiono e da quel momento, anche se la popolazione tende nuovamente a crescere, il numero dei cognomi rimane quello della generazione precedente.
L'analogia coi cognomi ci aiuta a comprendere l'effetto della deriva genetica sulla variabilità entro e tra popolazioni, che è esattamente contrario a quello della migrazione: infatti esso tende a far diminuire la variabilità all'interno di una popolazione - il numero dei cognomi (alleli) diminuisce, gli individui tendono ad avere lo stesso cognome (allele) e perciò a essere più simili tra loro - ma fa aumentare la variabilità tra popolazioni; se il numero dei cognomi (alleli) diminuisce all'interno di ogni popolazione e tra di esse non vi è stato contatto, è più probabile che i cognomi (alleli) siano diversi nelle due popolazioni, perché ognuna si è evoluta in modo indipendente dall'altra e in ognuna alcuni dei cognomi (alleli) si sono persi in modo casuale.
I quattro meccanismi che sono stati descritti per sommi capi (mutazione, selezione naturale, migrazione e deriva genetica) interagiscono nella loro evoluzione temporale, e il risultato del loro effetto congiunto si traduce nella distribuzione geografica delle frequenze dei vari geni che osserviamo oggi nelle diverse popolazioni. Per dare un quadro sintetico di tale variabilità è conveniente riferirci a un modello evolutivo, anche se logicamente tale modello è una conseguenza dell'analisi della distribuzione geografica delle frequenze geniche anziché costituirne una premessa. A grandi linee potrebbe essere il seguente.
1) I dati archeologici testimoniano che i primi insediamenti della nostra specie hanno coinvolto piccoli gruppi distribuiti su larghe superfici, con pochi scambi tra loro. È quindi ipotizzabile che nel periodo di tempo compreso tra la prima comparsa di Homo sapiens sapiens in Africa (circa 100.000 anni fa) e l'inizio del Neolitico (10.000 anni fa), la deriva genetica abbia avuto modo di causare le maggiori differenziazioni genetiche tra le popolazioni nel mondo. Ipotesi simili, ma riferite ad altri archi di tempo, si possono formulare relativamente agli insediamenti dell'uomo moderno in Europa e negli altri continenti.
2) In questo panorama di estrema differenziazione delle frequenze geniche hanno avuto luogo, in momenti critici della storia - generalmente associati a innovazioni tecnologiche di grande rilevanza per la produzione del cibo o per la conquista territoriale - movimenti di grandi masse di popolazioni, colonizzazioni, nuovi insediamenti, ecc. L'effetto di tali migrazioni, di segno contrario a quello della deriva genetica, è quello di smussare le differenze genetiche, creando gradienti continui di cambiamento. Questi gradienti, se le popolazioni non fossero soggette a nuove migrazioni, sarebbero destinati a rimanere stabili nel tempo, tanto che ancor oggi potremmo osservarne le tracce.
3) L'effetto congiunto, ma opposto, di deriva genetica e migrazione - la prima induce variabilità genetica, la seconda la rimuove - tende dunque a creare una situazione di relativa stabilità nei periodi in cui non avvengono nuovi movimenti migratori su larga scala. Intervenendo su tale ‛paesaggio' di variabilità genetica, la selezione naturale può modificarne il panorama: purtroppo non sappiamo come valutare il cambiamento. È lecito supporre che molti geni, essendo selettivamente neutrali, non ne rimangano influenzati e sui pochi che ne potrebbero subire gli effetti la variabilità agirebbe in modo diverso da gene a gene. Poiché il problema più delicato nell'interpretare la variabilità geografica delle frequenze geniche consiste proprio nella validità di quest'ultima ipotesi, si è cercato di separare l'effetto congiunto di deriva genetica e migrazione da quello della selezione naturale, elaborando metodi di analisi in grado di ‛filtrare' una pressione evolutiva dall'altra, e considerare per quanto possibile l'informazione globale fornita da tutti i geni esaminati.
3. Genetica delle popolazioni europee
a) L'effetto di un cambiamento tecnologico: rivoluzione agricola ed evoluzione biologica
La variabilità comune a molti geni è più verosimilmente quella legata all'effetto di deriva genetica e migrazione piuttosto che a pressioni selettive, e perciò dovrebbe essere la più idonea a darci informazioni dirette sulla nostra storia di migrazioni e insediamenti passati. La tecnica di elaborazione statistica adottata a questo scopo prende il nome di ‛analisi delle componenti principali'. L'idea fondamentale è quella di operare una trasformazione delle variabili originali (nel nostro caso frequenze geniche) rappresentandole con il minimo numero possibile di nuove variabili, conservando la variabilità dei dati originali. Nella nostra analisi dei dati genetici relativi alle popolazioni dell'Europa (v. Cavalli-Sforza e altri, 1993 e 1994) sono state sufficienti 10 nuove variabili (chiamate ‛componenti principali') per riprodurre il 90% della variabilità di 95 frequenze geniche. Ognuna di queste componenti si può considerare come una qualsiasi variabile continua (in realtà è una combinazione lineare delle 95 frequenze geniche osservate in un numero assai ampio di campioni geograficamente rappresentativi) e in quanto tale può essere rappresentata graficamente su una carta geografica, denominata ‛mappa sintetica'. Per definizione, la prima componente descrive la quantità maggiore di variazione, e le successive (seconda, terza, ecc.) componenti descrivono proporzioni di variabilità progressivamente più piccole, che al di sotto di una certa soglia riproducono fluttuazioni statistiche casuali.
La mappa sintetica dei valori della prima componente principale, calcolata dalle frequenze di 95 geni osservate in Europa, è riprodotta nella fig. 1: la rappresentazione sintetizza il 28,1% della variabilità delle frequenze geniche originali. In realtà è un fatto piuttosto straordinario che con una sola variabile si possa riassumere il 28% della variabilità contenuta in 95 variabili indipendenti: in questo caso, ciò è semplicemente dovuto a un avvenimento storico importante che ha indotto una variazione comune e parallela in tutte queste variabili.
La mappa della fig. 1 mostra un gradiente molto chiaro, con un estremo nel Medio Oriente e l'altro a nord-est, in Irlanda, Gran Bretagna settentrionale e Scandinavia. Una mappa molto simile, ma meno precisa perché ricostruita su un numero minore di frequenze geniche (38), è stata pubblicata già nel 1978 in un lavoro (v. Menozzi e altri, 1978) in cui si notava la corrispondenza tra il gradiente che si osserva nella fig. 1 e la distribuzione delle datazioni archeologiche relative alla coltivazione dei cereali introdotta dagli agricoltori neolitici in Medio Oriente e poi diffusa, nell'arco di 4.000 anni, in tutta l'Europa. La mappa di tali datazioni è riportata nella fig. 2.
La correlazione tra le due mappe è molto stretta (circa del 90%), e non è certamente casuale. È stato simulato il processo di transizione da un'economia basata su caccia e raccolta a un'economia agricola con diffusione degli agricoltori dal Medio Oriente verso l'Europa nordoccidentale, e si è trovata una precisa corrispondenza tra la distribuzione delle componenti principali relative ai dati simulati e alle frequenze geniche reali (v. Rendine e altri, 1986).
La mappa sintetica della fig. 1 è di estremo interesse per comprendere la struttura genetica delle popolazioni europee. Dimostra infatti che: 1) la diffusione dell'agricoltura in Europa nel Neolitico non riguardò solo le tecniche agricole, ma fu il risultato dello spostamento degli agricoltori stessi, spinti probabilmente da una esplosione demografica indotta dalla stessa innovazione tecnologica; 2) il processo che, iniziatosi 10.000 anni fa, nell'arco di 4.000 anni ha sostituito i cacciatori-raccoglitori con gli agricoltori (con tutti i nuovi insediamenti che ne sono conseguiti) è stato così rilevante per la struttura delle popolazioni europee, che ancor oggi possiamo identificarne le tracce genetiche: esse costituiscono una specie di ‛struttura portante' delle differenze genetiche osservate in Europa.
L'introduzione dell'agricoltura ha quindi innescato una rivoluzione culturale che ha coinvolto usi e costumi, regimi economici e lente espansioni demografiche il cui segno è ancor oggi riconoscibile. La mappa che sintetizza la distribuzione di tutti i geni oggi conosciuti nel mondo, anche se di lettura più articolata di quella illustrata per l'Europa, non è in disaccordo con l'idea che i luoghi dove hanno avuto origine nuove tecnologie di produzione del cibo sono anche quelli da cui si sono diffuse migrazioni di individui, oltre che di idee innovative.
b) Variabilità genetica e cambiamento linguistico
Quale lingua parlavano gli agricoltori neolitici medio-orientali che si sono diffusi in Europa, dai quali verosimilmente discende l'attuale struttura genetica delle popolazioni europee? L'archeologo Colin Renfrew (v., 1987) ha prospettato l'ipotesi che essi parlassero lingue indoeuropee e che le abbiano diffuse in Europa. Questa ipotesi è in accordo con quella prospettata dal linguista A. B. Dolgopolsky (v., 1988) - il quale ritiene che le lingue indoeuropee abbiano avuto origine in Anatolia - ma contrasta con l'opinione di molti altri linguisti. In realtà è molto difficile ricostruire una lingua parlata diecimila anni fa, nella totale assenza di documenti scritti. La scrittura ha poco più di cinquemila anni e la velocità di trasformazione delle lingue, essendo molto elevata, rende assai difficile ricostruire idiomi molto antichi. Le ipotesi finora formulate circa il luogo di origine delle lingue indoeuropee sono numerose e diverse (v. Mallory, 1989); quella prospettata dall'archeologa Marija Gimbutas (v., 1991), che ha incontrato un certo successo, pone tale origine in una zona situata a nord del Mar Nero e del Caucaso, nella Russia meridionale. Circa cinquemila anni fa (coll'inizio dell'età del bronzo) in questa area si è sviluppata una civiltà di pastori nomadi che riuscì ad addomesticare il cavallo, un animale della steppa che si estende quasi ininterrotta dalla Romania alla Manciuria: a questa civiltà, basata sullo sfruttamento del cavallo, è stato dato il nome di ‛cultura kurgan', dal nome delle tombe a monticello molto comuni nella pianura meridionale russa. Il cavallo, inizialmente usato come fonte di cibo, è poi divenuto di grande importanza come animale da trasporto e, insieme all'invenzione del carro su ruote (v. Anthony, 1995), ha permesso una rivoluzione della tecnica, anche di quella militare (v. Diamond, 1991). A partire da quattromila anni fa, i popoli del Medio Oriente e dell'Asia occidentale e meridionale, ove si erano saldamente affermati imperi e civiltà agricole molto importanti, si sono trovati esposti alle incursioni e alla conquista dei pastori nomadi della Russia meridionale. Questi pastori parlavano lingue indoeuropee che imposero in Iran, Afghanistan, Pakistan e India. È probabile, anche se non è accettato da tutti gli archeologi (in particolare dallo stesso Renfrew), che le migrazioni indoeuropee dei pastori della Russia meridionale siano state dirette anche verso l'occidente, la Grecia e l'Italia. È anche possibile che discendenti di questi pastori, rimasti per esempio in Ucraina e nella Russia meridionale fino in epoca classica (come gli Sciti, di cui parla a lungo Erodoto), abbiano dato origine ad altri popoli, successivamente protagonisti delle espansioni di cui oggi registriamo le tracce genetiche.
Oltre all'espansione degli agricoltori neolitici dall'Anatolia, che possiamo dedurre dalla rappresentazione sintetica più informativa delle frequenze geniche (v. fig. 1), abbiamo prove genetiche di altre espansioni, anche se purtroppo l'analisi non ci permette di datarle. In particolare, la mappa sintetica di un'altra componente genetica importante (v. fig. 3) riproduce un'espansione a partire dalla zona della cultura kurgan, che sembra quindi confermare l'ipotesi della Gimbutas.
Si deve però chiarire che non è strettamente necessario scegliere fra l'ipotesi di Renfrew e Dolgopolsky (i quali per ragioni profondamente diverse pongono le origini indoeuropee in Anatolia) e l'ipotesi di Gimbutas, perché possono essere vere tutte e due, ma riguardare tempi diversi. L'argomento sostenuto da Renfrew, secondo cui un popolo in espansione porta con sé la propria lingua, è storicamente ben documentato, specie se questo popolo si espande in una regione a bassa densità demografica. La diffusione dell'agricoltura ha favorito l'insediamento iniziale di gruppi neolitici anche nelle steppe eurasiatiche, da cui potrebbe essere derivato lo sviluppo della pastorizia e della civiltà associata alla cultura kurgan. Con l'età del bronzo e con lo sviluppo dell'impiego del cavallo per scopi militari è possibile che siano avvenute altre espansioni di pastori nomadi, tre o quattromila anni dopo l'inizio della diffusione dell'agricoltura. I nomadi parlavano lingue indoeuropee, certo diverse da quelle originali dato il tempo intercorso fra le due espansioni, e le diffusero in una parte dei territori ove si erano insediati i neolitici, forse sostituendo alcune delle lingue indoeuropee parlate in precedenza dagli agricoltori. Sia l'espansione neolitica, sia quella dei pastori nomadi sono potute avvenire grazie a una serie di importanti innovazioni che hanno determinato una maggiore disponibilità di cibo, e quindi crescita demografica e migrazioni. L'espansione dei pastori nomadi ha coinvolto un numero minore di persone di quella neolitica, anche perché è avvenuta in regioni che avevano già un'alta densità di popolazione, ma è stata avvantaggiata da un'organizzazione sociale e da una potenza militare relativamente avanzate.
4. La struttura genetica delle popolazioni umane
a) L'evoluzione biologica della nostra specie è punteggiata da espansioni demiche
L'analisi genetica delle popolazioni europee riassunta nel capitolo precedente ha dimostrato che un fattore importante, forse il più importante tra quelli che influiscono sull'evoluzione delle popolazioni umane, è la migrazione. I movimenti di individui, di gruppi e, raramente, di intere popolazioni sono generalmente causati da un'innovazione biologica o culturale; questa in un primo tempo favorisce l'aumento della popolazione nel luogo dove questa si è verificata, e in seguito, quando il carico demografico eccede la capacità portante del territorio, induce lo spostamento graduale, di solito lento, di un segmento della popolazione alla ricerca di nuovi territori. Questo meccanismo, distinto dalla semplice migrazione (che di per sé non implica un processo di crescita), è stato chiamato ‛espansione demica'. Da un esame particolareggiato della struttura genetica di tutte le popolazioni del mondo di cui siano disponibili dati relativi alle frequenze geniche, svolto in collaborazione con L. Cavalli-Sforza e P. Menozzi (v. Cavalli-Sforza e altri, 1994), abbiamo tratto la convinzione che l'evoluzione umana, almeno quale risulta attraverso i geni, è stata ‛punteggiata' (l'espressione è ripresa da un classico lavoro sulla speciazione di N. Eldredge e S. J. Gould del 1972) da una serie di espansioni demiche, la cui causa è di natura culturale (per lo più la ricerca di nuove tecnologie per disporre di maggior quantità di cibo). L'effetto dei fattori culturali è genetico, nel senso che ancora oggi si possono ricostruire le tracce di quelle onde di spostamento nella distribuzione non casuale dei geni portati dai discendenti delle popolazioni coinvolte nelle espansioni. La diversità biologica della nostra specie si esprime quindi in una geografia di geni che riflette processi prevalentemente culturali di origine preistorica e storica.
Le espansioni demiche che a nostro avviso hanno potuto maggiormente influire sulla nostra diversità genetica sono quelle avvenute in epoca preistorica, soprattutto durante il Paleolitico e il Neolitico, allorché la densità di popolazione era così ridotta da rendere prevalente l'effetto della deriva genetica (v. cap. 2) su tutte le altre pressioni evolutive. La geografia genetica generata dalla deriva genetica corrisponde a una distribuzione delle frequenze geniche ‛a mosaico', nel senso che regioni geografiche contigue potrebbero presentare discontinuità genetiche anche estreme. In una configurazione di questo tipo, l'effetto dell'espansione demica è quello di smussare le differenze nelle aree geografiche interessate e di generare gradienti di variazione continui, che si osservano ancora oggi.
Al fine di poterci riferire a eventi preistorici concreti, abbiamo riassunto nella tab. I le espansioni a nostro avviso più importanti (e le corrispondenti innovazioni tecnologiche) che hanno ‛punteggiato' la nostra evoluzione.
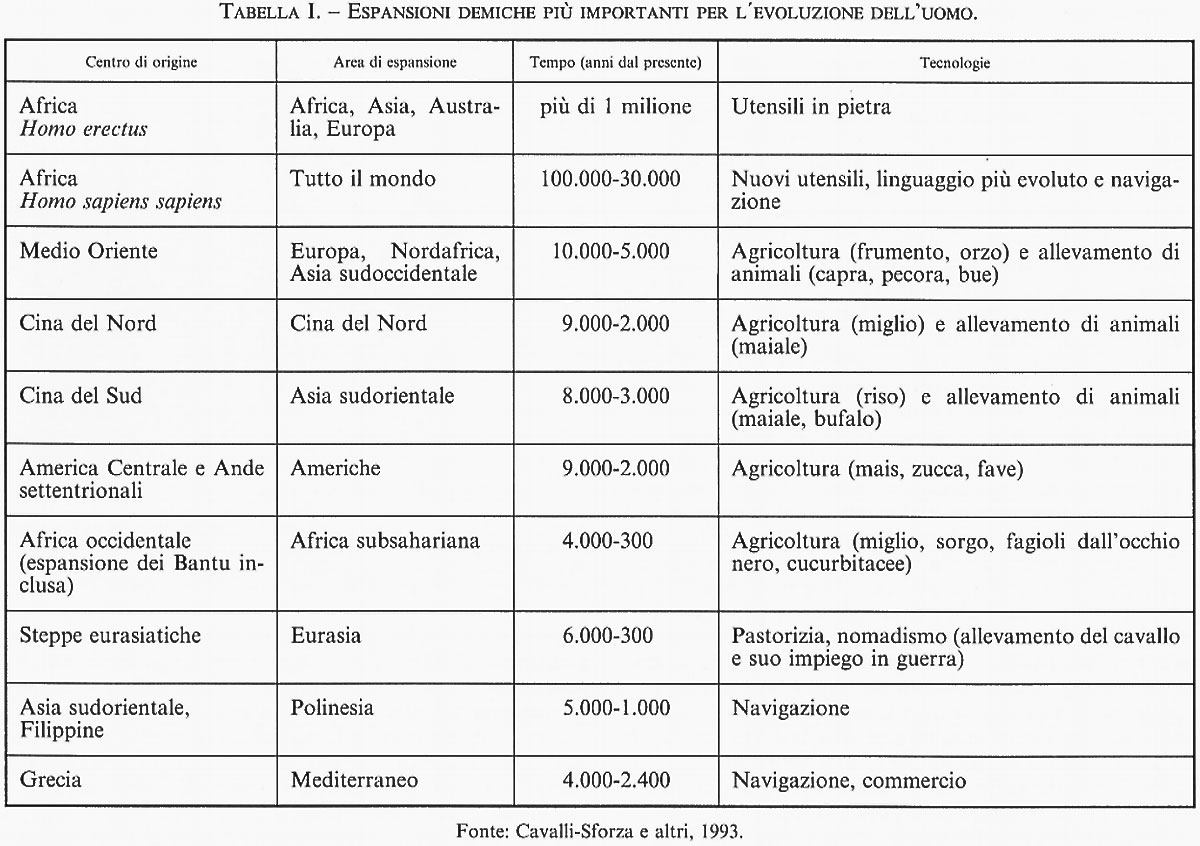
b) Espansioni nel Paleolitico
Le fonti archeologiche indicano che vi sono state almeno due espansioni di grande importanza per la storia del genere Homo. In un periodo compreso tra 1,7 e 1 milione di anni fa, Homo erectus ha incominciato a diffondersi dall'Africa, dove aveva avuto origine, nelle aree temperate dell'Asia e dell'Europa. Tra 100.000 e 60.000 anni fa ha avuto inizio l'espansione di Homo sapiens sapiens, cioè una specie anatomicamente moderna di Homo sapiens (v. Mellars e Stringer, 1989). L'origine africana di questa seconda espansione può forse essere messa in dubbio per il fatto che sono stati scoperti in Medio Oriente resti fossili di uomini anatomicamente moderni risalenti a più di 90.000 anni fa; tuttavia, sembra ormai accertata l'originaria provenienza africana di Homo sapiens, anatomicamente arcaico (v. uomo: origine ed evoluzione, vol. VII). Accanto a questa ipotesi monocentrica di entrambe le espansioni del genere Homo, alcuni paleoantropologi, di cui Wolpoff (v., 1989) è il più noto, preferiscono sostenere un'ipotesi policentrica della seconda espansione: l'uomo anatomicamente moderno sarebbe il risultato di evoluzioni locali svoltesi parallelamente in Asia, Europa e Australia e iniziate con la prima espansione africana (questa monocentrica) di Homo erectus. La validità di questa tesi sarebbe provata, secondo i suoi sostenitori, da una certa continuità morfologica dei reperti dell'Asia orientale in un arco di tempo di circa un milione di anni.
I risultati dell'analisi dei polimorfismi genetici (inclusi quelli del DNA nucleare e mitocondriale) riscontrati nelle attuali popolazioni aborigene concordano nel separare da tutte le altre le popolazioni africane attuali, come se fossero le più antiche. Il confronto di sequenze di DNA mitocondriale in popolazioni attuali non permette una ricostruzione filogenetica sufficientemente valida dal punto di vista statistico: tuttavia, pur lasciando in dubbio il luogo di origine (se in Africa oppure in Asia Minore) dell'uomo anatomicamente moderno, l'analisi mitocondriale ha contribuito in modo determinante ad accreditare l'ipotesi di una origine unica, in un'area geografica piuttosto circoscritta. Infatti, è difficile spiegare altrimenti l'osservazione che i tipi mitocondriali presentano un numero di mutazioni molto maggiore negli individui di origine africana che negli individui di altre origini, come ci si attenderebbe se i primi avessero avuto antenati molto più antichi e quindi maggior tempo per differenziarsi. Sulla base dei dati genetici attuali, mitocondriali e nucleari, non è comunque possibile escludere che i primi nuclei di uomini moderni provenienti dall'Africa o dal Medio Oriente arrivati in Asia centrale e orientale si siano poi in parte mescolati con i discendenti locali di Homo sapiens arcaico, il che si accorderebbe sia con l'ipotesi monocentrica, sia con la continuità delle forme fossili osservata in quelle regioni.
Per ciò che riguarda l'origine di questa seconda espansione, sia i dati relativi al DNA mitocondriale (calibrati sulla data di divergenza filogenetica tra lo scimpanzé e l'uomo anatomicamente moderno), sia i dati più recenti relativi ai polimorfismi dei microsatelliti del DNA nucleare (v. Goldstein e altri, 1995) suggeriscono che essa sia avvenuta dall'Africa a partire da 150.000 anni fa (il risultato più recente ottenuto saggiando i polimorfismi di 93 microsatelliti ha fornito una datazione assoluta della separazione degli Africani da tutte le altre popolazioni di 100.000 ±22.000 anni fa). Sia le datazioni ottenute dalla variabilità del DNA mitocondriale (associate tuttavia a un errore molto alto), sia queste ultime datazioni assolute, ottenute direttamente dal tasso medio di mutazione per anno dei microsatelliti saggiati, ben si accordano con le datazioni archeologiche (v. Mellars e Stringer, 1989).
Quali possono essere state le cause di queste due importanti espansioni della nostra specie dall'Africa? Devono essere stati fattori di cambiamento culturali e biologici, probabilmente associati a una ricerca e a una manipolazione del cibo più efficienti, nonché a una maggiore mobilità. Gli uomini che si sono diffusi con la prima espansione dall'Africa avevano un cervello di dimensioni superiori a quello dei loro predecessori, ed erano in grado di utilizzare una serie di utensili in pietra relativamente avanzati. L'evoluzione delle industrie litiche e l'uso controllato del fuoco (di cui si hanno testimonianze archeologiche che risalgono a circa 500.000 anni fa) riflettono cambiamenti culturali verosimilmente vantaggiosi in termini di disponibilità di cibo (e perciò di sopravvivenza) e opportunità di espandersi in nuovi habitat.
È probabile che una coevoluzione di cultura e biologia del cervello aiuti a spiegare l'origine dell'uomo anatomicamente moderno (Homo sapiens sapiens) e la sua successiva espansione in tutti i continenti. Il cervello umano, misurato dal volume endocranico, ha raggiunto le dimensioni attuali già nell'uomo anatomicamente arcaico (Homo sapiens) prima della seconda espansione dall'Africa. Durante questo periodo è possibile che il linguaggio abbia cominciato a evolversi fino a raggiungere il grado attuale di precisione e complessità, e abbia costituito il vantaggio culturale di maggior rilievo dell'uomo anatomicamente moderno, un vantaggio capace di indurre un'espansione così estesa e la sostituzione sostanziale (anche se avvenuta in tempi diversi da luogo a luogo) dell'uomo anatomicamente arcaico.
Anche se questa è per il momento un'ipotesi di lavoro, la documentazione archeologica testimonia una notevole evoluzione dei manufatti tra 100.000 e 60.000 anni fa, con un'altrettanto notevole differenziazione locale delle varie industrie litiche, come se la contemporanea differenziazione del comportamento e della comunicazione avesse favorito un processo prima di segregazione, poi di divergenza di tecnologie locali in rapida evoluzione.
Un progresso tecnologico innovativo che ha favorito la diffusione dell'uomo moderno è stata l'arte della navigazione. L'impiego di zattere e imbarcazioni doveva essere conosciuto almeno 60.000-55.000 anni fa, quando l'uomo anatomicamente moderno raggiunse l'Australia e la Nuova Guinea. Si può affermare che circa 40.000 anni fa tutto il Vecchio Mondo era colonizzato dalla nostra specie e le testimonianze archeologiche provano che attorno a quell'epoca si sono verificati grandi cambiamenti in ambito culturale, comportamentale e artistico. Sembra evidente che in un processo di espansione verso nuovi territori e di insediamento in nuove nicchie ecologiche un sistema di comunicazione verbale veloce e preciso debba aver conferito vantaggi non indifferenti per la sopravvivenza di chi ne era in possesso, favorendo così anche sotto il profilo biologico gli uomini anatomicamente moderni rispetto a quelli arcaici.
c) Espansioni neolitiche e post-neolitiche
Sebbene alla fine del Paleolitico il Mondo Antico fosse già interamente abitato dalla nostra specie, le dimensioni degli insediamenti erano ancora piuttosto piccole, con stime della popolazione totale che vanno da 1 a 10 milioni. Con simulazioni al calcolatore si è dimostrato (v. Rendine e altri, 1986) che i movimenti migratori avvenuti dopo il Paleolitico non hanno avuto il tempo di annullare le differenze genetiche generate in precedenza, ma hanno aggiunto nuovi segni, ancora oggi riconoscibili, alla composizione genetica delle popolazioni. Per valutare le impronte lasciate da quei segni, rispetto alle differenze geniche originali, e ricostruire così i movimenti migratori successivi al Paleolitico, occorre tuttavia ricorrere alle informazioni combinate di un numero sufficiente di geni, poiché la storia di un solo gene può descrivere eventi diversi da quelli migratori. Mappe geografiche ‛sintetiche', in grado cioè di riassumere in una sola immagine la distribuzione geografica delle frequenze di molti geni (v. cap. 3, § a), sono state introdotte appunto al fine di riscoprire tracce di espansioni demiche che i documenti archeologici o linguistici da soli non mettono chiaramente in evidenza. Tecnicamente le mappe sintetiche sono rappresentazioni geografiche delle linee di uguale valore delle ‛componenti principali' (intese in senso statistico) delle frequenze geniche rilevate in popolazioni comprese nell'area geografica considerata. Si è già detto che le mappe sintetiche della prima, seconda, terza, ecc. componente principale sono cartografie riassuntive, tra loro indipendenti, della distribuzione dei geni, che spiegano proporzioni sempre più piccole della variabilità genetica complessiva. Nei casi più semplici l'ordine delle componenti è associato al tempo cui la rappresentazione si riferisce, nel senso che la mappa della prima componente si riferisce comunemente a eventi di differenziazione genetica più antichi di quelli rappresentati dalla seconda, terza, ecc. In una mappa sintetica, il processo di espansione demica si ‛vede' come un ‛gradiente' (cambiamento graduale) continuo di valori che si sviluppa sotto forma di anelli concentrici a partire da un'area che indica approssimativamente il centro geografico dell'espansione. Dal momento che il processo di espansione è sempre accompagnato da altri fattori evolutivi che cambiano la distribuzione geografica della frequenza dei geni, il suo riconoscimento sotto forma di gradiente in una mappa sintetica è spesso oscuro o impossibile; tuttavia, quando il gradiente è chiaramente individuabile ed è statisticamente significativo (cioè non dovuto al caso) l'interpretazione più semplice è che esso rifletta la struttura genetica di popolazioni protagoniste di un processo di espansione demica importante.
Il quadro genetico dell'Europa è stato già discusso nel cap. 3 e illustrato nelle figg. 1-3. L'espansione dell'agricoltura ha avuto il suo riflesso genetico anche in Asia, nella cui prima mappa sintetica (v. fig. 4A) - che riassume l'informazione di 96 geni - si riconosce un chiaro gradiente da est verso ovest interpretabile come una semplice sintesi della progressiva diluizione del tipo genetico caucasoide nel tipo genetico orientale (o mongoloide) osservabile lungo la direzione ovest-est dell'Asia. Il gradiente non è regolare, ma vi è un cambiamento più brusco intorno a una linea che ha origine nella parte settentrionale degli Urali e scende verso sud-est per raggiungere la parte orientale dell'India: questa linea può essere considerata come il confine genetico tra tipo caucasoide e tipo orientale. È stato suggerito che le lingue dravidiche fossero state diffuse dagli agricoltori dell'area più orientale dell'Asia Minore e fossero state poi sostituite da lingue indoeuropee introdotte da popolazioni di pastori nomadi di tipo genetico caucasoide, che si sono infiltrate fino in India lasciandone intatta la parte meridionale. La mappa sintetica della seconda componente principale rappresenta un gradiente lungo la direzione nord-sud che forse può essere attribuito a un effetto selettivo del clima, anche se una traccia genetica delle migrazioni dal centro dell'Asia verso la Siberia non può essere esclusa. La fig. 4B illustra la terza componente della struttura genetica dell'Asia, con un chiaro e interessante centro di diffusione in Giappone. Le fonti archeologiche indicano un processo di crescita della popolazione giapponese che ha raggiunto il suo massimo (circa 300.000 individui) intorno a 4.000 anni fa, durante un periodo particolarmente florido nel quale l'economia ancora preagricola era basata essenzialmente sulla pesca. È probabile che a questo processo di crescita (archeologicamente documentata) sia corrisposta un'espansione (non ben documentata) le cui tracce sono riscontrabili nell'attuale quadro genetico.
La prima mappa sintetica dell'Africa, ricostruita sull'informazione fornita da 79 geni, illustra la distinzione tra le popolazioni di origine caucasoide e quelle di pelle nera che abitavano rispettivamente a nord e a sud del Sahara. Fino a circa 4.000 anni fa il Sahara non era un'area desertica e anzi fu teatro di importanti progressi, soprattutto nelle tecniche di allevamento del bestiame. Con la desertificazione del Sahara, da 4.000 a 3.000 anni fa, il bestiame fu obbligato a migrare verso sud, e con esso gli allevatori (nella maggior parte neri) che cominciarono a trasformarsi in coltivatori di piante, prediligendo il sorgo e il miglio nella regione del Sahel a sud del Sahara. Molte di queste innovazioni agricole ebbero luogo nell'Africa occidentale, un'area dalla quale cominciò a espandersi una corrente migratoria documentata verso est e verso sud. Questa diffusione è ben visibile nella seconda mappa sintetica, dalla quale si rileva anche che la parte sudoccidentale del Sudafrica è geneticamente diversa dall'Africa occidentale. La prima è oggi prevalentemente occupata da popolazioni che parlano linguaggi khoisan (Boscimani e Ottentotti) e che presentano qualche somiglianza genetica (ma non linguistica) con le popolazioni dell'Africa orientale. L'area geografica che nell'Africa occidentale è riportata con il tratteggio più chiaro definisce una regione che, da 5.000 a 4.000 anni fa, fu probabilmente critica per la diffusione iniziale dell'agricoltura in Africa, e anche per l'espansione meglio conosciuta delle popolazioni agricole di lingua bantu - originatesi nel Camerun vicino al confine con la Nigeria circa 3.000 anni fa - che si estese all'Africa centrale e alla maggior parte del Sudafrica.
Le mappe sintetiche delle Americhe sono più complicate da interpretare, anche se, come è ovvio, si riferiscono a popolazioni amerinde. Sui piccoli nuclei sopravvissuti allo sterminio dei colonizzatori europei ha agito in modo determinante la deriva genetica, che deve aver generato una estrema differenziazione locale. La mappa sintetica, ricostruita sull'informazione fornita da 72 geni, illustra le differenze genetiche dei tre gruppi etnici maggiori: gli Eschimesi a nord (corrispondenti alla zona con tratteggio più scuro); i Na-Dene nella zona contigua a sud degli Eschimesi; e gli Indiani d'America in tutte le altre aree con tratteggio più chiaro. Un risultato interessante di questa tripartizione è che essa corrisponde esattamente a quella suggerita dall'analisi linguistica di Greenberg (v., 1987), attualmente oggetto di accesi dibattiti tra i linguisti americani. Altre mappe sintetiche non mostrano gradienti, ma pongono in evidenza un'estrema eterogeneità genetica: in particolare, probabilmente a causa delle vicissitudini storiche delle popolazioni amerinde, non identificano i centri di diffusione dell'agricoltura ben conosciuti del Messico e delle Ande settentrionali (Ecuador e Perù), dove il clima era più favorevole. La documentazione archeologica indica la presenza di un'economia agricola in Messico e nelle Ande a partire da circa 9.000 anni fa e una diffusione molto più tardiva, iniziata circa 4.000 anni fa, verso il Nordamerica.
5. Genetica delle popolazioni umane e coevoluzione genetico-culturale
Se fossimo in grado di disegnare in un grafico le tappe della nostra civiltà, tracceremmo una linea che illustrerebbe dapprima la capacità di disegnare, poi quella di rappresentare i nostri comportamenti in simboli astratti, per giungere infine all'elaborazione di un sistema di scrittura in grado di riprodurre tutte le sfumature della lingua parlata. La transizione linguaggio-scrittura-cultura, pur nella sua semplificazione estrema, fornisce almeno un punto di riferimento per paragonare due diversi meccanismi evolutivi nell'uomo: quello biologico, mediato dai geni, e quello culturale, mediato dal linguaggio.
Viene naturale domandarsi: sono i geni a influenzare la cultura o è la cultura a influenzare i geni? Una risposta a questa domanda è relativamente semplice: entrambi i fenomeni sono possibili, ma è quasi impossibile capire quale dei due è più importante. Tuttavia, mentre è difficile trovare esempi di geni specifici che abbiano influenzato la cultura, abbiamo già trovato esempi di tratti culturali che hanno specificatamente influenzato la distribuzione dei geni nelle popolazioni umane. In una ricerca che ha suscitato molte discussioni (v. Cavalli-Sforza e altri, 1988) abbiamo tentato di ricostruire l'evoluzione dell'uomo integrando i dati genetici con quelli archeologici e linguistici: un risultato che emerge chiaramente da questa ricerca è la corrispondenza tra le grandi famiglie linguistiche in cui si suddividono le popolazioni umane e la loro evoluzione genetica. Tale corrispondenza è illustrata nella fig. 7: l'albero filogenetico è in accordo con i dati archeologici che pongono l'origine di Homo sapiens sapiens in Africa. La corrispondenza quasi biunivoca tra la struttura dei raggruppamenti genetici e quella delle famiglie linguistiche ci ha indotto a ipotizzare che uno dei vantaggi adattativi di maggior rilievo che può aver prodotto l'insediamento di Homo sapiens sapiens in tutte le parti abitate del nostro pianeta sia stato esattamente quello di un linguaggio completamente articolato.
La sostituzione repentina dell'uomo di Neanderthal da parte di una specie che era sulla Terra da almeno 50.000 anni, e la sua rapida estinzione, confermano l'ipotesi che il vantaggio della nostra specie sia essenzialmente consistito in un modo di comunicare più efficiente, che avrebbe migliorato le tecniche di caccia e di approvvigionamento del cibo, favorito più stretti legami sociali, reso più semplice la diffusione di informazioni e di persone. A sostegno, seppure indiretto, di questa ipotesi, si consideri che nella nostra società, fino a 100-150 anni fa, gli individui sordomuti avevano ben poche possibilità di riprodursi, a causa della discriminazione sociale cui erano sottoposti.
6. Il concetto di razza umana è scientificamente inconsistente
Da quanto discusso nei capitoli precedenti si evince che quando oggi si parla di diversità biologica tra individui o tra popolazioni di individui, occorre non cadere in equivoci che possono risultare fonte di pregiudizi diffusi ma completamente infondati. Se esaminiamo due individui scelti a caso, non parenti tra loro, il loro corredo genetico sarà, con certezza quasi assoluta, diverso, perché diversi saranno i nucleotidi delle catene di DNA che lo costituiscono: in altre parole, nel mondo biologico la diversità costituisce la regola, l'uniformità l'evento eccezionale. Di conseguenza, la tendenza quasi istintiva che ci porta a riconoscere razze diverse tra popolazioni umane di diverso insediamento geografico, non ha alcun fondamento biologico, ma è di natura esclusivamente culturale. Il ‛diverso' è classificato come tale in base a parametri che della biologia manifestano l'ignoranza più che la conoscenza: la persona di pelle scura è, da un punto di vista genetico, sì diversa dalla persona di pelle chiara, ma i geni che controllano il colore della pelle (non uno solo, ma dell'ordine di qualche unità) costituiscono una parte irrisoria della totalità dei geni (dell'ordine di 100.000) che sono differenti in due persone con lo stesso colore della pelle. La persona di pelle scura viene ‛vista' diversa dalla persona di pelle chiara, solo perché il colore della pelle è un carattere visibile, e in quanto tale viene culturalmente isolato rispetto ai caratteri la cui esistenza non è riconosciuta dai nostri sensi. Anche il colore degli occhi, la forma e le dimensioni del viso, il colore dei capelli, la statura, ecc., cioè caratteri che definiscono la morfologia o le dimensioni fisiche del nostro corpo, sono stati associati a differenze genetiche che in realtà hanno un peso molto minore di quello che sembra. È il caso poi di aggiungere che l'apparenza inganna non una, ma due volte: se ci si lascia guidare dai soli caratteri antropometrici, la storia delle loro differenze ha un vizio di origine, quello di essere prevalentemente legata non all'evoluzione storica degli individui che li portano, ma piuttosto alle caratteristiche fisico-ambientali dei loro insediamenti.
Le varie classificazioni delle popolazioni umane in razze che, dalla Bibbia sino alle opere della prima metà di questo secolo, sono state spesso il riflesso della nostra cultura classica o positivista più che della nostra conoscenza scientifica, si sono dimostrate un esercizio sterile per ragioni che erano chiare già a Darwin. Oltre che per i motivi precedentemente addotti, le razze umane sono entità estremamente instabili anche nelle mani dei tassonomisti moderni: a seconda degli algoritmi di classificazione, si possono distinguere da tre a più di sessanta cosiddette razze (v. Garn, 19713). Tale eterogeneità dipende in parte anche dalle diverse scuole tassonomiche le cui tendenze a unire (lumpers) o a diversificare (splitters) le popolazioni non sono definite da criteri univoci. Nel settimo capitolo della sua opera The descent of man and selection in relation to sex, Charles Darwin scriveva: ‟è senz'altro indifferente indicare le razze dell'uomo in questo modo o raggrupparle in specie o sottospecie, anche se il termine sottospecie sembra più appropriato. Possiamo concludere che quando i principî dell'evoluzione saranno generalmente accettati, come ci aspettiamo avvenga presto, la diatriba tra monogenisti e poligenisti morrà di una morte silenziosa cui nessuno farà caso" (v. Darwin, 1871, vol. I, p. 235). Questa previsione si è avverata solo in tempi molto recenti, avvalendosi dei risultati che i dati archeologici e molecolari hanno fornito in appoggio all'idea di una origine unica della nostra specie in Africa; l'analisi dell'evoluzione delle popolazioni umane odierne distingue vari gruppi etnici, nella maggior parte famiglie o isolati linguistici (v. fig. 7), ma osservando la figura dal centro verso sinistra, cioè andando a ritroso nella differenziazione genetica, si incontrano raggruppamenti sempre più ampi: una loro classificazione dipende dal livello gerarchico che si considera, ed è quindi completamente arbitraria.
Statisticamente si è dimostrato che la variazione genetica all'interno dei vari raggruppamenti, di solito definiti secondo criteri geografici o linguistici, è dello stesso ordine di grandezza di quella tra i raggruppamenti (v. Lewontin, 1972; v. Nei e Roychoudhury, 1982). Tale osservazione deriva dal fatto che in quasi tutte le popolazioni sono presenti, anche se con frequenze differenti, tutti i geni conosciuti e che tutte le popolazioni o gruppi di popolazioni possono essere posti in relazione con rapporti di affinità diversa a seconda del gene che si considera. Ne consegue che nessun singolo gene è sufficiente a classificare le popolazioni umane in categorie sistematiche. La variabilità che esiste in tutte le popolazioni, anche in quelle di piccole dimensioni, si è accumulata in tempi molto lunghi, forse dall'origine stessa del genere Homo, circa 700.000 anni fa: altrimenti non si spiegherebbe la presenza della maggior parte dei geni che conosciamo in quasi tutte le popolazioni. D'altra parte la differenziazione geografica dell'uomo anatomicamente moderno (Homo sapiens sapiens) è recente, risalendo a circa 100-150.000 anni fa (solo 1/5 del tempo trascorso dall'origine dell'uomo); perciò non vi è stato tempo sufficiente per una differenziazione, tra i molti gruppi etnici insediatisi in aree geografiche diverse, maggiore di quanto non lo sia all'interno di ciascun gruppo. Inoltre la nostra specie, come quella del nostro antenato più vicino Homo erectus, ha sviluppato un'attività migratoria molto intensa in tutte le direzioni, con fenomeni di ibridazione tra popolazioni magari separate da lungo tempo. Le mescolanze diminuiscono le differenze genetiche, introducendo gradienti continui di variabilità che rendono ancora più difficile la definizione di ‛confini' genetici, come è immediatamente constatabile osservando le mappe sintetiche illustrate nei capp. 3 e 4.
Dal punto di vista scientifico, il concetto di razza è perciò privo di fondamento. Si può obiettare che gli stereotipi razziali hanno una coerenza che permette anche ai profani di ‛classificare' le persone. Tuttavia, come già si è sottolineato, gli stereotipi più diffusi (colore della pelle, colore e struttura dei capelli, tratti del viso) riflettono differenze superficiali che analisi accurate non confermano essere di origine esclusivamente genetica. Con metodi statistici ormai raffinati, e confrontando la storia genetica di molti geni e di molte popolazioni, siamo in grado di distinguere gruppi di popolazioni diverse e di ordinarli in un albero di differenziazioni successive. Tali gruppi, tuttavia, non possono identificarsi con le razze, perché ciascun livello dell'albero separerebbe partizioni differenti, e non vi è alcuna ragione biologica per preferirne una in particolare. Inoltre, piccole variazioni nei geni o nei metodi di analisi impiegati possono spostare certe popolazioni da un gruppo all'altro. Pertanto, la speranza di realizzare una tassonomia delle popolazioni umane valida nel tempo è una causa persa: si tratta comunque di una perdita di scarso rilievo scientifico, che viene recuperata nella ricostruzione della storia evolutiva dell'uomo moderno, così come ci mostra l'analisi genetica. Questo argomento è di interesse ben maggiore, sia perché ipotesi evolutive sulla nostra specie possono essere verificate con l'ausilio di altri dati (archeologici, linguistici, ecc.); sia perché apre la strada metodologica per verificare, con problemi e dati concreti, la possibile interazione tra cambiamenti di origine genetica e cambiamenti di origine culturale. Riteniamo infatti che nella maggior parte dei casi siano state le grandi innovazioni culturali a segnare i momenti di maggiore sviluppo, anche demografico, della nostra storia: momenti che hanno determinato la struttura genetica attuale, e quindi la diversità biologica, delle popolazioni umane.
Rimane da esaminare il motivo per cui lo stereotipo della razza sia così difficile da estirpare. Vi è sicuramente una responsabilità della comunità scientifica, ormai ampiamente documentata almeno per quel che riguarda le generazioni passate (v. Müller-Hill, 1984). Permane tuttavia una contraddizione non ancora risolta tra l'evoluzione biologica che premia la variabilità, la diversità, che ci permette la sopravvivenza come specie, e l'evoluzione sociale che invece premia l'omogeneità sociale, il non essere diversi dagli altri quale garanzia di conservazione della struttura sociale esistente, l'identificarsi in un gruppo di uguali per potersi meglio riconoscere e difendere da altri gruppi. Forse non è preso abbastanza in considerazione il fatto che oltre alla variabilità biologica, anche la variabilità culturale possa essere una componente indispensabile per la nostra evoluzione. In questa tensione dialettica i genetisti sono chiamati a dare il loro contributo, almeno per sgombrare il campo da illazioni pseudo-scientifiche e per chiamare le cose con il loro nome. Nel 1959 il filologo G. Contini (v., 1959) aveva brillantemente individuato l'etimologia della parola razza nel francese antico haraz, ‛allevamento di cavalli, deposito di stalloni' di cui è rimasta anche in italiano l'espressione ‛cavallo di razza'. Sarebbe auspicabile restituire il termine alla sua etimologia originaria: il razzismo purtroppo esiste, ma la razza si addice all'allevamento di animali selezionati, e non ha nulla a che fare con la biologia della nostra specie.
BIBLIOGRAFIA
Ammerman, A. J., Cavalli Sforza, L., The neolithic transition and the genetics of populations in Europe, Princeton, N. J., 1984 (tr. it.: La transizione neolitica e la genetica di popolazioni in Europa, Torino 1986).
Anthony, D. W., Horse, wagon and chariot: Indo-European languages and archaeology, in ‟Antiquity", 1995, LXIX, pp. 554-565.
Bodmer, W. F., Cavalli-Sforza, L. L., Genetics, evolution, and man, San Francisco 1976 (tr. it.: Genetica, evoluzione, uomo, Milano 1977).
Cavalli-Sforza, L. L., Genes, peoples and languages, in ‟Scientific American", 1991, CCLXV, 5, pp. 72-78 (tr. it.: Geni, popolazioni e lingue, in ‟Le scienze", 1992, XLV, 281, pp. 18-25).
Cavalli-Sforza, L. L., Bodmer, W. F., The genetics of human populations, San Francisco 1971.
Cavalli-Sforza, L. L., Menozzi, P., Piazza, A., Demic expansions and human evolution, in ‟Science", 1993, CCLIX, pp. 639-646.
Cavalli-Sforza, L. L., Menozzi, P., Piazza, A., The history and geography of human genes, Princeton, N. J., 1994.
Cavalli-Sforza, L. L., Piazza, A., Menozzi, P., Mountain, J., Reconstruction of human evolution: bringing together genetic, archaeological, and linguistic data, in ‟Proceedings of the National Academy of Sciences", 1988, LXXXV, pp. 6002-6006.
Cohen, C., Les races humaines en histoire des sciences, in Aux origines d'Homo sapiens (a cura di J. J. Hublin e A.-M. Tillier), Paris 1991, pp. 7-47.
Contini, G., I più antichi esempi di ‛razza', in ‟Studi di filologia italiana", 1959, XVII, pp. 319-327.
Crow, J. F., Kimura, M., An introduction to population genetics theory, New York 1970.
Darwin, C., The descent of man and selection in relation to sex, 2 voll., London 1871 (tr. it.: L'origine dell'uomo e la selezione sessuale, Roma 1972).
Diamond, J. M., The earliest horsemen, in ‟Nature", 1991, CCCL, pp. 275-276.
Dolgopolsky, A. B., The Indo-European homeland and lexical contacts of proto-Indo-European with other languages, in ‟Mediterranean language review", 1988, III, pp. 7-31.
Eldredge, N., Gould, S. J., Punctuated equilibria: an alternative to phyletic gradualism, in Models in paleobiology (a cura di T. J. M. Schopf), San Francisco 1972, pp. 82-115.
Garn, S. M., Human races, Springfield, Ill., 19713.
Gimbutas, M., The civilization of the goddess, San Francisco 1991.
Gliozzi, G., Le teorie della razza nell'età moderna, Torino 1986.
Goldstein, D. B., Ruiz Limares, A., Cavalli-Sforza, L. L., Feldman, M. W., Genetic absolute dating based on microsatellites and the origin of modern humans, in ‟Proceedings of the National Academy of Sciences", 1995, XCII, pp. 6723-6727.
Greenberg, J. H., Language in the Americas, Stanford, Cal., 1987.
Hardy, G. H., Mendelian proportions in a mixed population, in ‟Science", 1908, XXVIII, pp. 49-50.
Lewontin, R. C., The apportionment of human diversity, in ‟Evolutionary biology", 1972, VI, pp. 381-398.
Lewontin, R. C., The genetic structure of populations, New York 1974.
Mallory, J. P., In search of the Indo-Europeans: language, archaeology and myth, London 1989.
Mellars, P., Stringer, C. (a cura di), The human revolution: behavioural and biological perspectives on the origins of modern humans, Princeton, N. J., 1989.
Menozzi, P., Piazza, A., Cavalli-Sforza, L. L., Synthetic maps of human gene frequencies in Europe, in ‟Science", 1978, CCI, pp. 786-792.
Müller-Hill, B., Tödliche Wissenschaft: die Aussonderung von Juden, Zigeunern und Geisteskranken 1933-1945, Reinbek b. Hamburg 1984 (tr. it.: Scienza di morte. L'eliminazione degli Ebrei, degli Zigani e dei malati di mente, 1933-1945, Pisa 1989).
Nei, M., Roychoudhury, A. K., Genetic relationships and evolution of human races, in ‟Evolutionary biology", 1982, XIV, pp. 1-59.
Piazza, A., Evoluzione biologica e cultura, in ‟Scientia", 1988, CXXIII, pp. 237-248.
Piazza, A., Rendine, S., Menozzi, P., Mountain, J., Cavalli-Sforza, L. L., Genetics and the origin of European language, in ‟Proceedings of the National Academy of Sciences", 1995, XCII, pp. 5836-5840.
Rendine, S., Piazza, A., Cavalli-Sforza, L. L., Simulation and separation by principal components of multiple demic expansions, in ‟American naturalist", 1986, CXXVIII, pp. 681-706.
Renfrew, C., Archaeology and language: the puzzle of Indo-European origins, London 1987 (tr. it.: Archeologia e linguaggio, Roma-Bari 1989).
Ruhlen, M., A guide to the world's languages, Stanford, Cal., 1987.
Vogel, F., Motulsky, A. G., Human genetics: problems and approaches, Berlin 1979, 19862 (tr. it.: Genetica umana, Milano 1988).
Weinberg, W., On the demonstration of heredity in man (1908), in Papers on human genetics (a cura di S. H. Boyer), Englewood Cliffs, N. J., 1963.
Wolpoff, M. H., Multiregional evolution: the fossil alternative to Eden, in The human revolution: behavioural and biological perspectives on the origins of modern humans (a cura di P. Mellars e C. Stringer), Princeton, N. J., 1989, pp. 62-108.
Wright, S., Evolution and the genetics of populations: a treatise, voll. I-II, Chicago 1968-1969.