
Chimica
Chimica
La chimica nella società contemporanea,
di Luciano Caglioti
SOMMARIO: 1 Introduzione. 2. Un po' di storia. 3. La chimica come punto di forza dell'economia: qualche cifra. 4. La chimica e l'alimentazione: a) l'uso di fertilizzanti e pesticidi in agricoltura; b) trasformazione, conservazione e trasporto dei cibi. 5. La chimica e la salute dell'uomo. 6. I nuovi materiali. 7. Chimica, energia e ambiente: a) chimica ed energia; b) chimica e ambiente. 8. L'informazione. 9. La ricerca. □ Bibliografia.
1. Introduzione
La scienza costituisce il naturale prolungamento dell'evoluzione culturale dell'uomo. Essa è alla base dello sviluppo economico-sociale, e al tempo stesso della cultura nel suo senso più lato. Nella scienza la chimica assume un ruolo centrale, essendo, come essa è, trasversale alle altre discipline, ed essendo, al tempo stesso, il canale di trasmissione attraverso il quale il progresso scientifico-tecnologico arriva all'uomo: chimica, infatti, sono gli alimenti, i medicamenti, i mezzi di trasporto, i delicatissimi chips dei sempre più sofisticati calcolatori, chimica sono le fotografie, i detersivi che usiamo, le fibre che indossiamo, le colle, le vernici, le lampade, le gomme.
Fondamentale è anche il ruolo che la chimica ha per la cultura dell'uomo. Chiunque, oggi, si ponga la stessa domanda - chi siamo noi - che si poneva Plotino, non può prescindere dalla conoscenza dei dati essenziali della chimica e della biochimica. Elementi fondamentali sono a nostra disposizione. È ormai saldamente acquisito il concetto della sostanziale unità del mondo biologico, in un contesto di molecole e di meccanismi ben inquadrabili nel quadro generale intuito da Darwin, e ora completo, a volte fin nei dettagli.
La teoria dell'evoluzione, contestatissima dal mondo religioso e da una parte del mondo scientifico, poggia adesso su inoppugnabili verifiche biochimiche. La teoria dell'informazione, unita alla termodinamica dei sistemi aperti, ci dà conto di come, in zone limitate, l'ordine possa scaturire dal caos, di come cioè la vita possa essere intesa come un fenomeno pienamente in accordo con le leggi fisiche: non vi è più bisogno di invocare la vis vitalis per spiegare il chimismo degli organismi, come coraggiosamente - per il suo tempo - intuì F. Wöhler. La sinergetica e la sociobiologia agitano inquietanti problematiche sul comportamento animale e umano, mentre le conoscenze via via più approfondite sul ruolo delle endorfine pongono interrogativi affascinanti e supremi sull'effettiva indipendenza del nostro atteggiamento da droghe esogene ed endogene.
Scrive infatti Sicuteri (v., 1983): ‟Il senso del benessere, di leggerezza del proprio corpo, lo spunto conciliante, la tolleranza verso l'ambiente, in sintesi l'atteggiamento psichico ottimistico e di benessere, sono gli effetti osservati nell'uomo dopo la somministrazione parenterale o intraspinale o intracerebroventricolare degli oppioidi endogeni". Stante il fatto che le endorfine sono sostanze normalmente presenti nel nostro organismo, il nostro ‛atteggiamento psichico' potrebbe, in parte più o meno grande, dipendere da contingenti fattori biochimici.
Se, pertanto, l'apporto della chimica alla cultura è ampio, importante e insostituibile, altrettanto dobbiamo dire per l'influenza che la chimica esercita, attraverso le tecnologie a essa legate, sulla società.
Può valere la pena di ricostruire in chiave storica alcune tappe dello sviluppo dell'uomo, per poterci meglio rendere conto dei motivi di fondo che sono alla base dell'attuale stato delle cose, e quindi per meglio comprendere quanto, di ciò che la chimica mette a nostra disposizione sia necessario e quanto invece superfluo, quanto dobbiamo a essa e a quali rischi essa ci espone, quali misure infine possiamo prendere per aumentare il rapporto fra i benefici e le controindicazioni.
2. Un po' di storia
Nei due grafici qui riportati Ricossa (v., 1984) ci fornisce informazioni fondamentali per comprendere la storia dell'umanità. Il primo grafico (v. fig. 1) riguarda la crescita della popolazione mondiale. E un po' il ‛senso' della storia dell'uomo. La popolazione oscilla, per qualche decina di millenni, fra due e dieci milioni. Alla fine del Neolitico ha inizio la rivoluzione agricola, e con essa si verifica un primo salto quantitativo: si passa da dieci a cento milioni, segno che la relativa abbondanza di alimenti ‛prodotti' (e non più faticosamente reperiti attraverso caccia, pesca e raccolta di vegetali) permetteva la sopravvivenza di una popolazione maggiore.
L'aumento della popolazione continuava con qualche oscillazione (in corrispondenza delle grandi epidemie, ad esempio, si registra un lieve regresso) fino alla rivoluzione industriale. In corrispondenza di questo punto, situato subito dopo il 1750, si verifica un salto drammatico che in circa due secoli ha portato la popolazione ai livelli attuali. In altre parole, tutto quel complesso di fenomeni e di avvenimenti che hanno caratterizzato, sul piano dell'industrializzazione, l'era moderna si è risolto in un'enorme facilitazione della vita umana, in termini di maggiore salute, aumento della vita media, minore mortalità infantile, ecc.
Il secondo grafico (v. fig. 2) ci informa di un altro fenomeno. Non solo la popolazione, in corrispondenza della rivoluzione industriale, è aumentata, ma si è anche ‛spostata', si è trasferita in città. La percentuale della popolazione urbana è infatti passata dal 18% del 1790 all'80% degli anni immediatamente precedenti il 2000. È questo il punto chiave che dobbiamo tenere ben presente nella nostra riflessione sull'importanza della tecnologia in genere, e della chimica in particolare, nello sviluppo della società moderna.
Una sola considerazione, fra tutte. Se l'uomo abbandona, in una percentuale che arriva all'80%, le campagne, ciò significa che meno persone coltivano i campi, o meglio che un numero basso di persone deve produrre il cibo per un numero alto di consumatori (se stessi e coloro che stanno in città). Ciò significa, ancora, che rispetto all'agricoltura patriarcale (nella quale il 95% della popolazione coltiva i campi) l'agricoltura moderna deve ‛produrre di più' per unità di coltivatore. Il caso limite è costituito dagli Stati Uniti, nei quali il 4-5% di lavoratori agricoli sopperisce ai bisogni dell'altro 95% e di un'altissima percentuale dell'esportazione mondiale di cereali, soia, ecc. Ciò significa, infine, che l'agricoltura moderna deve industrializzarsi, cioè usare fertilizzanti, pesticidi, ecc.
L'urbanizzazione dell'uomo è un fatto di dimensioni bibliche, certamente irreversibile. Piaccia o non piaccia, è così, malgrado gli sforzi di tutti i propagandisti della vita agreste che, in genere saldamente arroccati nel ceto medio urbano, non coincidono con coloro (gli altri) che dovrebbero vivere in campagna per trovarsi alle quattro del mattino dietro due buoi, anche con un tempo cattivo, ad arare un campo per guadagnarsi l'incerto reddito dell'agricoltura patriarcale.
È cambiato tutto: il modo di vivere, di vestirsi, di curarsi, di mangiare, i materiali che usiamo. Non vi è molto di concettualmente nuovo in questo: da sempre l'uomo ha usato materiali artificiali. Se si eccettuano la pietra, il legno, il cotone, la lana, qualche medicamento naturale, ciò che l'uomo usa da sempre è qualcosa di chimico, ottenuto attraverso procedimenti chimici. Il ferro, il bronzo, vocaboli che ci riportano ai primordi della nostra storia, provengono dalla chimica, così come oggi ne provengono i polimeri, le fibre sintetiche, le colle, l'infinita gamma di possibilità delle quali, quasi senza rendercene conto, usufruiamo.
Tutto questo si ottiene attraverso cultura tecnologica e materie prime. La rivoluzione industriale significò, all'inizio, carbone, macchine termiche, cotone. Dopo di allora il problema è stato, più che mai, quello di procurarsi materie prime, al miglior mercato possibile. Se le prime conquiste coloniali dell'Europa in espansione significarono spezie e oro, dopo il 1800 si verificò una caccia ai materiali più vari: il fosfato del Marocco, i metalli pregiati del Congo e del Sudafrica, lo zucchero, il cotone, via via fino all'attuale dopoguerra fatto di petrolio. In tutto questo la chimica ha avuto un ruolo di primissimo piano, nel bene e talvolta - nel male.
3. La chimica come punto di forza dell'economia: qualche cifra
Un paese moderno vive di tecnologia, di forza lavoro e di materie prime. I paesi più fortunati posseggono tutte e tre queste variabili: gli Stati Uniti, ad esempio, sono autosufficienti per molte delle materie prime più importanti, hanno costruito un'infrastruttura culturale potentissima e possono contare su una forza lavoro interna e di immigrazione. Non altrettanto può dirsi per l'Italia, che non possiede materie prime e ha una cultura tecnologica di medio livello.
L'industria chimica è una tipica industria di trasformazione, in cui le materie prime vengono trasformate in prodotti finiti di valore maggiore. Per ottenere questo risultato occorre ‛sapere': il ‛valore aggiunto' è un preciso derivato della cultura, tanto che si può formulare lo slogan ‟materie prime + cervello = guadagno". Nel linguaggio tecnico ricorrono due espressioni, riferite all'industria chimica: ‛chimica di base' e ‛chimica fine'. Come per tutte le definizioni, anche qui ci troviamo di fronte a un margine di incertezza: non sempre la divisione fra chimica di base e chimica fine è così netta. In generale per ‛chimica di base' si intende quel tipo di produzione rivolta a prodotti che sono alla base di altre produzioni, per esempio l'ammoniaca (che è alla base della produzione dei fertilizzanti), l'acido solforico (anch'esso usato nella produzione dei fertilizzanti e in numerosissimi altri processi), la soda, il cloro, l'etilene (alla base della chimica dei polimeri).
Per ‛chimica fine' si intende, invece, quel complesso di prodotti - e di produzioni - la cui caratteristica principale e ‛l'uso' che se ne può fare: un adesivo che resiste a condizioni estreme (quali, per esempio, quelle dei voli spaziali), una vernice anticorrosiva, un tecnopolimero adatto a essere inserito in una vena, un colorante, un principio attivo per cosmesi, un farmaco. L'esempio più idoneo a rappresentare concretamente quello che si intende per chimica fine, valore aggiunto, cultura tecnologica è la fotografia ad autosviluppo tipo Polaroid. Si scatta, e in una trentina di secondi si vedono comparire le immagini che vanno pian piano fissandosi per poi restare stabili. L'abitudine a queste cose - veri e propri miracoli della tecnologia - non deve farci dimenticare l'immane sforzo culturale che deve essere compiuto per risultati di questo genere.
L'industria chimica statunitense ha realizzato nel 1982 un fatturato di 170 miliardi di dollari, con un utile di 12 miliardi. Si tratta del 9% dell'intera industria manifatturiera statunitense, ed è importante rilevare che nel 1972 la chimica rappresentava il 7,5%. L'industria chimica impiega il 5% del personale dell'industria manifatturiera e l'11% degli addetti alla ricerca e allo sviluppo.
Anche in Italia il tasso di crescita dell'industria chimica è superiore a quello delle altre industrie: fra il 1973 e il 1983 esso è stato del 2,9% annuo, indice medio superiore a quelli dell'industria manifatturiera, meccanica, ecc. (v. tab. I). Se poi vogliamo renderci conto di quanto la chimica contribuisca alla ricchezza che si crea nel nostro paese attraverso il valore aggiunto, la fig. 3 ci rende edotti del fatto che il valore aggiunto dovuto alla chimica, in Italia, è il 7,2% dell'intera industria manifatturiera.

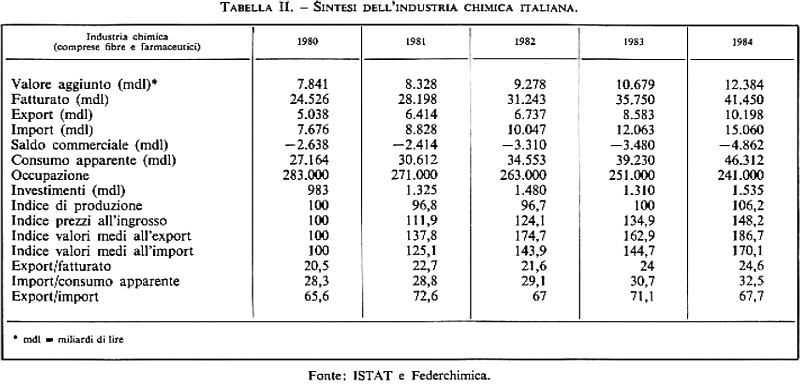
La fig. 3 ci dà inoltre la ripartizione della cifra d'affari nei singoli settori, mentre la tab. II ci dà la sintesi dei principali parametri che hanno caratterizzato la chimica italiana negli ultimi anni. Saltano subito all'occhio alcuni punti importanti: le dimensioni del fatturato, dell'occupazione, e anche il saldo negativo. Occorre dire che il saldo negativo nella chimica è una caratteristica della nostra industria. Per altri paesi (Germania, Francia, Gran Bretagna, Stati Uniti, Svizzera) con i quali siamo in continuo contatto commerciale l'industria chimica presenta da sempre un forte saldo attivo e costituisce pertanto un punto di forza dell'economia. Questa situazione anomala del nostro paese è legata a una serie di vicende aziendali, di scelte sbagliate culminate nella ‛guerra chimica' che i grossi gruppi industriali impegnarono fra loro verso la fine degli anni sessanta - e anche a una scarsa fiducia e uno scarso Impegno nella ricerca. Occorre però dire che a partire dal 1984 si sta verificando una svolta manageriale e imprenditoriale che potrebbe consentire una ripresa positiva.
4. La chimica e l'alimentazione.
La vita dell'uomo urbanizzato è caratterizzata dai grandi consumi. Egli riceve cibi freschi e trasformati dai luoghi di produzione, distanti talvolta migliaia di chilometri; si veste con grande varietà di stoffe e di colori, largamente al di là dello stretto necessario, si cura con numerosi medicinali, usa cosmetici in misura sempre maggiore, viaggia sempre più velocemente, usa automobili personali, mezzi di comunicazione pubblici, costruisce dimore sempre più comode. Potremmo proseguire - calcolatori, macchine fotografiche, elettrodomestici - ma il concetto è chiaro. Dopo l'età della pietra, del ferro, del bronzo, è venuta l'età dei polimeri, dei materiali speciali, dei cibi precotti. Benefici, e anche rischi. È chiaro che una tecnologia così invadente può creare perplessità, diffidenze, timori: non a caso la chimica è stata il principale bersaglio del movimento ecologico in tutto il mondo. Spesso, occorre dire, si è posta maggiore enfasi sui lati negativi che sugli aspetti positivi, ma con il calare delle emozioni e il prevalere dell'approccio razionale il problema del rapporto rischi/benefici si sta inquadrando in un'ottica più rispondente allo stato dei fatti. Qui di seguito riferiremo dettagliatamente sul ruolo di grande rilievo svolto dalla chimica per andare incontro alle esigenze alimentari dell'uomo.
a) L'uso di fertilizzanti e pesticidi in agricoltura
La fame è una vecchia compagna di viaggio dell'uomo, e non è stata certamente debellata. Anzi, la situazione appare drammatica, soprattutto per quei paesi che l'aumento del costo dell'energia ha messo sostanzialmente fuori causa per quanto riguarda la possibilità di sopperire con la produzione interna alle esigenze alimentari di popolazioni sempre crescenti.
L'agricoltura moderna si basa sull'impiego sempre maggiore di fertilizzanti, pesticidi, macchine agricole, energia. In tutti i paesi del mondo la tendenza è quella di industrializzare al massimo l'agricoltura e diminuire il numero degli addetti. Non fa eccezione la Repubblica Popolare Cinese, che pure fino al 1970-1975 costituiva un esempio di modello di sviluppo completamente diverso da quelli occidentale e sovietico. Negli Stati Uniti (ci riferiamo a quanto detto in apertura a proposito della crescente urbanizzazione della popolazione mondiale) gli addetti all'agricoltura e all'allevamento del bestiame sono (1982) il 4% della popolazione, mentre in paesi come l'India e il Pakistan si arriva al 70% circa. In Italia detta percentuale era, nel 1982, del 12,4%.
Si calcola che circa il 30-40% dell'aumento della produttività degli Stati Uniti sia da attribuire all'uso dei fertilizzanti. Tale aumento arriva al 50% per le nazioni in via di sviluppo. Riferendoci ancora al modello di agricoltura statunitense, al quale si ispirano i modelli agricoli europei, ricordiamo che un secolo fa i contadini americani adoperavano 320.000 t/anno di fertilizzanti, 50 anni orsono 7,2 milioni, oggi superano largamente i 40 milioni. Analogo andamento si è avuto in Italia, dove nel 1982 il consumo ha raggiunto i 4,4 milioni di t/anno. Tale consumo è comunque, a parità di superficie coltivata, inferiore a quello degli Stati Uniti e dei paesi della CEE. La fig. 4 riporta l'aumento di resa, in kg/ha, dei cereali e delle leguminose negli anni 1950-1972. La resa dei cereali è aumentata di molto (in seguito alla somministrazione di fertilizzanti azotati), mentre non altrettanto si è verificato per le leguminose. Ciò perché le leguminose provvedono naturalmente al loro fabbisogno di composti azotati mediante il processo di fissazione dell'azoto atmosferico (cioè la trasformazione dell'azoto gassoso in composti utilizzabili dalla pianta) operato da particolari microrganismi che vivono in simbiosi sulle loro radici.
Oltre all'azoto, i vegetali richiedono fosforo, potassio e altri elementi. I fertilizzanti devono essere forniti sotto forma di composti assimilabili, alcuni dei quali sono presenti in natura, mentre altri si ottengono per trasformazioni operate dall'industria chimica. Si trovano in natura i fosfati e i sali di potassio. Le rocce fosfatiche si trovano in quantità tale da assicurare il fabbisogno agricolo per secoli; la loro distribuzione terrestre non è tuttavia omogenea: si trovano nell'Africa nordoccidentale (soprattutto in Marocco, che ne possiede il 50% del totale), negli Stati Uniti e nell'Unione Sovietica, ma anche in Sudafrica, in Sudamerica, in Australia, nel Medio Oriente e in varie isole dell'Oceano Pacifico e dell'Oceano Indiano; scarseggiano nel continente asiatico, la cui sovrappopolazione richiederebbe invece una più abbondante produzione agricola. Anche per i sali di potassio non ci sono timori di esaurimento delle riserve naturali, almeno in tempi medio-brevi. Per l'azoto, invece, essendo assai scarsi i relativi composti naturali utilizzabili dalle piante, è necessaria una massiccia produzione di sintesi a partire dall'azoto atmosferico, che viene trasformato in ammoniaca, nitrati, urea, calciocianammide, ecc. La materia prima azoto è inesauribile, ma l'idrogeno che serve per trasformarla in ammoniaca è praticamente assente in natura e viene fornito dalla petrolchimica (gas naturale e nafta) e dal carbone (attraverso il ‛gas d'acqua'), e il petrolio e il carbone non sono inesauribili.
Bisogna inoltre considerare che le sintesi chimiche richiedono spesso grossi quantitativi di energia e quindi il prossimo esaurirsi di certe fonti energetiche pone problemi anche da questo punto di vista. Già l'aumento del costo del greggio, provocato dalla crisi del petrolio (1973), ha fatto diminuire la produzione agricola; l'effetto si è fatto sentire soprattutto nei paesi - quali l'India e il Bangladesh - in cui erano state introdotte, come strumenti contro la fame, varietà ad alta produttività ottenute dalle pazienti ricerche di N. A. Borlaug, che richiedono però quantità particolarmente elevate di concimi chimici. I paesi a più alto reddito, invece, possono acquistare fertilizzanti in abbondanza e ne utilizzano oggi l'86% della produzione mondiale, mentre la loro popolazione è solo il 39% di quella mondiale; dispongono perciò di una produzione eccedentaria di derrate alimentari di origine vegetale, che destinano in gran parte all'alimentazione del bestiame da carne. È questo un grosso, tragico problema sociale di oggi, che tuttavia non rientra nel campo della chimica. Per quanto riguarda, invece, l'impiego dei concimi chimici, che molti ecologisti vorrebbero eliminare a favore dei concimi naturali (letame), occorre dire che il letame non viene prodotto in quantità sufficiente e inoltre non permette di utilizzare, per ogni tipo di coltura, un concime mirato, cioè contenente il rapporto più adatto fra i principali elementi necessari: azoto, fosforo, potassio. D'altra parte, se non si desse al terreno un congruo apporto dei minerali utilizzati dalle piante, si avrebbe uno sfruttamento eccessivo del terreno stesso, con conseguente drastico abbassamento della fertilità. Non si può negare che i fertilizzanti presentino anche effetti negativi: uno è l'eutrofizzazione delle acque che ricevono, per dilavamento, l'eccesso dei fertilizzanti; l'altro, meno appariscente ma da non trascurare, è l'arricchimento in certi metalli pesanti, presenti come impurezze nei concimi chimici ma estranei ai processi biologici. Gli effetti negativi possono essere minimizzati facendo dei fertilizzanti un uso accorto, il che corrisponde anche a un principio di economia.
Un altro gruppo di composti chimici che ha arrecato enormi vantaggi all'agricoltura ma che presenta, più dei fertilizzanti, problemi di tipo ecologico, con riflessi diretti sulla salute dell'uomo, è quello dei pesticidi. Sotto questo nome (v. pesticidi) si suole indicare un insieme di composti impiegati per la lotta agli organismi, generalmente parassiti, che danneggiano la produzione agricola e zootecnica: prima di tutto insetti, e poi acari, molluschi, vermi, roditori, erbe, ecc. e vari tipi di microrganismi (batteri, virus, muffe).
Tali organismi nocivi (pests) appartengono a numerosissime specie (solo di insetti ne sono state classificate oltre 10.000) e agiscono sulla vegetazione agricola in tutte le fasi del suo sviluppo, sugli animali d'allevamento, sulle derrate alimentari. Il problema è sempre esistito; per la lotta agli insetti - la categoria maggiore di parassiti - si usavano un tempo gli estratti di piretro e di tabacco, poi si sono trovati più efficaci prodotti chimici di sintesi. Il loro capostipite è il DDT (diclorodifeniltricloroetano), le cui proprietà insetticide furono scoperte dal chimico svizzero P. H. Müller nel 1939. Il DDT, usato massicciamente nel dopoguerra soprattutto per la lotta alle zanzare, ha permesso l'eradicazione della malaria in molti paesi (fra cui l'Italia) e una drastica riduzione in molti paesi del Terzo Mondo in cui la malaria era endemica.
Al DDT sono seguiti altri insetticidi dello stesso gruppo chimico (organoclorurati): l'esaclorobenzene (lindano o gammaesano), il clordano, l'aldrin, il dieldrin, l'eptacloro e altri. Oltre a essere molto efficaci, questi composti sono assai stabili: permangono nell'ambiente per mesi o addirittura per anni, in quanto la loro degradazione è molto lenta. Se questo è un vantaggio per la loro azione insetticida, è invece uno svantaggio per l'ambiente, che ne viene contaminato per molto tempo; per di più molti organismi hanno la tendenza ad accumularli e quindi la loro concentrazione cresce man mano che si sale nella catena alimentare, fino all'uomo. I danni a lungo termine riscontrati in alcuni animali da laboratorio, insieme con alcuni ritrovamenti che colpirono l'opinione pubblica (per esempio il suo rinvenimento negli orsi polari e nel latte di donna), provocarono una campagna contro il DDT e contro gli insetticidi in genere. Antesignana di questa campagna fu Rachel Carson, autrice nel 1962 di un libro cui arrise un grande successo: Silent spring. Negli anni settanta l'impiego agricolo del DDT è stato vietato in diversi paesi, fra i quali l'Italia, pur fra opinioni discordi; in effetti il provvedimento fu preso senza considerare molto quali possibili alternative venissero offerte agli agricoltori.
Il bando del DDT coinvolse anche molti altri insetticidi che furono vietati o sottoposti a limitazioni, alcuni perché molto stabili (come gli organoclorurati), altri perché molto tossici, anche se degradati abbastanza rapidamente dall'ambiente: tali sono gli organofosforici (parathion, malathion, ecc.) e i carbammati (carbaryl o Sevin, ecc.).
Accanto ai danni ecologici gli insetticidi sintetici presentano anche l'inconveniente di potere dar luogo a varietà di insetti resistenti: l'azione dell'antiparassitario, infatti, può selezionare quegli insetti che per qualche motivo siano insensibili a esso. I chimici hanno cercato di ovviare all'inconveniente sintetizzando nuovi composti, ma la selezione di individui resistenti, se continuata con più insetticidi e per più anni, può portare a vaste popolazioni di insetti resistenti agli insetticidi chimici: ci troveremmo, in tal caso, davanti a un più grave problema.
Per diverse considerazioni, quindi, a partire dagli anni sessanta venne a formarsi un generale movimento di rifiuto dei prodotti chimici e innanzitutto degli insetticidi; tale movimento, sorto all'interno della ‛società del benessere' e condotto pertanto in un'atmosfera privilegiata, fece rapidamente grossi passi avanti pur in mezzo ad alcune perplessità e anche a opinioni contrarie. La tesi prevalente vedeva questi prodotti chimici soprattutto sotto il profilo del rischio, ma vi era anche chi ne apprezzava i benefici. Fra questi Norman A. Borlaug, premio Nobel per la pace per la sua azione nell'ambito della ‛rivoluzione verde', che in una conferenza tenuta nel 1971, parlando della campagna di stampa contro i pesticidi, diceva: ‟Se l'agricoltura dovesse fare a meno dei mezzi chimici (fertilizzanti e pesticidi) a causa di una legislazione aberrante, auspicata da un potente gruppo di pressione nel quale militano certi maniaci dell'ambiente che terrorizzano il mondo predicendo che morirà avvelenato, allora sì che il mondo perirà, ma di fame". Riferendosi poi al libro della Carson, Borlaug affermava: ‟La campagna odiosa e isterica condotta attualmente da ambientalisti seminatori di panico e irresponsabili contro i prodotti agrochimici trae le sue origini da un bestseller [...]. L'autrice non menziona il DDT come uno dei mezzi per difendere le derrate, o per salvare l'umanità dal paludismo". E inoltre: ‟L'innocuità del DDT si é rivelata veramente rimarchevole. Nel periodo di produzione massima ne sono state utilizzate in agricoltura 400.000 t/anno. Benché centinaia di milioni di uomini siano stati in contatto col prodotto in modo prolungato, e molti per motivi professionali siano stati esposti a esso, i soli casi di danni alla salute dell'uomo sono stati il risultato di incidenti o di ingestione per suicidio. Non esiste alcuna prova che il DDT provochi il cancro o modificazioni genetiche nell'uomo [...]. Quando la campagna di disinfestazione con DDT cominciò a Ceylon, nel 1950, vi erano 2 milioni di casi di malaria. Nel 1962 essi erano scesi a 31, nel 1963 a 17. A quella data, per motivi economici, si sospese la campagna. Nel 1967 vi erano 3.000 casi, nel 1968 16.000. Alla fine del 1969 2.000.000 di casi".
La necessità di conciliare la lotta ai parassiti con la salvaguardia dell'ambiente, e in particolare della salute umana, ha stimolato gli studi chimici e biologici, nonché di tecnica agronomica.
I primi hanno sostanzialmente due obiettivi:
1) preparare altri pesticidi di tipo tradizionale, aventi efficacia possibilmente elevata, ma con controindicazioni più limitate;
2) studiare pesticidi di concezione completamente nuova.
Per quanto concerne il secondo punto, si é cercato di riconoscere e produrre per sintesi alcune sostanze che si comportano come ‛ormoni' degli insetti e presiedono ai fenomeni della crescita, della muta e dell'impupamento delle larve. La somministrazione di queste sostanze in quantità anomala per l'organismo provoca la morte degli insetti. Sempre sul piano delle sostanze chimiche, possono essere sintetizzati e adoperati nella lotta agli insetti alcuni prodotti che agiscono come richiamo sessuale. Si tratta di sostanze relativamente semplici, che gli organismi di un sesso emettono per attrarre gli organismi di sesso opposto. Per mezzo di esse possono essere attivate delle vere e proprie trappole, convogliando in uno spazio limitato gli insetti, attirati dal richiamo dell'altro sesso, per ucciderli con un insetticida convenzionale (v. lotta biologica).
Gli studi biologici seguono le vie della selezione genetica e dell'ingegneria genetica. Si stanno selezionando piante resistenti agli insetti o ad agenti patogeni sia attraverso normali pratiche di ibridazione, sia avvalendosi di tecnologie di ingegneria genetica, ossia immettendo in piante ‛normali' materiale genetico di altre piante che conferisce loro la resistenza a un insetto o a una malattia. Si tratta di ricerche assai promettenti, anche se di non semplice attuabilità su scala generalizzata, che avranno certamente notevole importanza pratica in tempi medi.
Queste nuove vie per la lotta agli insetti e ai microrganismi patogeni presentano il vantaggio di poter agire selettivamente su di essi senza fare uso di sostanze dotate di una nocività generalizzata, con notevoli vantaggi sia sul piano dell'efficacia che sul piano strettamente ecologico.
Accanto ai predetti studi vanno ricordati quelli più propriamente tecnologici riguardanti le modalità di somministrazione: bisogna scegliere accortamente il pesticida (o la miscela) da usare in relazione all'obiettivo specifico che si vuole raggiungere; attenersi alla dose minima efficace e scegliere i tempi più adatti (tali da assicurare una protezione ottimale, senza inquinare il raccolto); talvolta può essere utile ricorrere a tecniche nuove di somministrazione, per esempio l'incapsulamento del pesticida in una membrana porosa che ne regola un lento rilascio, e va invece evitata la somministrazione mediante veicoli aerei, che provoca un'indiscriminata diffusione. È molto importante che siano adeguatamente istruiti gli utilizzatori, non solo per quanto riguarda un corretto impiego, ma anche per quel che concerne le precauzioni da prendere durante la manipolazione, onde salvaguardare la salute dell'operatore stesso.
La legislazione italiana, in continuo aggiornamento parallelamente a quella della CEE, detta norme per la manipolazione e l'impiego dei vari pesticidi e fissa per ognuno di essi le quantità massime che possono essere presenti nelle varie derrate alimentari.
Fra i pesticidi bisogna ricordare anche gli erbicidi. Difendere i raccolti dalle erbe parassite è un problema di sempre, per il contadino. ‟Non seminare zizzania" è una raccomandazione evangelica che deriva, con diverso significato, da una diffusa problematica del momento.
L'impiego degli erbicidi (iniziato nel 1945) ha indubbiamente segnato una svolta in agricoltura e anche nella gestione di pascoli, boschi e parchi. Accanto agli evidenti vantaggi non si possono tuttavia ignorare alcuni svantaggi. Uno riguarda il terreno che, se eccessivamente liberato dall'erba, è più soggetto all'erosione causata dalle acque piovane. L'altro riguarda gli animali e l'uomo, nei confronti dei quali gli erbicidi hanno una certa nocività come tali, soprattutto in seguito a fenomeni di accumulo, ma possono anche essere letali se accompagnati da qualche impurezza molto tossica: quest'ultimo fatto è stato drammaticamente messo in evidenza dalla tragedia di Seveso (10 luglio 1976) determinata dalla cosiddetta diossina (tetraclorodibenzoparadiossina, TCDD), che può essere presente come impurezza in alcuni erbicidi del gruppo dell'acido fenossiacetico.
Roditori e funghi patogeni costituiscono un altro inquietante problema. I topi sono un vero e proprio flagello, e rispondono con sorprendente furbizia ai rimedi che l'uomo riesce a escogitare. Una volta somministrato un veleno, c'è da attendersi che i topi ‛imparino' a evitarlo. Per questo motivo i rimedi più efficaci sono veleni che agiscono molto tempo dopo la somministrazione: ciò rende più difficile per gli animali il collegamento fra la causa e l'effetto. Si adoperano anticoagulanti, derivati della scilla, fluoroacetammide, derivati della stricnina, derivati del tallio, ecc. In ambienti chiusi si adoperano gas tossici come ossido di carbonio, diossido di zolfo, cianuro di idrogeno, ecc. Per quanto concerne i funghi, essi vengono combattuti con agenti tradizionali come la miscela bordolese (a base di solfato di rame e idrossido di calcio) e lo zolfo, o con agenti di più recente ritrovamento, come i derivati dell'acido ditiocarbammico (Zineb, Ziram, ecc.). Altri composti usati sono taluni antibiotici (ad esempio la streptomicina contro la peronospora del tabacco), derivati guanidinici, ecc.
b) Trasformazione, conservazione e trasporto dei cibi
L'uomo urbanizzato riceve gli alimenti necessari al suo sostentamento dai luoghi di produzione lontani chilometri (talvolta migliaia di chilometri). Si tratta di cibi prodotti altrove, che quindi devono essere trasportati e idoneamente conservati. A ciò aggiungasi che l'industria alimentare ha creato prodotti di nuova concezione, adatti alla diminuzione dei tempi disponibili per la cucina e per i servizi domestici e alla necessità di mangiare in intervalli di tempo ristretti.
Cibi in scatola, liofilizzati, surgelati, precotti, avvolti in fogli di plastica sono all'ordine del giorno: è innegabile che l'industria alimentare ha saputo rispondere alle esigenze - belle o brutte che siano - dei tempi moderni. La chimica ha un suo preciso ruolo in questo contesto, sia che si tratti di produrre membrane idonee per l'ultrafiltrazione dei succhi di arancia e di pomodoro o per l'industria lattiero-casearia, sia che si tratti di composti chimici che inibiscono la fermentazione, l'ossidazione, la degradazione in genere dei cibi, o ne ripristinano le caratteristiche organolettiche originali, andate perdute o deteriorate durante la lavorazione. Queste sostanze vanno sotto il nome di ‛additivi alimentari', con cui si indicano secondo la legge italiana (decreto ministeriale del 31.3.1965) ‟quelle sostanze, prive di potere nutritivo o impiegate a scopo non nutritivo, che si aggiungono, in qualsiasi fase della lavorazione, alla massa o alla superficie degli alimenti per conservare nel tempo le caratteristiche chimiche, chimico-fisiche, fisiche, per evitare l'alterazione spontanea, o per impartire a essi, o per esaltare favorevolmente, particolari caratteristiche di aspetto, di sapore, di odore, di consistenza".
A differenza dagli inquinanti, gli additivi sono quindi sostanze chimiche introdotte intenzionalmente negli alimenti per svolgere una precisa funzione.
A seconda di questa, possiamo distinguere tre categorie principali di additivi:
a) conservanti;
b) emulsionanti;
c) addensanti e gelificanti.
Assimilabili agli additivi, anche se regolamentati diversamente, sono i coloranti, gli aromatizzanti e gli edulcoranti.
I conservanti (antimicrobici, antiossidanti, ecc.) preservano gli alimenti dall'azione dei microrganismi e degli agenti ossidanti, che sono le più importanti cause del loro deterioramento. In assenza di sostanze protettive, naturali o sintetiche, si formano nella maggior parte degli alimenti, più o meno rapidamente, sostanze con sapore e odore sgradevoli, talvolta dotate di maggiore o minore tossicità. L'ossidazione è provocata dall'aria ed è spesso catalizzata dalla luce, da tracce di certi metalli (ferro, rame), da enzimi presenti in microrganismi. Essa avviene soprattutto a carico delle sostanze grasse (oli, burro, margarine, ecc.), di cui provoca l'irrancidimento. Esistono antiossidanti e antimicrobici naturali e sintetici (questi ultimi generalmente più efficaci dei primi) che, avendo la proprietà di rallentare o inibire l'alterazione degli alimenti, sono di estrema importanza tecnologica ed economica nell'industria alimentare.
Per quanto riguarda gli emulsionanti, si tratta di sostanze tensioattive che hanno la proprietà di stabilizzare le emulsioni, cioè le fini dispersioni di un olio nell'acqua o dell'acqua in un olio. Possono essere naturali (per esempio le lecitine) o semisintetici (per esempio i sucresteri).
Gli addensanti e gelificanti hanno la proprietà di rigonfiarsi con l'acqua, conferendo all'alimento cui vengono aggiunti una particolare consistenza; sono ampiamente usati per succhi di frutta, creme, budini, marmellate, gelati, formaggi fusi. Possono essere naturali (gomme, pectine, gelatine, ecc.) o semisintetici (carbossimetilcellulosa, polifosfati, ecc.).
Per migliorare il colore o il sapore di certi alimenti si usano numerose sostanze, naturali o sintetiche: abbiamo così i coloranti, gli aromatizzanti, gli edulcoranti. Questi ultimi vengono impiegati per sostituire lo zucchero in alimenti che potrebbero dar luogo a fermentazioni indesiderate o in alimenti dietetici destinati a un'alimentazione particolare, ipocalorica; in questo caso si tratta di sostanze, generalmente sintetiche (per esempio la saccarina), che hanno un forte sapore dolce ma praticamente non forniscono calorie all'organismo.
Sono note le violente polemiche spesso sorte sugli additivi alimentari. Non sempre, soprattutto fino agli anni settanta, si è prestata la necessaria attenzione alle possibili controindicazioni che l'uso degli additivi presentava, per cui talvolta ci si è resi conto dopo anni di impiego della nocività di alcuni prodotti. Da una ventina di anni, tuttavia, le agenzie internazionali e le autorità sanitarie dei vari paesi preposte alla tutela della salute hanno decisamente rivolto la loro attenzione verso questo tipo di problemi, inasprendo le normative da rispettare prima di mettere in commercio prodotti destinati all'alimentazione.
5. La chimica e la salute dell'uomo
L'avanzamento della conoscenza dei meccanismi fondamentali della biologia e della fisiologia e lo sviluppo della chimica analitica e sintetica hanno determinato un grande progresso nella medicina. Malattie che una volta erano molto temute sono state debellate, sono stati identificati i principi attivi come le vitamine e gli ormoni, sono stati chiariti i motivi di alcune malattie da carenza: si può dire che uno dei benefici di cui gode l'uomo moderno è la possibilità di usare determinate medicine.
I punti più qualificanti della produzione farmaceutica sono i sulfamidici, gli antibiotici, l'isoniazide (che ha debellato la tubercolosi), gli ormoni corticosurrenali, alcuni psicofarmaci, gli antistaminici, gli antidiabetici, gli anestetici, i materiali per protesi, i contraccettivi, i vaccini.
Possiamo ricordare brevemente alcuni punti salienti dello sviluppo dei farmaci. Fino al 1920 i medicinali erano soprattutto di origine naturale: estratti di chinino per la malaria, estratti di coca per le anestesie, decotti di erbe. Fra il 1920 e il 1930 si è cominciato a ricorrere alla sintesi e nel 1935 si ebbero i sulfamidici. Fu la prima grande svolta nella cura delle malattie infettive. La seconda fu determinata, all'inizio degli anni quaranta, dalla penicillina, capostipite degli antibiotici prodotti per fermentazione. Nel campo dei principi naturali, il ritrovamento di vitamine (v. vitamine) e ormoni (v. ormoni nei vertebrati) ha costituito un compito arduo per i chimici, sotto il profilo sia delle analisi che della sintesi. La ‛pillola' costituisce il primo approccio razionale al controllo delle nascite. Un salto qualitativo immenso è atteso, per la medicina, dagli sviluppi dell'ingegneria genetica (v. genetica: Applicazioni della genetica; v. biotecnologie, suppl.). Fattori di crescita, fattori del sangue, fattori immunitari, vaccini sintetici: sono questi i principali progressi che si attendono.
Se si considera come un indice di successo l'allungamento della vita media e la diminuzione della mortalità connessa con la malattia, la medicina può vantare la forte diminuzione dei casi di morte per malattie infettive nei paesi tecnologicamente avanzati. Negli Stati Uniti, dove nel 1900 morivano per malattie infettive circa 500 persone l'anno su 100.000, questo numero si è ora abbassato a 50. Anche in Italia la mortalità per malattie infettive è drasticamente diminuita negli ultimi decenni. È stata debellata la sifilide; è stata sconfitta la tubercolosi; la difterite non terrorizza più come un tempo; le infezioni gastrointestinali sono fortemente diminuite e sono in genere curabili (v. morbilità, suppl.).
L'uso delle medicine, dunque, se fatto in modo razionale, può portare all'uomo enormi vantaggi. L'abuso delle medicine è invece una malattia sociale molto seria. La ‛iatrogenesi' (termine con cui si intende l'insorgenza di disturbi a causa dell'uso improprio di medicamenti) provoca negli Stati Uniti dal 3 al 5% dei casi di ricovero. Il 18% dei ricoverati, a sua volta, ha una reazione patologica a un medicamento. Può accadere inoltre che talvolta, malgrado le precauzioni, qualche farmaco venga introdotto in terapia senza che ne siano state accertate fino in fondo le eventuali controindicazioni. Un esempio fra tutti è costituito dal talidomide, un tranquillante che, assunto dalle gestanti provocava gravi anomalie nel feto.
La salute dell'uomo dipende anche dall'igiene: nei paesi sviluppati una delle voci che sono in continua espansione nei bilanci individuali è quella della cosmesi: ogni abitante di un paese minimamente civilizzato usa ogni giorno per la sua pulizia personale uno o due prodotti e molti arrivano a usarne una dozzina. Le condizioni igieniche sono straordinariamente migliorate rispetto al cosiddetto ‛buon tempo antico' e ciò ha portato un contributo non indifferente all'aumento della vita media e del benessere in generale.
6. I nuovi materiali
Un terzo settore nel quale la chimica ha un ruolo fondamentale, oltre quello dell'alimentazione e quello della salute, riguarda i nuovi materiali di sintesi (v. materiali, suppl.) Non vi è forse nulla, come l'avvento delle materie plastiche, che abbia inciso in modo così profondo sulla vita dell'uomo contemporaneo. Non vi è oggetto che noi tocchiamo, si può dire, che non sia in qualche modo legato agli alti polimeri (v. polimeri) e sono ora accessibili a buon mercato oggetti di uso comune che una volta erano a disposizione di pochi. Ciò ha un importante significato sociale, di fronte al quale hanno ben poco valore i rimpianti per il crescente abbandono di certi materiali tradizionali come la lana, la seta, i metalli, il legno. La sostituzione di quest'ultimo con materie plastiche è anche vantaggiosa dal punto di vista ecologico: è evidente, infatti, che per ottenere il legno necessario a fabbricare tutti gli oggetti che oggi si realizzano con le materie plastiche si dovrebbe attingere massicciamente al patrimonio forestale mondiale.
Le materie plastiche prendono origine dalla petrolchimica; i principali prodotti base sono l'etilene (20 milioni di tonnellate l'anno), lo stirene, il propilene, i derivati acrilici. Il fatturato annuo delle materie plastiche negli Stati Uniti supera i cinque miliardi di dollari. Telefoni, maniglie, interruttori, pezzi di automobili, posaterie, vernici protettive, adesivi, isolanti: sono tutti oggetti della nostra vita quotidiana realizzati con materiali plastici.
Una vera e propria rivoluzione industriale si è avuta anche nel campo dei tessuti con lo sviluppo delle fibre tessili artificiali. Se ci riferiamo al mondo occidentale, a partire dalla fine degli anni sessanta le fibre tessili artificiali hanno sorpassato in quantità il cotone e la lana, e soprattutto la seta. L'ampia disponibilità di fibre artificiali (nailon, poliacrilico, poliestere, raion) ha cambiato le abitudini individuali per quanto riguarda il vestiario: il guardaroba si è arricchito, vi sono molti abiti estivi e invernali, il vestiario viene cambiato per motivi legati alla moda più che per un vero e proprio consumo, sono sempre più disponibili tendaggi e tappeti, il materiale è più facile da trattare e più resistente, la varietà di colori è maggiore.
Stando alle statistiche, l'individuo occidentale può ottenere, spendendo il 9% del suo reddito, una disponibilità di tessuti migliore, in qualità e quantità, di quella che otteneva trent'anni orsono spendendo il 12% del reddito. Nel Terzo Mondo coprirsi non è più un privilegio di pochi.
Ausiliari chimici dell'industria tessile permettono inoltre di confezionare tessuti no iron, resistenti all'infiammabilità e all'infeltrimento e impermeabilizzati, tutte cose che stanno divenendo ovvie nei nostri consumi.
Di profondo significato per il benessere dell'uomo sono le applicazioni delle materie plastiche alla medicina, che riguardano soprattutto due settori: le protesi e le tecnologie di somministrazione. Per quanto riguarda le protesi, le materie plastiche forniscono i materiali più adatti per la fabbricazione di pezzi di tessuto, di vene, di organi o di strutture artificiali. Sono stati risolti, anche con contributo italiano, i problemi legati alla trombogenicità dei materiali estranei una volta entrati in contatto col sangue, e si prevede per questo settore uno sviluppo importante. Vi è poi tutta la problematica del lento rilascio dei medicamenti: sono state studiate, dapprima negli Stati Uniti e quindi negli altri paesi tecnologicamente avanzati, delle formulazioni che vedono l'inglobamento di medicinali in materiali polimerici che li rilasciano lentamente una volta in circolo. Questo approccio è importantissimo ai fini di evitare i sovradosaggi o la discontinuità nei dosaggi, ed è particolarmente adatto in tutti quei casi in cui occorre somministrare quantità piccole, ma costanti, di un medicamento (ad esempio sostanze ad azione contraccettiva o taluni medicamenti indicati per malattie cardiache).
Come per gli argomenti trattati precedentemente, anche nel caso delle materie plastiche dobbiamo considerare le controindicazioni. Queste sono dirette e indirette. Sono dirette le controindicazioni dovute allo smaltimento dei rifiuti di materie plastiche, che sono difficilmente degradabili, si trovano dappertutto e infestano laghi, fiumi, mari, boschi, città. Lo smaltimento di questi rifiuti pone seri problemi, in quanto non si è ancora giunti alla convenienza delle tecniche di riciclo, e il loro incenerimento può provocare, come nel caso del polivinildoruro, la formazione di sostanze tossiche. Le controindicazioni indirette sono di tipo sanitario: recenti studi hanno accertato la pericolosità di molti dei monomeri con i quali si producono le materie plastiche più importanti: vinildoruro, acrilonitrile, stirene, benzene (precursore, quest'ultimo, dello stirene).
Sempre collegata con la petrolchimica è la produzione di detersivi. Questo settore ha completamente rivoluzionato le nostre abitudini per quanto riguarda il lavaggio della biancheria, alcuni procedimenti industriali, la cura della persona, e anche, indirettamente, l'industria degli elettrodomestici. I saponi di una volta, inadatti alle lavatrici automatiche, sono stati obbligatoriamente sostituiti dai detersivi, che non danno sali insolubili con acque ricche di calcio e magnesio, e sono additivati di fosfati che impediscono l'intasamento dei tubi delle lavatrici. Anche in questo caso le controindicazioni sono svariate. Riguarda direttamente noi italiani il potere eutrofizzante dei fosfati: si pensa che una causa non trascurabile dell'inquinamento da alghe dell'Adriatico sia proprio la presenza di fosfati nei detersivi.
L'industria della carta, l'industria dei metalli, i materiali fotografici e per fotocopie, i materiali per i transistor e per le celle solari, i nuovi materiali compositi per costruzione: infinite sono le vie attraverso cui la chimica influisce sullo sviluppo e sull'economia della moderna società.
7. Chimica, energia e ambiente
La chimica ha dovuto affrontare in questi ultimi anni due diverse crisi - la crisi energetica e la crisi ambientale - in quanto le nazioni industrializzate si trovano a fronteggiare il passaggio da un'economia basata su risorse illimitate e sul libero sfruttamento dell'ambiente a un'economia basata su risorse limitate e sul rispetto dell'ambiente.
a) Chimica ed energia
L'energia costituisce il cardine dell'intera problematica delle risorse. Essa entra dappertutto: non vi è processo chimico che non preveda un dispendio più o meno grande di energia.
Nel campo dell'industria chimica organica l'aumento del prezzo del petrolio ha creato un doppio problema: infatti il greggio petrolifero è per l'industria chimica organica fonte di energia e fonte di materie prime. Si calcola che il 90% della chimica industriale organica utilizzi come materia prima derivati del petrolio.
L'agricoltura moderna è anch'essa basata su un larghissimo impiego di energia. La resa per ettaro di molte coltivazioni è aumentata, la qualità del cibo è migliorata, la fatica dell'uomo è diminuita, il numero di addetti è calato contemporaneamente all'aumento della produzione, ma ogni caloria di cibo che arriva sulle nostre tavole costa 7-8 calorie di energia termica, il che equivale a dire che coi nostri sistemi alimentari ognuno di noi consuma per alimentarsi 0,8 tonnellate di greggio all'anno. L'energia serve come combustibile, per i macchinari, per i fertilizzanti, per i pesticidi, per cuocere, trasportare, conservare. La fatica dell'uomo nell'agricoltura moderna è il 5% dell'energia impiegata, mentre nei sistemi patriarcali arriva al 95%.
L'aumento del prezzo del petrolio verificatosi dopo il 1973 ha colpito l'agricoltura dei paesi del Terzo Mondo - e del Quarto - togliendo loro la possibilità di acquistare i fertilizzanti necessari per portare avanti la produzione agricola.
Nel settore dei metalli il consumo di energia è direttamente proporzionale alla diminuzione in percentuale dei metalli presenti nei minerali. Man mano che vengono ad esaurirsi i minerali più ricchi, si deve ricorrere a minerali sempre più poveri, la cui purificazione richiede lavoro ed energia in quantità enormi.
L'energia consumata pro capite, d'altronde, è un indice del livello di vita ed è strettamente legata al reddito: Stati Uniti, Svezia, Canada, Belgio presentano al tempo stesso un elevatissimo reddito individuale e un altissimo consumo pro capite di energia, mentre paesi come India, Brasile, Turchia uniscono al basso reddito individuale un altrettanto basso consumo di energia. L'oscillazione dei prezzi e l'instabilità politica di alcune zone (Medio Oriente, Sudamerica) fanno dell'energia una variabile assai importante per la chimica mondiale.
b) Chimica e ambiente
La crisi ambientale ha colpito la chimica in maniera intensa. Alla fine degli anni sessanta ci si è cominciati a rendere conto degli effetti che la diffusione incontrollata di prodotti chimici esercitava sull'ambiente e sugli esseri viventi. La resipiscenza, benché tardiva e disordinata, è stata importante. Si può affermare che il 60-80% del cancro ha origine ambientale. Prima causa il fumo, poi le radiazioni naturali, quindi l'esposizione a composti chimici, sia naturali che sintetici. Il cancro è passato fra il 1900 e il 1970 dall'ottavo posto, come causa di morte, al secondo. I più esposti sono ovviamente i lavoratori dell'industria chimica, poi i loro familiari e i consumatori di prodotti contaminati da certi composti chimici. Molte sostanze inoltre (mercurio, piomho, metalli pesanti in genere, asbesto, benzene, ecc.) hanno una specifica attività nociva.
Se vogliamo riesaminare l'evoluzione della mentalità generale a proposito delle complesse relazioni che intercorrono fra la produzione chimica e l'ambiente, dobbiamo osservare che vi è stata una prima fase nella quale le iniziative ufficiali, gli studi, i provvedimenti delle agenzie internazionali erano la conseguenza diretta o indiretta di un avvenimento grave, e pertanto erano soggetti a una generalizzata tensione emotiva. Dopo il famoso episodio di Minamata - ci riferiamo all'intossicazione da mercurio cui andarono incontro alla fine degli anni cinquanta gli abitanti della baia giapponese di Minamata a causa degli scanchi di mercurio di una ditta produttrice di acetaldeide - tutte le grandi organizzazioni si sono occupate del mercurio. Allo stesso modo, dopo analoghi episodi, si occuparono del cadmio, dei PCB (policlorobifenili), ecc. Si procedeva in modo casuale, episodico, con spreco di forze, studi sovrapposti e non coordinati, al di fuori di una reale scala di priorità. A questa fase è seguita, da qualche anno, una serie di iniziative e di provvedimenti di largo respiro, volti a riparare i danni provocati dall'immissione sul mercato - e pertanto nell'ambiente - di prodotti non sufficientemente controllati sotto il profilo della nocività, e a evitare, per il futuro, che tale immissione possa avvenire.
Sono state emesse normative alle quali dovrà attenersi chiunque voglia distribuire sul mercato un composto nuovo: si prevedono saggi sulla nocività, sulla persistenza, sulla cancerogenicità, sulle caratteristiche chimico-fisiche, sul comportamento nell'ambiente dei prodotti, anche in relazione al loro impiego. All'idea del beneficio che la società o l'individuo si attendono da un nuovo prodotto si è aggiunta quella del rischio che l'uso del prodotto comporta.
Ciò è avvenuto, lo ricordiamo, per alcuni insetticidi, per alcuni medicamenti, per alcuni additivi alimentari, per diserbanti, coloranti, adesivi. Dopo polemiche, campagne di stampa, movimenti ecologici, si è pervenuti alla presa di coscienza dell'impossibilità di ottenere composti chimici a rischio zero. L'opinione pubblica, nella fase meno emotiva e più razionale, ha preso atto di un fatto incontrovertibile: ogni azione umana è soggetta a un margine - più o meno ampio - di rischio. Occorre quindi abbandonare utopistiche speranze di avere solo vantaggi dallo sviluppo tecnologico, e agire per far sì che il rapporto fra benefici e rischi sia ottimale. Si è verificata quindi un'evoluzione positiva della questione ambientale, che ha visto l'opinione pubblica più cauta nelle reazioni ad avvenimenti dolorosi, le agenzie internazionali molto attive nei controlli e nelle prescrizioni, e gli imprenditori più accorti (in molti casi potremmo dire meno disinvolti) nel determinare, con le loro decisioni e le loro valutazioni di bilancio, il margine di rischio delle loro produzioni. Come è avvenuto per il fattore energetico, anche per il problema ambientale si è arrivati a creare le premesse per un generale miglioramento: l'energia è divenuta così un parametro di cui tenere ben conto e i problemi legati alla sicurezza del lavoro e dell'ambiente fanno parte delle considerazioni che sono alla base di una produzione nuova o di una iniziativa commerciale. Se nel settore dei farmaci le precauzioni sono - tradizionalmente - in continuo affinamento per renderli idonei alle situazioni e agli scopi (ciò nonostante ogni tanto qualche prodotto viene ritirato dal mercato a seguito dei controlli e degli studi epidemiologici), analoghe precauzioni vengono prese per gli insetticidi, i cosmetici e i prodotti chimici in genere. Oltre a ciò, una sempre maggiore attenzione viene posta, sia dalle autorità che dagli imprenditori, al problema costituito da rifiuti nocivi, per impedire il ripetersi di inquietanti episodi di intossicazione o di inquinamento di falde acquifere a seguito di una malaccorta - se non incosciente - gestione dei rifiuti pericolosi.
Queste misure e queste cautele comportano costi aggiuntivi, talvolta rilevanti, ma è tuttavia importante che si stabiliscano e/o si rafforzino i collegamenti fra il mondo che produce tecnologie e il mondo che le utilizza, perché la scienza possa assolvere ancor meglio il compito di sempre, che è quello di generare cultura, tecnologia, soluzioni ai problemi dell'uomo.
8. L'informazione
La sempre maggiore partecipazione del largo pubblico alle decisioni politiche e tecniche è un indice di democrazia. Perché questa partecipazione sia - tuttavia - elemento di progresso e non di confusione è necessario che tutti coloro che partecipano siano correttamente e sufficientemente informati. Ciò vale anche - vorremmo dire soprattutto - per le scelte tecnologiche, che implicano la conoscenza di concetti non ovvii e la capacità di apprezzare anche sul piano quantitativo le dimensioni dei fenomeni.
È molto importante che gli operatori politici e sindacali, o quelli preposti ai mezzi di informazione, siano al corrente del fatto - per fare qualche esempio - che l'energia è essenziale per la fabbricazione dei fertilizzanti e per lo sviluppo dell'agricoltura e, quindi, per la soluzione del problema della fame, così come è importante che si sappia che il 40-5o% dei raccolti, se non si interviene con i pesticidi, viene distrutto dagli insetti o dai roditori. È essenziale che ognuno sappia che buona parte di ciò che indossa è costituito da materiali sintetici, che quando gira in macchina utilizza materiali metallici e plastici e contribuisce col suo scappamento all'inquinamento dell'aria, che l'enorme produzione chimica serve a soddisfare la somma delle esigenze individuali. In genere, ora come ora, tutti sono d'accordo nell'individuare i beni ai quali ‛gli altri' devono rinunciare. E invece la responsabilità dell'inquinamento, della degradazione ambientale, è la somma di responsabilità singole, alle quali ognuno di noi è tutt'altro che estraneo.
È anche essenziale che si conoscano a fondo i dati quantitativi che riguardano il problema energetico: tutti ritengono ovvio avvalersi dei benefici che la disponibilità di energia consente, ma pochi (in senso relativo) si rendono conto della necessità di impiantare nuove centrali.
L'informazione è necessaria anche per evitare l'incongruenza. E le incongruenze sono sotto gli occhi di tutti. Consideriamo, ad esempio, il fumo. Esso è considerato come la causa di gran lunga maggiore dell'insorgenza dei tumori polmonari, e tuttavia chiunque può acquistare sigari e sigarette, salvo qualche restrizione recente che regolamenta la possibilità di fumare nei locali pubblici. Attenzione molto superiore è stata data dalle autorità sanitarie a qualche sostanza certamente meno pericolosa, ad esempio ciclammati e saccarina, utilissima ai diabetici, il cui uso è stato limitato o proibito a seguito di controverse e discusse risultanze. Qualcosa del genere accade per gli alcolici.
Vi è poi un'altra incongruenza, dalle dimensioni planetarie, costituita dalla grande attività - e dalle grandi spese - nel settore delle armi. Le polemiche o le considerazioni sui rischi e sui benefici del progresso tecnologico riguardano in genere la scienza e la tecnologia promosse dalle università e dalle industrie, che concernono l'agricoltura, la salute, i materiali, l'energia; poca attenzione si pone invece al continuo sviluppo delle armi in genere, e anche delle armi chimiche. Mentre noi discutiamo e rallentiamo, per giusta cautela, le ricerche su qualche insetticida o farmaco sotto il largo controllo dell'opinione pubblica, nel segreto d'acciaio dei laboratori militari vengono concepite armi tremende. Valga per tutti l'esempio dell'energia atomica: è stato più facile riempire l'Est e l'Ovest di missili a testata atomica che costruire utilissime centrali nucleari.
In altri termini, di ogni branca della scienza noi abbiamo, di sicuro, le applicazioni volte al male, anzi in genere le scoperte più importanti sono possibili proprio perché già sviluppate nell'ambito militare. Su questo non possiamo dire nulla, ma quando si tratta di applicare per la qualità della vita queste scoperte, allora sorgono polemiche e discussioni. Il male è certo, il bene è solo possibile.
9. La ricerca
La chimica è stata, e sarà ancora, un punto di forza essenziale per lo sviluppo della società. La ricerca è a sua volta la base dello sviluppo della chimica. Le nuove frontiere della biologia e della scienza dei materiali stanno aprendo prospettive immense. La chimica ha permesso di identificare principi attivi presenti nel nostro organismo, di sintetizzarli, di mutare il patrimonio genetico di microrganismi, creando le premesse per una nuova medicina basata su sostanze regolatrici che fanno già parte dei normali cicli biologici. Per contro, nuovi materiali speciali, spesso creati su misura per una precisa esigenza, stanno rivoluzionando gli usi e aprendo la via a mutamenti radicali. Tutto questo è dovuto alla ricerca, che assume sempre più quel carattere di interdisciplinarità che è essenziale per un effettivo progresso. Né va dimenticato che il possesso di nuove tecnologie significa anche nuovi mestieri, prospettive importanti per il lavoro di coloro che oggi sono giovani.
Bibliografia
American Chemical Society, Chemistry and economy, Washington 1973.
Bosschère, G. de, Les deux versants de l'histoire. Perspectives de la décolonisation, Paris 1969 (tr. it.: I due versanti della storia. Storia della decolonizzazione, Milano 1973).
Boyd, E. M., Toxicity of pure foods, Cleveland 1973.
Caglioti, L., I due volti della chimica, Milano 1979.
Carson, R., Silent spring, Boston 1962 (tr. it.: Primavera silenziosa, Milano 1963).
Green, M. E., Turk, A., Safety in working with chemicals, New York 1978.
Marei, S., Risorse, popolazione, produzione agricola, in ‟Scienza e tecnica", 1976, LXXVI.
Ricossa, C., L'economia in 100 grafici, Milano 1984.
Selleri, R., Botré, C., Orzaleri, G., Chimica e tecnologia dei prodotti cosmetici, Roma 1977.
Sicuteri, F., Gli oppioidi endogeni come modulatori della omeostasi, Milano 1983.
Travaglia, S., Maledetta industria, Milano 1986.
Elettrochimica,
di Roger Parsons
SOMMARIO: 1. Origini. 2. Conduttanza e reazioni agli elettrodi. 3. Celle elettrochimiche. 4. Cinetica delle reazioni elettrodiche. 5. Struttura della regione interfacciale. 6. Elettroanalisi. 7. Fonti di energia elettrochimica: a) celle primarie; b) celle secondarie; c) celle a combustibile. 8. Fotoelettrochimica. 9. Elettrolisi industriale. 10. Finitura e lavorazione dei metalli. 11. Corrosione. 12. Bioelettrochimica. □ Bibliografia.
1. Origini
L'elettrochimica, quale importante branca della scienza, nacque alla fine del XVIII secolo: la sua nascita può essere datata quasi esattamente con la lettera di Volta alla Royal Society di Londra nel 1800 (v. Volta, 1800), dove egli descriveva la generazione di elettricità da parte di quella che è conosciuta come la ‛pila di Volta'. Questa consisteva in una pila di coppie di dischi di zinco e di rame (o di zinco e di argento), intramezzate da dischi di cartone imbevuto di soluzione salma. Solo pochi mesi dopo Nicholson e Carlisle (v., 1800) usarono tale pila per generare elettricità, con la quale decomposero l'acqua nei suoi elementi: idrogeno e ossigeno. È questo processo a due vie, la produzione di elettricità da una reazione chimica e la produzione di modificazioni chimiche mediante il passaggio di corrente elettrica, ciò di cui si occupa sostanzialmente l'elettrochimica, sebbene le sue ramificazioni nella scienza moderna siano molto ampie.
La Scoperta di Volta fu il culmine di alcuni decenni di ricerche sull'‛elettricità animale'. La capacità di certe specie marine, come la torpedine e l'anguilla elettrica, di produrre scosse elettriche suscitava grande interesse, ma fu l'osservazione di Galvani, pubblicata nel 1791 (v. Galvani, 1791), dell'eccitazione elettrica di zampe di rana isolate dal corpo, che spinse Volta alla ricerca che portò alla pila. In breve, Galvani sosteneva che l'‛elettricità animale' era un fenomeno puramente biologico, mentre Volta affermava che essa era essenzialmente identica all'elettricità statica (da strofinio) e dimostrò che poteva essere generata senza la partecipazione di sistemi viventi. Le ricerche successive hanno confermato la conclusione di Volta, ma hanno anche confermato l'importante ruolo dei processi elettrochimici nei sistemi biologici.
È possibile che processi elettrochimici fossero usati empiricamente nell'antichità. Scavi effettuati vicino a Baghdad, nel 1930 (v. Waterman, 1931), hanno portato alla luce vasi di argilla contenenti bacchette di ferro, che sono stati ritenuti batterie primitive (v. Gray, 1963). Essi risalgono al Il secolo a. C. Si è immaginato che tali batterie potessero essere utilizzate per la doratura elettrolitica. Simili processi potrebbero essere stati usati anche dagli antichi Egizi.
Il termine ‛elettrochimica' fu introdotto da sir Humphry Davy nel 1807 (v. Hartley, 1966), verso la fine di uno straordinario periodo di attività presso la appena creata Royal Institution a Londra, durante il quale egli dette solide basi alla nuova scienza. In particolare egli utilizzò metodi elettrochimici per scoprire una serie di nuovi elementi: potassio, sodio, calcio, stronzio, bario, berillio e alluminio. Il suo assistente e successore, Michael Faraday (v. Williams, 1965), dimostrò l'identità di tutte le forme di elettricità e trovò la relazione quantitativa fra la quantità di elettricità passata attraverso una cella elettrolitica e la quantità di sostanza modificata chimicamente (leggi di Faraday). Oltre a queste eleganti ricerche sperimentali, Faraday propose la nomenclatura elettrochimica usata ancora oggi: ‛elettrodo', ‛elettrolita', ‛catodo', ‛anodo', ‛anione', ‛catione', ecc. (v. Ross, 1961).
2. Conduttanza e reazioni agli elettrodi
Le modificazioni chimiche che sono causate dal flusso di corrente elettrica, o lo causano, avvengono nella zona di contatto tra due materiali differenti - spesso un metallo e una soluzione - che devono entrambi condurre l'elettricità. Per capire il processo elettrochimico è prima necessario capire come avvenga il passaggio di elettricità attraverso tali differenti materiali. La comprensione di questo fenomeno si approfondì nel corso del XIX secolo e raggiunse una forma soddisfacente verso la fine dello stesso secolo, con la teoria ionica della conduzione elettrolitica, proposta da S. Arrhenius nel 1887, e la scoperta dell'elettrone da parte di J. J. Thomson nel 1897.
La corrente elettrica è sempre dovuta a un flusso di particelle elementari aventi carica di −1,602 × 10-19 C e massa di 1,661 × 10-24 g. Gli ioni sono atomi o gruppi di atomi (molecole) che hanno acquistato o ceduto elettroni. Essi hanno una carica, negativa nel primo caso e positiva nel secondo, che in genere è un multiplo piccolo della carica dell'elettrone. La loro massa è oltre 103 volte quella dell'elettrone; infatti lo ione più leggero, il protone (H+), ha una massa 1.840 volte quella dell'elettrone.
L'importante differenza tra i due tipi di conduzione è che nei conduttori elettronici non c'è trasferimento di materia, mentre nei conduttori ionici il movimento degli ioni implica trasferimento di materia. I metalli e i semiconduttori sono i principali conduttori elettronici, mentre i sali fusi (materiali fusi ionici) e le soluzioni di acidi, sali o basi, in solventi come l'acqua, sono i principali conduttori ionici; essi sono chiamati elettroliti.
Nei plasmi, o gas ionizzati, sono presenti sia ioni sia elettroni, ma gli elettroni, essendo leggeri, tendono a prevalere nella conduzione, a causa della loro elevata mobilità. La conduzione mista può avvenire anche in alcuni solidi.
Per buona parte del XIX secolo si pensò che il processo di conduzione fosse un tipo di processo ‛a relè', secondo un meccanismo proposto da Grotthuss (v., 1806), ma a partire dal lavoro di Arrhenius è stata ben accertata la stabilità degli ioni. Questa stabilità deriva soprattutto dalle loro interazioni elettrostatiche con le molecole di solvente, sebbene molte sostanze che formano cristalli ionici (come gli alogenuri alcalini) si presentino ionizzate anche allo stato fuso. Il meccanismo ‛a relè' è responsabile solo di pochi processi di conduzione, fra i quali è importante la conduzione di acidi e basi in soluzione acquosa. Il protone è stabile in tali soluzioni solo a causa della forte interazione con il solvente, tuttavia dimostra una mobilità molto maggiore di quella che ci si aspetterebbe se la specie in movimento fosse un protone solvatato: ciò perché il protone salta da una molecola di solvente all'altra. Questo è particolarmente importante nelle soluzioni acquose, data la natura anfotera dell'acqua. L'acqua, in altre parole, può agire come un acido, donando un protone,
H2O → H+ + OH-, (1)
o come una base, accettando un protone,
H2O + H+ → H3O+. (2)
Le reazioni elettrochimiche avvengono alla superficie di separazione, all'interfaccia, tra un conduttore elettronico e un conduttore ionico, e sono perciò il risultato di questo cambiamento nel modo di conduzione. Un tipico esempio è un pezzo di rame (Cu) in contatto con una soluzione acquosa di solfato di rame (CuSO4), che contiene ioni rameici solvatati e ioni solfato solvatati (SO42- (aq)).
All'interfaccia tra questi due conduttori la modificazione del meccanismo di conduzione avviene mediante la reazione all'elettrodo
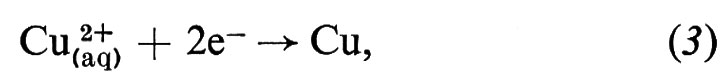
in cui gli ioni rameici sono neutralizzati dagli elettroni provenienti dal metallo per formare atomi di rame che si depositano sul metallo stesso. Su questo tipo di reazione è basato il processo di placcatura elettrolitica. Esso avviene quando la corrente di elettricità positiva fluisce dalla soluzione al metallo. L'elettrodo è allora un ‛catodo' e i ‛cationi' (Cu2+) si muovono verso l'interfaccia: la corrente è detta ‛catodica'. Se la corrente viene invertita, avviene il processo inverso: il rame passa in soluzione per formare ioni rameici e gli elettroni fluiscono dall'interfaccia verso il metallo. L'elettrodo è ora un ‛anodo' e la corrente è detta ‛anodica'. Il processo di addizione di elettroni a una specie è un processo di riduzione, mentre il processo inverso è un'ossidazione. Quindi una riduzione avviene sempre a un catodo e un'ossidazione sempre a un anodo. La comprensione di un processo elettrochimico elementare in termini di una reazione come la (3) lascia immediatamente capire la relazione quantitativa espressa dalle ‛leggi di Faraday dell'elettrolisi', in quanto due elettroni devono fluire attraverso il circuito esterno per formare un atomo di rame. Quindi ci dev'essere sempre una esatta relazione tra la quantità di elettricità passata attraverso il circuito e la quantità di sostanza modificata chimicamente. Bisogna notare che non è necessario che la corrente nell'elettrolita sia portata totalmente (o anche parzialmente) dalla specie che reagisce all'interfaccia. Nell'esempio visto sopra solo una frazione della corrente è portata dal catione, mentre il resto è portato dagli anioni solfato.
3. Celle elettrochimiche
La pila di Volta era un insieme di unità, ognuna delle quali consisteva in due dischi di metalli diversi, separate da un disco di cartone imbevuto di soluzione salma. Questo è un esempio di cella elettrochimica, che ha sempre due elettrodi. Un esempio semplice di cella potrebbe essere l'elettrodo di rame sopra descritto, combinato con un elettrodo di zinco (v. fig. 1); la reazione a questo secondo elettrodo sarebbe
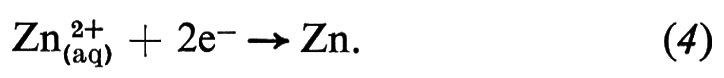
È la diversa reattività dei due metalli, Cu e Zn, con l'elettrolita a fornire la differenza di potenziale che fa passare la corrente elettrica attraverso la cella. La combinazione delle reazioni (3) e (4) nella forma
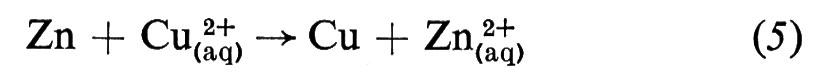
dà la reazione globale nella cella, che produce elettricità, poichè questa reazione è spontanea. Se però viene applicata alla cella una differenza di potenziale esterna (ΔE) sufficientemente grande, la reazione può essere invertita; cioè la reazione chimica procederà in direzione opposta a quella spontanea mostrata nella reazione (5), come risultato del passaggio forzato della corrente elettrica. Ciò può essere rappresentato mediante un diagramma del flusso di corrente in funzione del potenziale applicato esternamente (v. fig. 2). Per un particolare valore di ΔE, in corrispondenza del quale la corrente è zero, la reazione nella cella è bilanciata dalla differenza di potenziale esterna. Questa ΔE è una grandezza caratteristica della cella, nota come f.e.m. ('forza elettromotrice'), termine che ha perso il suo significato originario, sebbene la grandezza in sé sia di grande importanza.
H. von Helmholtz (v., 1881) e J. W. Gibbs (v., 1875) hanno dimostrato che la f.e.m. (E) di una cella in equilibrio è correlata all'energia libera di Gibbs (ΔG) della reazione che avviene nella cella mediante la relazione
E = − ΔG/nF, (6)
dove n è il numero di elettroni che attraversano il circuito esterno per ogni ione che reagisce all'elettrodo (nell'esempio visto sopra n = 2, come risulta dalle reazioni parziali (3) e (4)) e F è la costante di Faraday (96.484 C mol-1), che equivale alla carica di una ‛mole' di elettroni. La (6) è un'equazione chiave, poiché costituisce il ponte fra la termodinamica e le proprietà delle celle elettrochimiche in equilibrio. Queste proprietà possono ora essere interpretate in termini di reattività chimica.
Uno dei numerosi risultati dell'applicazione dell'equazione (6) è la deduzione di come la f.e.m. di una cella dipenda dalla concentrazione dei reagenti nella cella. L'energia libera di Gibbs, e di conseguenza la f.e.m., dipendono logaritmicamente dalle attività delle specie chimiche che partecipano a una reazione, secondo la seguente equazione:
E = E0 − (RT/nF) Σi νi ln ai, (7)
dove R è la costante dei gas (8,31441 JK-1 mol-1), T è la temperatura termodinamica, ai è l'attività della specie i e νi è il suo coefficiente stechiometrico nella reazione che avviene nella cella (i prodotti di reazione hanno valori positivi di νi e i reagenti valori negativi); E0 è la f.e.m. quando tutti i reagenti hanno attività unitaria (tutte le specie nel loro stato standard).
Nel caso particolare della reazione (5), n = 2, νCu2+ = − 1, νZn2+ = 1, e aZn = aCu = 1, perché i metalli puri sono nel loro stato standard; sicché si ha:
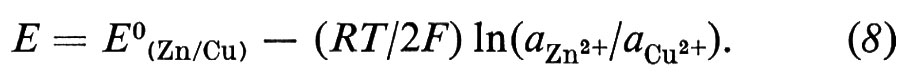
L'equazione (7) fu ricavata da W. Nernst (v., 1889) ed è nota come ‛equazione di Nernst'.
Poiché le energie di Gibbs sono proprietà additive delle reazioni, anche la f.e.m. standard è una proprietà additiva. Perciò è possibile compilare una tabella delle f.e.m. standard delle celle in cui ogni elettrodo è collegato con un elettrodo standard; quindi si possono calcolare le f.e.m. di tutte le altre combinazioni.
Per celle con elettroliti acquosi (e altri elettroliti in cui il protone solvatato è stabile) l'elettrodo standard è l'elettrodo a idrogeno, che consiste in un metallo cataliticamente attivo (generalmente platino), sul quale si fa gorgogliare idrogeno, immerso in una soluzione acida avente attività degli ioni idrogeno unitaria (v. Latimer, 1938; v. Bard e altri, 1985). L'elenco dei potenziali standard dei metalli in ordine progressivo è spesso chiamato ‛serie delle forze elettromotrici'; esso dà un'indicazione sulla reattività relativa dei metalli in un dato solvente.
Per buona parte del XIX secolo e durante i primi anni del XX l'origine della f.e.m. in una cella elettrochimica fu oggetto di controversie. I sostenitori della teoria ‛chimica' affermavano, in base all'equazione (6), che l'origine della f.e.m. stesse nelle reazioni chimiche all'interfaccia metallo/elettrolita. Invece i sostenitori della teoria ‛fisica' affermavano che la f.e.m. della cella dipendesse dal contatto fra i diversi metalli dei due elettrodi (la cella completa della fig. 1, per esempio, implica un contatto metallo/metallo fra il terminale di rame e l'elettrodo di zinco); si sapeva che in corrispondenza di queste giunzioni metallo/metallo si determina una differenza di potenziale di contatto della stessa entità della f.e.m. della cella. Un'analisi completa basata sui concetti introdotti da A. N. Frumkin (v., 1917) e da E. Lange (v. Lange e Koenig, 1933) concilia queste due vedute estreme e mostra che, in effetti, le differenze di potenziale a ognuna delle giunzioni danno un contributo alla f.e.m. totale, che è anche, naturalmente, correlata quantitativamente all'energia di Gibbs della reazione che avviene nella cella.
4. Cinetica delle reazioni elettrodiche
Il fatto che la corrente che passa attraverso la cella non diventi infinita, quando il potenziale applicato esternamente differisce dalla f.e.m. di equilibrio, indica l'esistenza di processi lenti implicati nel passaggio di questa corrente. Nel metallo e nell'elettrolita questi processi sono espressi dalla resistenza elettrica dei materiali, dovuta al fatto che gli ioni possono muoversi solo spingendo ‛da parte' altre particelle, mentre il movimento di elettroni è impedito dal moto termico del reticolo metallico, nonché dai difetti presenti in questo. Comunque queste caratteristiche di resistenza non spiegano il comportamento di una cella elettrochimica al passaggio della corrente: essa mostra una resistenza maggiore, che è non lineare, nel senso che la corrente non è una funzione lineare del potenziale applicato, come sarebbe se gli unici effetti presenti fossero quelli della resistenza di massa. Effetti addizionali sono dovuti alla lentezza della stessa reazione elettrodica e al fatto che essa è una reazione eterogenea, nel senso che lo scambio dei portatori di carica avviene solo all'interfaccia.
Formalmente, il tipo più semplice di reazione elettrodica è quello in cui il reagente e il prodotto sono due diversi stati di ossidazione di uno stesso ione, entrambi presenti nell'elettrolita, e la reazione consiste nello scambio di un elettrone con l'elettrodo metallico. Un esempio di questo tipo di reazione potrebbe essere quello tra ione ferroso e ione ferrico:
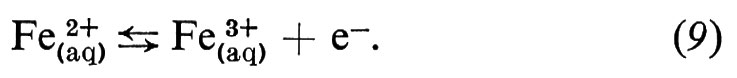
La cinetica di questa reazione segue le leggi della normale cinetica chimica. La (9) è del primo ordine in entrambe le direzioni: verso destra (anodica) e verso sinistra (catodica); cioè la velocità di reazione (v) proporzionale alla prima potenza della concentrazione del reagente:
υ→ = k→[Fe2+], (10)
υÁ = kÁ[Fe2+], (11)
dove [Fe2+] e [Fe3+] sono le concentrazioni ‛locali' rispettivamente di Fe(aq)2+ e Fe(aq)3+ nel luogo della reazione. La caratteristica delle equazioni di velocità delle reazioni elettrodiche è che le costanti di velocità k→ e kÁ dipendono dal potenziale dell'elettrodo: nel caso più semplice l'energia di attivazione è una funzione lineare del potenziale dell'elettrodo. Questo conduce a una dipendenza esponenziale delle costanti di velocità dal potenziale:
k→ = k0 exp[(1 − α)nEF/RT], (12)
kÁ = k0 exp[αnEF/RT]. (13)
La costante di proporzionalità α è detta ‛coefficiente di trasferimento' e il suo valore è normalmente compreso fra zero e uno. (Segue dal principio di reversibilità microscopica che la somma degli argomenti dei due esponenziali in (12) e (13) dev'essere nEF/RT; perciò è richiesto un solo coefficiente di trasferimento). Questa caratteristica unica delle reazioni elettrodiche significa che c'è un modo semplice per controllare la loro velocità e che, a causa della forma esponenziale, questo controllo può essere esercitato entro un intervallo molto ampio di velocità.
La velocità netta della reazione, υ→ − υÁ, può essere espressa in termini di una corrente, poiché per ogni gradino elementare n elettroni sono trasferiti al (o dal) metallo (n = 1 nel caso della reazione 9). Questa è un'espressione delle leggi di Faraday.
Poiché la reazione avviene all'interfaccia tra due fasi, la grandezza che interessa è la densità di corrente j e le velocità υ→ e υÁ sono espresse anche come velocità per unità di superficie dell'interfaccia. Si ha
j = nF(υ→ − υÁ). (14)
La combinazione delle equazioni (10)-(14) dà un'equazione della curva potenziale-corrente mostrata nella fig. 2, che è interpretabile direttamente come un'espressione della velocità della reazione elettrodica in funzione del potenziale dell'elettrodo. Si tratta di una doppia curva esponenziale; da una parte e dall'altra del punto di equilibrio (j = 0) la funzione ha un andamento esponenziale semplice. Questa relazione fu trovata empiricamente nel 1905 da J. Tafel (v., 1905) ed è conosciuta nella sua forma logaritmica come ‛legge di Tafel'. L'incremento esponenziale della velocità non può continuare all'infinito, man mano che il potenziale dell'elettrodo si allontana dal valore di equilibrio. Benché la stessa reazione elettrodica sia caratterizzata da una velocità massima, un altro processo incomincia a controllare la velocità prima che sia raggiunto tale limite: il trasporto dei reagenti da e verso l'interfaccia. Esso dipende dalla geometria dell'interfaccia, dalle condizioni idrodinamiche nell'elettrolita e dalla composizione dell'elettrolita stesso. Se l'elettrodo ha una forma semplice e l'elettrolita è ben mescolato, un semplice modello ideato da Nernst (v., 1904) può essere usato per descrivere il trasporto di massa nel caso che il reagente non sia carico, o sia presente in un elettrolita con grande eccesso di ioni non reagenti.
Nernst suppose che la concentrazione del reagente fosse uniforme nell'elettrolita fino a una distanza δ dalla superficie dell'elettrodo e che, entro δ, il trasporto di massa seguisse la legge di diffusione di Fick, secondo cui la velocità di diffusione è proporzionale al gradiente di concentrazione tramite una costante di proporzionalità D, il coefficiente di diffusione. In effetti Nernst suppose pure che la concentrazione variasse linearmente dal suo valore in δ (uguale alla concentrazione nella massa) al suo valore all'interfaccia dell'elettrodo. Perciò per lo ione Fe2+ nella reazione (9) la velocità di trasporto può essere scritta
j/nF = D{[Fe2+]m − [Fe2+]}/δ, (15)
dato che questa velocità per unità di superficie può essere anche espressa come densità di corrente. Nella (15) m indica la concentrazione nella massa. Se la reazione elettrodica è accelerata in direzione anodica variando il potenziale verso valori positivi rispetto al potenziale di equilibrio, vicino all'elettrodo si consumano ioni ferrosi; perciò la concentrazione locale [Fe2+] decresce. Ciò causa un aumento della velocità di diffusione data dalla (15), che va di pari passo con la reazione elettrodica. Tuttavia, quando [Fe2+] diventa molto piccola rispetto a [Fe2+]m, la (15) mostra che si è raggiunta una velocità limite, cioè j raggiunge il valore limite jl.
Dai tempi di Nernst sono state sviluppate molte espressioni matematiche per descrivere il trasporto di massa agli elettrodi, specialmente per condizioni in cui lo stato stazionario non è stato raggiunto e dev'essere risolto un problema di diffusione dipendente dal tempo (v. Macdonald, 1977). Queste condizioni sono di particolare interesse, perché dispositivi elettronici permettono il controllo dipendente dal tempo del potenziale dell'elettrodo in numerosi modi periodici o non periodici, che sono importanti per la soluzione di problemi particolari riguardanti il meccanismo delle reazioni elettrodiche. In alcune di queste soluzioni il risultato può essere formulato come nell'equazione (15), con un valore di δ dipendente dal tempo. Questi metodi transitori elettrochimici furono i primi metodi di ‛rilassamento' usati per studiare le reazioni ‛veloci' (v. Frumkin e altri, 1940; v. Brdička e Wiesner, 1943). In modo simile si può trovare, per il problema dello stato stazionario, nel caso di particolari configurazioni di elettrodi, una soluzione più esatta, ma sempre espressa nella forma semplice dell'equazione (15). Un esempio importante è l'elettrodo a disco rotante, che consiste in un disco che ruota intorno al suo asse centrale. Levič (v., 1962) mostrò, sulla base di precedenti teorie idrodinamiche, che
δ = 1,62(D/ν)1/3(ν/ω)1/2, (16)
dove ν è la viscosità cinematica dell'elettrolita e ω è la velocità angolare del disco.
Oltre al trasferimento di carica e al trasporto di massa, ci sono altri processi associati a reazioni elettrodiche più complesse, che ne possono influenzare la velocità globale: può trattarsi di reazioni che non implicano un trasferimento di carica interfacciale (reazioni ‛chimiche') e che avvengono o alla stessa interfaccia o durante il trasporto dei reagenti verso o dall'interfaccia. Possono anche verificarsi processi di formazione di nuove fasi: nucleazione e crescita. La cinetica formale di questi processi elementari è ora compresa abbastanza bene, ma l'analisi di una reazione elettrodica complessa che comprende parecchi tipi diversi di gradini elementari può essere ancora compito difficile, che richiede un gran numero di tecniche sofisticate.
5. Struttura della regione interfacciale
La reazione elettrochimica elementare di trasferimento di carica avviene al confine tra due diversi tipi di conduttori e perciò in una regione che (nella direzione perpendicolare al piano dell'interfaccia) è di dimensioni atomiche. L'ampiezza approssimata di questa regione fu stimata da lord Kelvin (v. von Helmholtz, 1879) verso la fine del XIX secolo, ma lo studio dettagliato della sua struttura si è sviluppato durante il XX secolo. H. von Helmholtz (ibid.) ipotizzò una separazione di cariche elettriche a livello dell'interfaccia, cioè una configurazione costituita da due strati paralleli di cariche di segno opposto, situati l'uno di fronte all'altro a distanza di solo pochi diametri atomici. Questa configurazione è stata chiamata ‛doppio strato elettrico', termine che è stato mantenuto nonostante ora si sappia che la struttura è più complessa.
Nella prima metà del XX secolo gli studi sul doppio strato elettrico erano basati per lo più sulla termodinamica classica, utilizzando le equazioni formulate da Gibbs (v., 1875) per analizzare misure della tensione interfacciale in funzione della composizione dell'elettrolita e del potenziale dell'elettrodo. Ciò includeva la derivazione dell'equazione proposta per primo da Lippmann (v., 1875) per spiegare la dipendenza della tensione interfacciale y tra un metallo liquido e un elettrolita dal potenziale applicato al metallo:
(∂γ/∂Ε) = − σ. (17)
Questa relazione è conosciuta come equazione di Lippmann e σ può essere identificata con la densità di carica dello strato di cariche sul lato metallico dell'interfaccia. La curva γ = f(E) per un dato sistema è chiamata ‛curva elettrocapillare' ed è approssimativamente parabolica, con un massimo dove è σ = 0, come segue dall'equazione (17). Nei primi due decenni del XX secolo Gouy (v., 1903, 1906 e 1917) eseguì una lunga serie di misure della curva elettrocapillare del mercurio in contatto con una vasta gamma di elettroliti. Queste misure condussero alla formulazione dei modelli elaborati dallo stesso Gouy, da Chapman, da Stern, da Frumkin e da Butler (v. Parsons, 1954), che forniscono una descrizione più dettagliata della struttura della regione interfacciale. Un ulteriore studio dettagliato fu condotto da Grahame (v., 1947), che eseguì delle misure precise della capacità differenziale
C = (∂σ/∂E), (18)
usando un ponte a corrente alternata. Confrontando la (17) con la (18), si vede che la capacità differenziale è la derivata seconda della curva elettrocapillare; essa fornisce perciò un'informazione simile. La capacità varia fortemente con E, ma il suo valore è sempre di diverse decine di μF cm-2. Se essa è interpretata come un semplice condensatore piatto parallelo, corrisponde a un rapporto fra spessore e permittività relativa di circa 0,1 nm, cioè vicino a un raggio atomico. Questo mostra che le proprietà dell'interfaccia sono dominate da una regione spessa all'incirca quanto uno strato monomolecolare. Ciò spiega anche perché queste proprietà (incluso il comportamento cinetico discusso nel capitolo precedente) sono estremamente sensibili a tracce di impurezze. Una quantità di queste ultime dell'ordine di 10-9 moli è sufficiente per riempire questo monostrato e perciò per cambiare totalmente la sua composizione. Per questo motivo molti dei primi studi su elettrodi accessibili erano del tutto irriproducibili (il lavoro di Gouy è un'eccezione, perché egli usò elettrodi contenuti in un capillare sottile, che ritardava l'accesso delle impurezze provenienti dall'elettrolita). Della necessità di usare sistemi altamente purificati si rese conto Frumkin negli anni trenta, ma fino agli anni cinquanta la maggior parte delle misure attendibili furono fatte usando l'elettrodo a goccia di mercurio, introdotto da Heyrovský (v., 1923). Misure con elettrodi solidi richiedono molta più cura e solo nell'ultimo quarto del XX secolo è diventato possibile ottenere risultati per superfici ben caratterizzate sia dal punto di vista della composizione sia da quello della struttura (v. Hansen e altri, 1983).
Secondo i modelli correnti, la regione interfacciale metallo/elettrolita si divide in tre parti: 1) la carica sul metallo, molto vicina alla superficie metallica e distribuita su una piccola regione perpendicolare alla superficie, perché gli elettroni possono uscire dal metallo al di là dei nuclei atomici caricati positivamente; 2) una regione a monostrato nella quale ioni e molecole sono soggetti a un'interazione specifica a corto raggio con la superficie metallica; 3) una regione in cui gli ioni sono distribuiti nella direzione perpendicolare all'interfaccia sotto l'azione del campo elettrostatico della carica sul metallo e sulla regione 2). L'ultima regione è importante nella discussione della stabilità dei colloidi liofobi e per quel che riguarda gli effetti elettrocinetici. I dettagli di questo modello sono ancora controversi, ma la maggior parte delle interpretazioni attuali trattano la 1) come un piano di cariche ideale, la 2) come uno schieramento bidimensionale di ioni e molecole di solvente e la 3) come una nuvola diffusa di ioni in un dielettrico senza struttura (modello di Gouy-Chapman). Modelli migliori saranno probabilmente elaborati come risultato di una trattazione dei problemi interfacciali con metodi meccanico-statistici avanzati (v. Rowlinson e Widom, 1982) e anche come risultato di metodi sperimentali più diretti, quali la spettrometria Raman (v. Fleischmann e altri, 1974) e la spettrometria infrarossa (v. Bewick e altri, 1980), che hanno cominciato a fornire informazioni sulle proprietà vibrazionali delle molecole nella regione interfacciale.
6. Elettroanalisi
I primi impieghi dell'elettrochimica per scopi analitici erano basati sulla deposizione di cationi e sulla pesata del deposito metallico o sulla misura della corrente e sull'uso delle leggi di Faraday per ottenere la quantità di reagente (‛coulometria'). Entrambe queste tecniche sono di limitata applicazione, perché richiedono una reazione elettrodica semplice (senza reazioni secondarie), e sono piuttosto lente. La moderna chimica elettroanalitica ebbe inizio nel 1922 con l'invenzione del ‛polarografo' da parte di Heyrovský (v., 1923) a Praga. Egli impiegò un elettrodo a goccia di mercurio (v. fig. 3), che formava una nuova goccia ogni 3-4 secondi. Ciò forniva una superficie dell'elettrodo pulita e riproducibile e, nello stesso tempo, un ben definito campo di diffusione attorno all'elettrodo. Sebbene le condizioni di quest'ultimo siano più complesse di quelle che conducono all'equazione (15), si ottiene un tipo analogo di corrente limite, che è pure direttamente proporzionale alla concentrazione della specie reattiva nella massa della soluzione e può quindi essere usata per una rapida analisi quantitativa. Nello stesso tempo il potenziale al quale avviene la reazione di una data specie è caratteristico della specie stessa, sicché diventa possibile anche un'analisi qualitativa. La caratteristica forma della curva corrente-potenziale è mostrata nella fig. 4. Ogni reazione elettrodica dà origine a una cosiddetta ‛onda polarografica' e, in casi favorevoli, è possibile analizzare una miscela in un unico esperimento. L'intervallo di potenziali che si può usare è limitato dalla decomposizione dell'elettrodo stesso (dissoluzione anodica del mercurio) e del solvente (sviluppo catodico di idrogeno). Quest'ultimo processo è molto lento sul mercurio, sicché l'intervallo di potenziale è notevolmente esteso nella direzione negativa. Heyrovský ideò anche un polarografo registratore automatico (v. fig. 3), che fu il primo apparecchio analitico automatizzato. Il potenziometro cilindrico che faceva variare il potenziale applicato era fatto girare da un motorino (inizialmente un meccanismo a orologeria), che faceva ruotare simultaneamente un foglio di carta sul quale veniva registrata la corrente. Tutta la curva corrente-potenziale poteva così essere registrata direttamente in circa 4 minuti. Fin verso il 1950 la registrazione veniva fatta fotograficamente, ma poi divennero di impiego più generale i registratori a penna scrivente. A Heyrovský fu assegnato il premio Nobel nel 1959.
Le due principali limitazioni di questa polarografia ‛classica' erano l'impossibilità di studiare reazioni che avvengono a potenziali positivi rispetto a quello di dissoluzione del mercurio, e la sua scarsa sensibilità. La prima limitazione può essere superata mediante l'uso di altri materiali per l'elettrodo e, sebbene non sia stata trovata una soluzione d'impiego generale, si sono escogitate diverse soluzioni molto adatte per casi specifici. La seconda limitazione deriva dal fatto che nell'elettrodo a goccia di mercurio si genera una piccolissima corrente dovuta alla continua formazione di nuova superficie dell'elettrodo. Questa corrente ha origine dal fatto che il doppio strato elettrico di questa nuova superficie deve essere caricato allo stato adatto al potenziale dell'elettrodo. Di conseguenza la corrente per una reazione elettrodica deve essere rivelabile in presenza di questa ‛corrente di carica'. Ciò limita la sensibilità del metodo a concentrazioni di poche μmoli dm-3.
Per estendere i limiti di sensibilità è stata elaborata tutta una serie di nuovi metodi polarografici; molti di questi sono stati ideati da G. C. Barker. Si è fatto ricorso a due metodologie generali. La prima consiste nell'uso di tecniche transienti: per esempio, nella polarografia a impulsi, è stato applicato all'elettrodo un breve impulso rettangolare di potenziale. Siccome la costante di tempo per caricare il doppio strato è molto più breve di quella di un processo elettrodico controllato dalla diffusione, la corrente misurata verso la fine dell'impulso è dovuta essenzialmente a quest'ultimo. La seconda metodologia consiste nell'impiego della preconcentrazione in situ: per esempio, nella voltammetria di ridissoluzione anodica (Anodic Stripping Voltammetry, ASV), uno ione metallico viene depositato su una goccia di mercurio statica a un potenziale negativo e la corrente dovuta alla sua dissoluzione viene misurata durante una rapida deviazione lineare del potenziale verso valori più positivi. Sono ora disponibili numerosi metodi di questo tipo (v., per esempio, Bond, 1980; v. Bard e Faulkner, 1980), che hanno portato i limiti di rivelabilità a valori dell'ordine delle ppb (parti per bilione), quindi paragonabili a quelli della spettrometria di assorbimento atomico.
Questi nuovi metodi polarografici hanno comunque il vantaggio di potere spesso essere usati su campioni come l'acqua di mare senza trattamento preliminare, e inoltre di essere sensibili allo stato chimico di metalli in traccia (per es. il tipo di legante), in quanto questo influenza il potenziale a cui avviene la reazione.
Un tipo del tutto diverso di dispositivo elettroanalitico è basato sulla misura di un potenziale di equilibrio e sull'impiego di un'equazione come la (8) per determinare la concentrazione dello ione con cui l'elettrodo è in equilibrio. Sebbene questa possibilità sia esistita fin dall'inizio del XX secolo, essa fu sfruttata ampiamente solo nella forma dell'elettrodo a vetro usato per misurare il pH (= − log[H+]). Questo elettrodo è costituito da un sottile bulbo di vetro entro il quale si trovano una soluzione di acido cloridrico e un elettrodo ad argento-cloruro d'argento (v. fig. 5). Il vetro ha la struttura di un silicato contenente ioni di metalli alcalini che possono essere sostituiti da altri ioni, in particolare da ioni idrogeno provenienti da una soluzione in contatto con l'elettrodo. Questo processo di scambio, che avviene sulla superficie esterna del vetro, dà origine a una differenza di potenziale (misurabile fra l'elettrodo posto entro il bulbo di vetro e un elettrodo di riferimento a contatto con la soluzione in esame) che dipende logaritmicamente dalla concentrazione idrogenionica, secondo l'equazione di Nernst. Modificando opportunamente la composizione del vetro, l'intervallo di pH a cui l'elettrodo ancora funziona può essere esteso fino a 11 (ambiente notevolmente alcalino), oppure l'elettrodo può essere reso sensibile all'uno o all'altro ione di metallo alcalino. Questi furono i primi elettrodi ionici selettivi (Ion Selective Electrodes, ISE), ai quali più di recente se ne sono aggiunti molti altri (v., per es., Koryta e Stulik, 1983). Il primo elettrodo di questa nuova serie che dette buoni risultati fu l'elettrodo selettivo per gli ioni fluoruro (1966), a base di LaF3 solido. L'anno seguente furono realizzati dei pratici elettrodi a scambio ionico liquidi, costituiti da una fase liquida, immiscibile con la soluzione in esame (normalmente acquosa), che contiene un agente complessante, altamente specifico per lo ione da analizzare e insolubile nella fase acquosa. Ne risulta una distribuzione dello ione in esame (e degli altri ioni), che ubbidisce alle leggi dell'equilibrio di Donnan (v., 1911) e che dà luogo a un ‛potenziale di membrana', dipendente dalla concentrazione ionica nella soluzione nel modo descritto dall'equazione di Nernst. Il principio basilare è simile a quello dell'elettrodo a vetro, ma il reticolo di silicato solido è sostituito da un agente complessante in soluzione. L'efficienza di questi elettrodi dipende dalla selettività dell'agente complessante e dalla possibilità che hanno altri ioni di attraversare l'interfaccia, in relazione anche alla loro concentrazione nella soluzione. Un recente sviluppo è stato la combinazione di elettrodi selettivi e di dispositivi semiconduttori, i cosiddetti ‛transistori selettivi ionici a effetto di campo' (Ion Selective Field Effect Transistors, ISFETS). Questi elettrodi ionici selettivi, benché misurino ioni in soluzione, possono essere adattati ad altri tipi di misure, per esempio all'analisi di gas, facendo in modo che il gas si sciolga in uno strato sottile di soluzione e produca una variazione rivelabile in una composizione ionica. In modo simile, per combinazione con un enzima, si può rivelare un componente biologico nella forma del suo prodotto di decomposizione enzimatica. Infine bisogna notare che le tecniche elettroanalitiche sono particolarmente adatte ad analisi di routine e ad analisi continue, poiché possono essere facilmente controllate mediante microcalcolatori.
7. Fonti di energia elettrochimica
Le fonti di energia elettrochimica (v. Bagotsky e Skundin, 1980) possono suddividersi in celle primarie, celle secondarie e celle a combustibile.
a) Celle primarie
Dentro una cella primaria si formano prodotti chimici, mentre altri vengono consumati, con produzione di elettricità; una volta che i secondi siano esauriti, la cella cessa di funzionare. La maggior parte delle primitive sorgenti di energia elettrochimica, compresa la pila di Volta, erano celle primarie. Quella che ha avuto più successo finora è la cella a zinco/diossido di manganese, inventata da G. L. Leclanché nel 1867. La cella originale conteneva una soluzione acquosa di cloruro d'ammonio, ma nei successivi anni ottanta l'elettrolita fu sostituito da una pasta e si ottenne la cosiddetta ‛pila a secco'. Queste pile sono oggi largamente usate come sorgente di corrente per apparecchi portatili elettrici ed elettronici, quali le radio. La produzione mondiale annua si aggira intorno agli 8 miliardi di esemplari. Ogni singola pila ha un voltaggio iniziale di circa 1,5 V e una capacità che può andare da 0,01 Ah fino a 600 Ah. Una tipica piccola pila è mostrata nella fig. 6. La reazione all'anodo consiste nella dissoluzione di zinco:
Zn → Zn2+ + 2e-; (19)
la reazione al catodo è complicata; all'inizio probabilmente essa è:
MnO2 + H+ + e- → MnOOH. (20)
Sono state costruite delle varianti del modello originale: le più comuni sono la pila con un elettrolita alcalino e quella con anodo di magnesio; ambedue queste pile hanno una densità di energia più alta di quella della pila originale di Leclanché.
Numerosi altri elettrodi sono stati combinati in vario modo per costruire pile destinate a scopi speciali: per esempio, le pile non acquose a litio/iodio usate nei pacemakers cardiaci e le pile a pasticca usate per calcolatori tascabili, orologi, ecc.
b) Celle secondarie
Le celle secondarie si comportano come le primarie, salvo che in esse i prodotti chimici, dopo che si sono consumati, possono essere rigenerati in situ facendo passare una corrente in direzione opposta: viene così ripristinato lo stato iniziale della cella. Una buona cella secondaria può essere rigenerata più volte in tal modo.
La cella secondaria che ha avuto più successo, inventata da G. Planté nel 1860, è la cella a piombo/acido, che è usata come batteria d'accensione praticamente in tutte le automobili (se ne consumano 100 milioni di esemplari all'anno), nonché come principale fonte di energia nella stragrande maggioranza dei veicoli elettrici (furgoncini per il trasporto del latte, carrelli elevatori a forca, ecc.) e in parecchi altri sistemi che richiedono accumulo di elettricità.
Il funzionamento di questo tipo di cella dipende dall'esistenza di tre stati di ossidazione stabili, tutti presenti come composti solidi relativamente insolubili in acido solforico concentrato. L'elettrodo positivo è costituito da una pasta di diossido di piombo supportata da una griglia di piombo. Durante la scarica esso viene ridotto a solfato di piombo (o a miscela di questo con PbO):
PbO2 + 2H+ + H2SO4 + 2e- → PbSO4 + 2H2O. (21)
L'elettrodo negativo è costituito da un'analoga griglia di piombo che supporta una pasta di piombo finemente suddiviso. Durante la scarica il piombo viene ossidato a solfato di piombo:
Pb + H2SO4 → PbSO4 + 2H+ + 2e-. (22)
Quando la cella si è parzialmente o completamente scaricata, è possibile invertire queste reazioni applicando una fonte di elettricità esterna con un voltaggio più alto di quello prodotto dalla cella stessa. In condizioni ottimali si possono ottenere circa 400 cicli completi, se la scarica è quasi completa a ogni ciclo. Il voltaggio di una cella completamente carica è di 2,2 V.
Altri tipi affermati di celle secondarie sono la cella al nichel/cadmio e quella al nichel/ferro. La loro densità di energia è paragonabile a quella della batteria al piombo/acido (≃ 25 Wh/kg), ma esse hanno un ciclo vitale molto lungo (diverse migliaia di cicli). La prima fu inventata da W. Jungner nel 1899 e l'altra da Th. A. Edison nel 1901. In entrambe la reazione all'elettrodo positivo, durante la scarica, è
NiOOH + H2O + e- → Ni(OH)2 + OH-, (23)
mentre la reazione all'elettrodo negativo è la formazione dell'idrossido bivalente del metallo per reazione con l'elettrolita alcalino. Il voltaggio a circuito aperto è di circa 1,3 V per la cella al Ni/Cd e da 20 a 50 mV più alto per quella al Ni/Fe. Queste celle sono utili particolarmente per le applicazioni a bassa temperatura, perché le loro prestazioni non diminuiscono tanto quanto quelle di altre celle. È stata usata una batteria sigillata al Ni/Cd, per esempio, nel primo satellite per telecomunicazioni, il Telstar.
Sono state fatte molte ricerche su altre celle, come la cella al sodio/zolfo, introdotta dalla Ford Co. nel 1966; questa cella ha sodio fuso come elettrodo negativo, β-allumina come elettrolita solido, nel quale la corrente è portata dagli ioni sodio mobili, e una bacchetta di carbonio, che pesca in zolfo fuso, come elettrodo positivo. Le reazioni che avvengono nella scarica sono:
Na → Na+ + e-, (24)
nS + Na+ + e- → NaSn, (25)
dove n è 5/2 o 3/2 secondo il livello di scarica. La temperatura deve essere mantenuta nell'intervallo fra 300 e 375 °C, per evitare una rottura del fragile tubo di β-allumina, che tiene separati il sodio liquido e lo zolfo liquido. Celle di questo tipo potrebbero essere impiegate per la propulsione di veicoli elettrici di grandi dimensioni, come le locomotive, ma in pratica non si è ancora realizzata un'applicazione del genere.
c) Celle a combustibile
Entrambi i tipi di celle finora visti contengono una carica limitata di reagenti, che ovviamente limita la quantità di elettricità prodotta prima che la cella debba essere sostituita (cella primaria) o ricaricata (cella secondaria). In una cella a combustibile i reagenti sono forniti continuamente, di modo che la cella funziona in uno stato stazionario, limitato solo dalla stabilità dei suoi componenti. Così, in linea di principio, una cella a combustibile potrebbe sostituire i generatori convenzionali, fondati su una combinazione di combustione/macchina termica/dinamo, e avrebbe una maggiore efficienza, non perché il processo venga completato in un unico stadio, ma perché evita l'impiego di una macchina termica, la cui efficienza ha un limite superiore assoluto espresso dall'efficienza di una macchina termica ideale di Carnot.
La prima cella a combustibile fu descritta da sir W. Grove nel 1839. In essa la reazione anodica era l'ossidazione dell'idrogeno,
H2 → 2H+ + 2e-, (26)
e la reazione catodica era la riduzione dell'ossigeno,
O2 + 4H+ + 4e- → 2H2O. (27)
Entrambe le reazioni avvenivano su elettrodi di platino. La cella era quindi la stessa usata per l'elettrolisi dell'acqua, per es. da Nicholson e Carlisle (v., 1800), ma con il flusso di corrente in direzione opposta. La reazione complessiva,
2H2 + O2 → 2H2O, (28)
è la combustione dell'idrogeno. Sebbene Grove fosse riuscito a illuminare un'aula per conferenze con primitive lampade a incandescenza alimentate dalla sua cella a combustibile, l'applicazione pratica delle celle aventi come combustibile l'idrogeno si realizzò soltanto nella seconda metà del XX secolo e fu possibile soprattutto in seguito al lavoro pionieristico di F. T. Bacon. La prima applicazione si ebbe nei veicoli spaziali con equipaggio, dove l'uso di un combustibile leggero e facilmente disponibile, nonché la produzione di acqua, molto utile, fecero di queste celle a combustibile la soluzione ideale per il problema energetico. Gli elettrodi di Bacon erano di nichel poroso, con pori di due diverse dimensioni, che permettevano un intimo contatto fra gas ed elettrolita senza ingolfamento dell'elettrodo. In modelli successivi si è impiegato un foglio poroso di Teflon/carbonio che fa da supporto a un catalizzatore di metallo, spesso platino finemente suddiviso, per catalizzare le reazioni elettrodiche.
Si è tentato di usare altri combustibili nella speranza di poter sostituire con celle a combustibile i motori a combustione interna, ma solo la costosissima idrazina ha dato buoni risultati a temperatura ambiente. Il problema è che le reazioni elettrodiche di combustibili come il metanolo e, ancor di più, degli idrocarburi sono lente (le costanti di velocità, analoghe a quelle delle reazioni (9) e (10), sono troppo piccole). Un problema simile esiste, in parte, anche per la reazione di riduzione dell'ossigeno. Come tutte le reazioni chimiche, queste reazioni elettrodiche possono essere accelerate innalzando la temperatura: le celle a idrocarburi possono esser fatte funzionare in carbonati fusi a 550-700 °C. Sono stati anche provati elettroliti solidi che conducono per trasporto di O2- a temperature ancora più alte (850-1.000 °C).
Nei recenti tentativi di impiegare celle a combustibile per la produzione di energia su vasta scala si è però adottata un'altra soluzione: vengono decomposti idrocarburi combustibili e l'idrogeno ottenuto viene poi usato in una cella a combustibile a idrogeno/ossigeno. Impiegando questo principio, la United Technologies ha costruito due unità da 4,8 MW, una a Manhattan e una a Tokyo. Si sostiene che il rendimento globale sia del 40% circa, cioè più elevato di quello di una centrale energetica convenzionale. Un ulteriore vantaggio è che una centrale del genere opera senza inquinare e senza far rumore; ciò ne fa un impianto generatore accettabile in un ambiente urbano. Perché la combustione diretta di combustibili più convenzionali divenga una possibilità pratica, sono necessari ulteriori progressi, sia nella comprensione delle complesse reazioni elettrodiche, sia nella tecnologia degli elettrodi.
8. Fotoelettrochimica
Sebbene l'effetto fotovoltaico fosse stato scoperto da A.-C. Becquerel in una ricerca iniziata nel 1839, una chiara comprensione dell'effetto non si raggiunse finché non si realizzarono materiali semiconduttori ben definiti, come gli elettrodi di germanio usati nel 1955 nel lavoro pionieristico di Brattain e Garrett (v., 1955). Similmente il fenomeno di fotoemissione di elettroni dai metalli ebbe la sua prima chiara spiegazione nel lavoro di Barker (v. Barker e Gardner, 1965). Usando l'elettrodo di mercurio ben definito, egli poté mostrare che gli elettroni possono essere fotoemessi dal mercurio verso la soluzione a intervalli di pochi nm. Questi elettroni ‛asciutti', ad alta energia, erano termalizzati e solvatati e potevano o ritornare all'elettrodo o essere catturati da ‛catturatori di elettroni'; queste ultime reazioni erano molto simili a quelle studiate in radiochimica. Una cattura completa può essere ottenuta con N2O, il cui prodotto primario N2O- è trasformato immediatamente dalla reazione
N2O- + H2O → N2 + OH + OH-, (29)
e il radicale OH è rapidamente ridotto all'elettrodo. Perciò ogni fotone assorbito produce un flusso di 2 elettroni nel circuito esterno (effetto di raddoppio della corrente, o resa quantica 2).
Barker usò la dipendenza di questa fotocorrente Y dalla lunghezza d'onda e la legge di Fowler della fotoemissione (enunciata per la fotoemissione nel vuoto),
Y = cost. (hω − hω0)2, (30)
per dedurre la frequenza di soglia ω0 che dà la funzione di lavoro effettivo del metallo. Brodskij e altri (v., 1970) hanno eseguito un'analisi teorica dell'emissione in un mezzo condensato, che suggeriva una dipendenza da (hω − hω0)5/2 piuttosto che la dipendenza quadratica della legge di Fowler. La corretta interpretazione del fenomeno è ancora in discussione, ma gli studi dettagliati di Pleskov e collaboratori (v. Brodskij e altri, 1971) hanno mostrato che la soglia dipende solo dal potenziale e non dalla natura del metallo, cioè dalla posizione del livello di Fermi. Essi hanno anche elaborato il metodo come un modo di studiare la struttura del doppio strato e, con Barker, di studiare la cinetica delle reazioni elementari all'elettrodo.
Bisogna notare che le correnti prodotte da elettroni fotoemessi da metalli sono molto piccole (nA cm-2) a causa dell'inefficienza del processo di assorbimento primario. L'assorbimento della luce è molto più efficiente nei semiconduttori e la coppia lacuna-elettrone prodotta può essere prontamente separata spazialmente nella carica spaziale prodotta vicino alla superficie del semiconduttore. Questo è il principio fondamentale sul quale è basato l'uso di elettrodi semiconduttori come convertitori fotoelettrici. Le idee basilari dell'elettrochimica dei semiconduttori furono enunciate, sulla base dei lavori di Brattain e Oarrett, da Dewald (v., 1959) e particolarmente da Gerischer (v., 1961). La bassa densità dei portatori di carica (elettroni o lacune) nei semiconduttori significa che, quando viene stabilito un doppio strato all'interfaccia dell'elettrodo, la carica sul lato del semiconduttore non è limitata a una regione di dimensioni atomiche, ma è distribuita in una carica spaziale spessa, sovente, migliaia di nm. Questa distribuzione obbedisce a equazioni molto simili a quelle della parte diffusa del doppio strato nell'elettrolita. È il campo in questa regione di carica spaziale che separa le coppie lacuna-elettrone.
Cariche spaziali simili sono prodotte in dispositivi allo stato solido puro mediante drogaggio graduale, che richiede un controllo molto accurato. Viceversa, nella situazione elettrochimica, la carica spaziale insorge semplicemente per contatto con l'elettrolita. Nel caso di un semiconduttore di tipo n, che è polarizzato in modo che la superficie sia caricata positivamente (esaurimento dei portatori maggioritari), le lacune migrano verso la superficie e gli elettroni verso la massa del semiconduttore. Se le lacune riescono a ossidare una specie in soluzione (rimuoverne elettroni), allora una corrente elettrica è indotta a fluire attraverso la cella: così si può convertire energia luminosa in elettricità. La reazione chimica può anche produrre una sostanza utile, per es. idrogeno. Comunque, esistono sempre processi concorrenti: le lacune possono ossidare lo stesso semiconduttore, cioè corroderlo, e gli elettroni e le lacune possono ricombinarsi, con degradazione della loro energia di eccitazione in calore. Non si è ancora trovata una soluzione soddisfacente di questi problemi, nonostante i notevoli progressi fatti negli anni settanta e nei primi anni ottanta, progressi che hanno portato alla costruzione di celle che convertono l'energia solare con un rendimento di circa il 12%.
9. Elettrolisi industriale
Il processo elettrochimico industriale impiegato su più vasta scala è l'elettrolisi di una soluzione di cloruro di sodio per produrre cloro e idrossido di sodio. Nel 1983, in tutto il mondo, sono state prodotte oltre 4 × 107 tonnellate di cloro. Il processo di base è semplice: all'anodo si produce cloro secondo la reazione
2Cl- → Cl2 + 2e-, (31)
mentre al catodo si producono idrogeno e alcali secondo la reazione
2H2O + 2e- → H2 + 2OH-. (32)
Una buona produzione di Cl2 e NaOH dipende dall'esclusione della reazione anodica concorrente
2H2O → O2 + 4H+ + 4e-, (33)
e dalla presenza di un mezzo di separazione dell'alcali dalla salamoia originale. Questi problemi furono risolti originariamente da H.Y. Castner e K. Kellner, che, nel 1894, elaborarono un processo a due stadi. L'elettrolisi veniva fatta in una cella con un catodo di mercurio e un anodo di carbone, e dava cloro e amalgama di sodio. Quest'ultimo veniva decomposto in una cella separata, chiamata denuder, originariamente per via elettrochimica, ma più recentemente mediante un tipo di reazione di corrosione, in cui lo svolgimento di idrogeno è catalizzato da un metallo di transizione supportato su sfere di grafite.
Il processo Castner-Kellner, benché sia tuttora un procedimento efficiente per la produzione di cloro e alcali, richiede uno speciale trattamento dell'effluente, per evitare inquinamento dell'ambiente da mercurio. Questa è una delle ragioni principali della sua sostituzione, dal 1950, particolarmente negli Stati Uniti e in Giappone, con il processo, a un solo stadio, che si verifica nella cella a diaframma. La cella in questione è divisa in due compartimenti da un diaframma fatto di asbesto impregnato di polimero; in effetti il diaframma è attaccato al catodo, ragion per cui il compartimento catodico è piccolo e l'NaOH prodotto è estratto attraverso la rete catodica. Il diaframma non è selettivo, sicché parte degli ioni cloro attraversa il diaframma verso il catodo e l'alcali è sempre contaminato da NaCl. Per evitare un'eccessiva contaminazione, la reazione non è portata fino a un'elevata concentrazione di NaOH ed è necessaria una fase di concentrazione separata. La cella a membrana rappresenta un ulteriore miglioramento, in quanto il diaframma è rimpiazzato da una membrana selettiva per i cationi o addirittura per il sodio: questo permette la produzione di una concentrazione più elevata di NaOH. Le membrane sono polimeri perfluorurati con catene laterali terminanti in gruppi solfonici (Nafion della du Pont) o carbossilici (Flemion della Asahi). Le proprietà di queste membrane al presente impongono un limite alle dimensioni delle unità, ma esse sono in via di rapido sviluppo.
Il materiale anodico era originariamente grafite e sebbene questo favorisse la cinetica dello sviluppo di cloro rispetto a quello di ossigeno, il processo era relativamente lento e comportava un notevole spreco di energia elettrica. Inoltre il cloro corrodeva la grafite e spesso gli anodi dovevano essere rimodellati o sostituiti. Un progresso sostanziale venne fatto da Beer (v., 1972 e 1973) con l'introduzione di anodi di titanio rivestiti di RuO2, che sono catalizzatori migliori per la produzione del cloro e hanno una velocità di corrosione insignificante. Gli ‛anodi stabili dimensionalmente' (Dimensionally Stable Anodes, DSA) hanno sostituito completamente il carbone, con un risparmio del 10% circa nel consumo di energia.
L'elettrolisi dell'acqua è un procedimento per ottenere ossigeno e idrogeno puri ed è usata nella produzione di idrogeno per l'idrogenazione di molecole organiche su piccola scala. Si è suggerito di usare l'elettrolisi come metodo per immagazzinare l'elettricità in eccedenza durante i periodi di minore consumo: la rigenerazione avviene nella cella fatta lavorare in direzione opposta, come una cella a combustibile. In particolare questo sistema, in combinazione con l'energia nucleare, è stato proposto come futura fonte di energia: su questa combinazione di metodi produttivi si basa la cosiddetta ‛economia a idrogeno'.
Poiché gli isotopi più pesanti reagiscono un po' più lentamente di quelli più leggeri, l'acqua, in un elettrolizzatore ad acqua, si arricchisce in D2O. Questa fu l'unica fonte di D2O fino al 1943 e rimane spesso uno stadio di prearricchimento.
Il fluoro è prodotto solo tramite elettrolisi, usando KF + HF fusi, secondo il procedimento con cui H. Moissan per primo isolò il fluoro nel 1886. Il catodo è di acciaio dolce e l'anodo è di un tipo di carbonio preparato in modo speciale. In tutto il mondo vengono prodotte fino a 2 × 104 tonnellate di F2 l'anno e sono usate per produrre UF6, utilizzato per la separazione degli isotopi dell'uranio e per la preparazione di SF6 e di composti organici fluorati, come il Teflon.
L'ossidazione elettrolitica è usata per produrre gli agenti ossidanti NaClO3, K2Cr2O7, KMnO4 e vari perossiacidi, nonché il diossido di manganese per le celle Leclanché e l'ossido rameoso.
Dopo quello per produrre cloro e alcali il processo elettrochimico industriale impiegato su più vasta scala è l'estrazione dell'alluminio, la cui produzione mondiale è stata di circa 1,4 × 107 tonnellate nel 1983. Il minerale usato è la bauxite, che dopo la purificazione consta di allumina, Al2O3, ossido che fonde a 2.020 °C. La scoperta, fatta nel 1886 da C. M. Hall e P. L. T. Héroult, che l'allumina si scioglie nella criolite (Na3AlF6), per dare, intorno ai 1.000 °C, un fluido conduttore di elettricità, rese possibile la produzione industriale di alluminio su vasta scala. Entrambi gli elettrodi sono di carbonio; il catodo costituisce il fondo della cella, su cui si accumula l'alluminio: in realtà è quest'ultimo che funziona da catodo effettivo. L'anodo si consuma man mano che reagisce con l'ossigeno e dev'essere ripristinato continuamente. Perciò la reazione globale della cella è
2Al2O3 + 3C → 4Al + 3CO2. (34)
Si è cercato di sostituire il processo Hall-Héroult con un processo che utilizza la miscela NaCl + LiCl + AlCl3, la quale fonde a temperatura più bassa (processo Alcoa), ma non si sono finora riscontrati vantaggi tali da indurre a sostituire il vecchio metodo.
Altri processi di preparazione di metalli per elettrolisi di sali fusi sono quelli del magnesio, del sodio, del litio e del Mischmetal (miscela di metalli delle terre rare). Si è anche cercato di realizzare l'estrazione elettrolitica di metalli come il titanio, ma finora non si è riusciti a mettere a punto un processo in grado di sostituire il processo termico di Kroll.
Per l'estrazione del rame e dello zinco si usano soluzioni acquose, ma il metodo elettrolitico viene impiegato solo per il 10% della produzione del primo e per il 50% di quella del secondo. Anche quantità più piccole di altri metalli sono prodotte elettroliticamente. Processi simili sono usati per la raffinazione dei metalli Cu, Ni, Co, Pb, Zn (in soluzione acquosa) e Al (allo stato fuso).
Anche alcuni composti organici sono prodotti elettroliticamente. Il processo più usato è il metodo Monsanto per l'idrodimerizzazione dell'acrilonitrile ad adiponitrile, un intermedio nella produzione del nailon:
2CH2=CHCN + 2H2O + 2e- → CN(CH2)4CN + 2OH-. (35)
Questo processo fu ideato da Baizer nel 1963 (v., 1964). Originariamente il catodo era di piombo e l'anodo di diossido di piombo, ma ora questi materiali sono stati sostituiti, rispettivamente, col cadmio e con l'acciaio. Per favorire il processo si usa un elettrolita acquoso con un'alta concentrazione di acrilonitrile e un tensioattivo; questo viene adsorbito sul catodo e forma una regione in gran parte non acquosa che ritarda simultaneamente le reazioni concorrenti di sviluppo di idrogeno e di protonazione dell'acrilonitrile. Perciò è favorita la dimerizzazione di quest'ultimo.
Molte altre ossidazioni e riduzioni organiche sono ottenute per via elettrolitica su piccola scala industriale. In alcune di esse si usa un sistema intermediario inorganico di ossidoriduzione: è quest'ultimo che in realtà reagisce all'elettrodo, mentre la reazione desiderata è completata come un processo omogeneo.
La natura specifica delle reazioni elettrolitiche, insieme al fatto che altri reagenti possono spesso essere esclusi, le rende adatte alla preparazione di prodotti puri; c'è quindi da aspettarsi un'espansione dell'elettrochimica industriale in questa direzione.
(Per un approfondimento dell'argomento trattato in questo capitolo si rinvia il lettore a Pletcher, 1982).
10. Finitura e lavorazione dei metalli
I metodi elettrochimici sono ampiamente usati per preparare metalli in uno stato di diretta utilità o di aspetto attraente. Ciò può essere fatto o depositando il metallo o rimuovendolo, e sia in grandi quantità sia solo superficialmente.
La deposizione di un oggetto metallico completo è chiamata ‛elettroformatura' ed è usata per produrre lamine sottili, reticelle, tubi o bande senza giunzioni, e oggetti più complessi, come le guide d'onda. Si ricorre alla precisione dell'elettrodeposizione nella preparazione di matrici per dischi per grammofono, attraverso la deposizione di rame sulla matrice. Gioielli e medaglie d'argento e d'oro sono formati per questa via, ma la maggior parte dell'elettroformatura è eseguita con nichel. La quantità di metallo depositata nell'elettroplaccatura è di gran lunga minore di quella depositata nell'elettroformatura. Generalmente l'applicazione di uno strato sottile serve semplicemente per ottenere proprietà di superficie migliori, senza un grande aumento dei costi. Esempi tipici sono la cromatura (che, generalmente, consiste nella deposizione di uno strato di cromo molto sottile su uno strato di nichel più spesso) e la placcatura in oro di contatti di molti componenti elettronici, di conduttori nelle tavole dei circuiti e di alcuni condensatori. Per assicurare una buona aderenza, l'elettroplaccatura deve essere effettuata su una superficie pulita e questa spesso è ottenuta elettroliticamente.
Il processo di deposizione spesso comincia con la deposizione di un monostrato di atomi, seguita da nucleazione e crescita degli strati successivi. Gli aspetti fondamentali di questi processi sono stati molto studiati e la loro cinetica è stata analizzata in termini di stadi elementari. Questo tipo di analisi dev'essere fatto in condizioni accuratamente controllate, che sono piuttosto lontane da quelle di un bagno di placcatura pratico, dove l'obiettivo è generalmente quello di formare sull'oggetto un deposito microcristallino uniforme, spesso rifinito con una lucidatura a specchio. La capacità di un bagno di placcatura di formare un deposito uniforme è chiamata ‛potere ricoprente'. Un bagno con buon potere ricoprente darà un deposito uniforme su un oggetto di forma complessa.
In linea di principio una densità di corrente uniforme si ottiene lavorando sulla parte della curva corrente-potenziale che è quasi piatta, dove cioè la densità di corrente è indipendente dal potenziale, cosicché una distribuzione di potenziale complessa su un oggetto, per es., a spirale non condurrà a un deposito non uniforme. Sebbene questo risultato possa essere ottenuto lavorando in condizioni limitate dalla diffusione, queste spesso non conducono a una buona forma di deposito, ma producono dendriti o polveri: ha maggior successo l'uso preliminare di una reazione chimica, come la dissociazione di un complesso. Ciò promuove anche la dissoluzione dell'anodo, che mantiene costante la composizione del bagno. Frequentemente si usano complessi cianidrici e allo stesso tempo agenti tensioattivi, allo scopo di modificare le condizioni della deposizione, per adsorbimento sulla superficie. Questi tensioattivi di solito sono realizzati empiricamente per ottenere lucentezza, livellamento, compensazione degli sforzi nel deposito e per facilitare la rimozione di idrogeno, che potrebbe, altrimenti, formare bolle aderenti alla superficie. Si tratta generalmente di molecole organiche a volte con gruppi contenenti zolfo, che le rendono capaci di attaccarsi alla superficie dell'elettrodo.
Le leghe possono, in alcuni casi, essere elettroplaccate, controllando attentamente le condizioni del potenziale e la composizione del bagno. Oggetti non metallici possono essere placcati dopo essere stati rivestiti di un materiale conduttore, come la grafite. Viceversa si possono rivestire superfici metalliche con materiali non metallici attraverso un processo di elettroforesi. Questo metodo è usato per la protezione dalla corrosione delle carrozzerie delle automobili. La vernice o i componenti della pellicola di polimero sono ridotti allo stato di sospensione colloidale, cioè una sospensione di particelle portatrici di carica elettrica. Esse migrano in un campo elettrico come ioni e aderiscono a una superficie metallica pulita. Dopo l'essiccamento, la pellicola è trattata come un normale strato di vernice.
Metalli in tracce possono essere rimossi da un effluente per placcatura elettrolitica. È richiesto un catodo a superficie molto ampia, che generalmente si ottiene usando fasci di fibre di carbonio o un panno di carbonio.
Si può ricorrere a processi anodici su metalli per ottenere la rimozione di strati metallici o la formazione di pellicole protettive. Nella lavorazione per elettroerosione si rimuovono quantità macroscopiche di metallo e, usando un catodo di forma appropriata, si possono produrre forme abbastanza complesse. A differenza dell'elettroplaccatura, questo procedimento è stato messo a punto di recente, per la necessità di lavorare leghe molto dure, come quelle di titanio per i motori degli aerei. In questo processo si usano alte correnti e un flusso abbondante di elettrolita, e si ottiene un'ottima finitura, che non richiede ulteriore lavorazione.
Quantità ben inferiori di metallo vengono rimosse nella politura elettrolitica. In questo caso il potenziale del metallo e la natura del bagno sono tali che la dissoluzione del metallo avviene rapidamente attraverso una membrana anodica poco aderente: si ottiene così una superficie a elevata politura, che può essere molto liscia perfino su scala atomica.
Il trattamento anodico di un metallo ha spesso come risultato la formazione di una pellicola protettiva. Sottili pellicole di ossido proteggono metalli come l'alluminio e il cromo, il che spiega la stabilità di questi metalli nell'uso quotidiano. Comunque, la pellicola sull'alluminio può essere inspessita e le sue proprietà possono essere modificate per fini ornamentali: variando lo spessore, si possono produrre colori d'interferenza, o, alternativamente, si possono incorporare nella pellicola, inizialmente porosa, dei pigmenti, che poi vengono fissati chiudendo i pori con un trattamento in acqua bollente. Molti articoli d'alluminio e di sue leghe sono trattati in questo modo: così se ne migliora l'aspetto e se ne aumenta la resistenza alla corrosione.
11. Corrosione
La corrosione è il processo attraverso cui i metalli reagiscono con sostanze presenti nel loro ambiente per diventare composti, generalmente ossidi, perdendo così la loro forza, spesso con risultati catastrofici. L'esempio più noto è l'arrugginimento del ferro. La corrosione comporta uno spreco molto costoso di materiale: per esempio si è calcolato che le perdite dovute alla corrosione sono costate agli Stati Uniti, nel 1975, circa 7 × 1010 dollari, pari al 4,5%, circa, del PNL.
Sebbene la reazione complessiva possa essere scritta nella forma di una reazione chimica, già Davy si accorse che essa è la somma di due reazioni elettrochimiche, come la reazione in una cella elettrochimica. Davy applicò questo concetto, nel 1824, quando fu consultato dall'ammiragliato britannico a proposito della corrosione delle navi di legno rivestite di rame. Egli mostrò che si poteva impedire la corrosione attaccando al rame un pezzo di un metallo più ossidabile (ferro, zinco), noto come ‛anodo di sacrificio'; esso si scioglie nell'acqua di mare a un potenziale più negativo del rame, mantenendo questo a un potenziale a cui è stabile; così il rame diventa il luogo di una reazione catodica, che normalmente è la riduzione dell'ossigeno sciolto nell'acqua di mare.
Questa combinazione di una reazione di corrosione anodica in una certa zona e di una reazione catodica in un'altra zona può avvenire anche su scala più piccola, quando due diversi metalli sono a contatto fra loro in un ambiente umido, o anche quando zone diverse di uno stesso metallo si trovano in condizioni diverse, per esempio in differenti condizioni di tensione o di esposizione all'ossigeno. In generale, questo fenomeno è noto come corrosione per ‛azione locale'.
Può anche accadere che la reazione di corrosione e la reazione catodica a essa accoppiata avvengano nella stessa zona di un metallo omogeneo. In condizioni di stato stazionario si stabilisce un potenziale stazionario noto come ‛potenziale misto'. Questo concetto fu introdotto contemporaneamente, nel 1932, da Frumkin (v., 1932) e da Hammett e Lorch (v., 1932) per spiegare la cinetica di dissoluzione di amalgami di metalli alcalini; fu poi sviluppato, nel 1938, da Wagner e Traud (v., 1938) e applicato alle reazioni di corrosione in generale.
Le condizioni per la corrosione non dipendono solo dall'attività del metallo e dall'aggressività dell'ambiente, ma anche dalla natura dei prodotti di corrosione: se sono solubili, la dissoluzione del metallo può continuare incontrollata; nel caso che, invece, si formi un prodotto insolubile la situazione è più complessa: se tale prodotto forma uno strato isolante aderente, la corrosione può fermarsi rapidamente. Questa è la ragione della stabilità dell'alluminio e del magnesio in molti ambienti, a dispetto della loro alta reattività, come accennato nel capitolo precedente. Il ferro, d'altro canto, è meno reattivo, ma l'ossido (ruggine) che si forma è, normalmente, poroso, conduttore e poco aderente, sebbene in certe condizioni possa accrescersi in una forma aderente come una pellicola sottile, rendendo il ferro ‛passivo'. Questa spiegazione fu data da Faraday e, sebbene molto contestata, si sa ora che è corretta. Il cromo forma una pellicola passiva migliore e la presenza di una piccola quantità di cromo nel ferro (con nichel) conferisce le sue proprietà protettive all'acciaio inossidabile.
L'uso di pellicole protettive, di pellicole passive o di pellicole di vernice o di un altro metallo (rame, zinco) è uno dei modi per ritardare o prevenire la corrosione. Tuttavia questo sistema può causare problemi se la pellicola è perforata, perché in questo caso la corrosione si concentra in una piccola superficie. Questo e altri tipi di inomogeneità causano corrosione per ‛vaiolatura' o corrosione ‛interstiziale'. Se inoltre il metallo è sotto sforzo, la corrosione può essere accentuata dalla formazione di ‛cricche per corrosione sotto sforzo'. L'uso dello zinco nella ‛galvanizzazione' è un modo per combattere questi fenomeni: se avviene la perforazione, lo zinco agisce da ‛anodo di sacrificio', mentre il ferro esposto funge da zona catodica.
Si può evitare la corrosione anche modificando il potenziale, o con l'uso di un ‛anodo di sacrificio', oppure utilizzando una fonte di potenziale esterna e un anodo non corrodibile: questo procedimento è noto come ‛protezione catodica'. Poiché sul metallo da proteggere può svilupparsi idrogeno, c'è il rischio che questo possa sciogliersi nel metallo facendolo diventare fragile. Un'alternativa è quella di usare un potenziale più positivo, per formare una pellicola passiva anodicamente. Questa ‛protezione anodica' a sua volta richiede un controllo attento del potenziale, per mantenere la superficie nella regione di potenziale in cui la pellicola passiva è stabile, altrimenti la corrosione può perfino essere accentuata.
Le condizioni generali per la corrosione possono essere predette su base termodinamica, in quanto la termodinamica fornisce informazioni riguardo agli ambiti di potenziale e di composizione dell'ambiente nei quali il metallo è stabile. In altre regioni può avvenire la corrosione. Queste predizioni furono chiaramente riassunte da Pourbaix (v., 1966) nei diagrammi potenziale-pH, che forniscono un utile punto di partenza per una discussione sulla corrosione, ma naturalmente non prendono in considerazione la cinetica delle reazioni; inoltre le condizioni reali sono generalmente molto più complesse di quelle rappresentate nei diagrammi di Pourbaix. La tecnica dettagliata della corrosione rimane un fatto sperimentale, necessario per una comprensione teorica del fenomeno.
(Per un approfondimento dell'argomento trattato in questo capitolo si rinvia il lettore a Fontana e Greene, 1967).
12. Bioelettrochimica
Molti processi biologici fondamentali possono essere considerati sostanzialmente elettrochimici, poiché implicano trasferimento di carica alle interfacce, sebbene non comportino sempre lo stesso tipo di cambiamento del portatore di carica, tipico delle reazioni discusse sopra. Le interfacce sono membrane, la cui struttura è complessa. Esse consistono, tipicamente, di uno strato bimolecolare di fosfolipidi in cui le catene idrocarburiche (idrofobe) sono dirette all'interno e i gruppi fosfolipidici (idrofili) formano le due superfici esterne. In questa struttura sono inserite proteine e altre grandi molecole che svolgono le funzioni specifiche della membrana. Strutture simili sono state prodotte artificialmente e sono conosciute come ‛membrane lipidiche a doppio strato' (Bilayer Lipid Membranes, BLM). Esse sono ottenute spargendo una goccia di una soluzione di un lipide su un piccolo foro (~ 1 mm di diametro) praticato in un foglio di Teflon che divide una cella elettrolitica. A mano a mano che la goccia si asciuga, la membrana che ottura il foro si assottiglia, come si può vedere dal cambiamento dei colori d'interferenza (è più o meno quel che succede quando una bolla di sapone si asciuga). Infine la membrana diventa nera: il doppio strato sopra descritto si è formato.
Un doppio strato lipidico puro ha una resistenza molto alta, tra 108 e 1010 ohm cm-2, che, però, può essere abbassata addizionando varie sostanze che diminuiscono anche la resistenza delle membrane biologiche. Sono stati identificati tre meccanismi di trasporto di piccoli ioni inorganici attraverso queste membrane: 1) il moto termico delle molecole può determinare la formazione temporanea di una lacuna di dimensioni molecolari attraverso cui può passare uno ione. Questi eventi sono rari, sicché la resistenza intrinseca della membrana è alta; 2) il piccolo ione può complessarsi con una specie neutra, solubile nella membrana, passare attraverso la membrana e quindi essere rilasciato dal complesso dall'altra parte della membrana. L'agente complessante o trasportatore (carrier) può, in effetti, essere presente solo nella membrana. Un tipico carrier è la molecola macrociclica valinomicina, che si adatta ai cationi alcalini più grossi, ma che non può spostare il rivestimento di idratazione di Li+ o di Na+; 3) un terzo meccanismo è quello delle molecole capaci di formare canali. Ne è un esempio tipico la gramicidina A, un peptide con struttura a elica e con la parte esterna lipofila, sicché è stabile nella membrana. Se l'elica è orientata con il suo asse perpendicolare al piano della membrana, gli ioni possono passare lungo quest'asse e pertanto attraversare la membrana. La gramicidina in effetti permette il trasferimento di protoni preferenzialmente rispetto ad altri ioni monovalenti.
Una volta che il trasporto di uno ione sia reso possibile dalla presenza nella membrana di un carrier o di un formatore di canali, esso può essere indotto da un'adatta forza motrice, come un aumento di concentrazione o di potenziale da un lato della membrana rispetto all'altro. Questo è il cosiddetto ‛trasporto passivo'. In molte situazioni biologiche, tuttavia, il trasporto sembra avvenire contro un gradiente di concentrazione o di potenziale e viene chiamato ‛trasporto attivo'. Il trasporto attivo è il risultato dell'accoppiamento del trasporto con una reazione metabolica connessa con l'ossidazione di un ‛combustibile' biologico oppure con l'uso di energia immagazzinata in un legame polifosforico dell'adenosintrifosfato (ATP), che viene rotto per formare adenosindifosfato (ADP) e H2PO4-. Questi processi sono catalizzati da enzimi. L'esatta natura di questo accoppiamento non è ancora compresa. L'energia deriva originariamente dall'ossidazione di materiale organico durante la respirazione. Se questa ossidazione coinvolgesse direttamente l'ossigeno molecolare respirato dall'organismo, l'energia sarebbe trasformata in calore e non sarebbe, quindi, disponibile per promuovere altri processi. L'ossidazione, quindi, deve avvenire attraverso una serie di reazioni che sono sempre vicine all'equilibrio. Così l'energia è disponibile per compiere lavoro utile, all'incirca come lo è in una cella elettrochimica. Una serie di reazioni accoppiate di ossidoriduzione, che formano una catena trasportatrice di elettroni, si verificano nella membrana interna di un organulo (una regione specializzata della cellula) chiamato ‛mitocondrio'. Secondo Mitchell (v., 1968) l'energia prodotta viene immagazzinata sotto forma di un'aumentata attività dei protoni nei mitocondri.
Una serie quasi simile di reazioni avviene nella fotosintesi, ma in questo caso, naturalmente, l'energia iniziale è fornita dalla luce incidente. Questa viene assorbita dalla clorofilla con formazione di una coppia elettrone-lacuna, che viene poi separata, tramite reazione con sistemi adiacenti di ossidoriduzione, in un organulo chiamato ‛tilacoide'. Ci sono due fotosistemi paralleli ed è richiesto un totale di 4 quanti di luce per ridurre ogni molecola di CO2. Molte delle singole molecole coinvolte in queste catene trasportatrici di elettroni sono state studiate con tecniche elettrochimiche nel tentativo di capire i dettagli di questi processi estremamente complessi, che sono ancora oggetto di molte discussioni.
Dal tempo di Galvani si conosce l'intima relazione fra attività neuromuscolare ed elettricità. Si sospettava che un qualche tipo di conduzione fosse responsabile della propagazione dell'informazione attraverso i nervi (v. Moore, 1977). Originariamente si credeva che la velocità di trasmissione degli impulsi nervosi fosse infinita, ma Helmholtz mostrò, nel 1850, che essa in effetti è di circa 30 ms-1, cioè molto inferiore alla velocità di conduzione elettronica in un metallo: il fenomeno, d'altronde, non può essere spiegato in termini di semplice conduzione ionica, perché in tal caso richiederebbe campi elettrici molto elevati. La spiegazione fu trovata in seguito a esperimenti effettuati verso la metà del XX secolo, soprattutto da parte di Hodgkin e Huxley (v., 1947), i quali usarono microelettrodi inseriti nelle fibre nervose (assoni) del calamaro gigante, che sono molto grandi. Si trovò che, nello stato di riposo, la composizione all'interno dell'assone era piuttosto differente da quella all'esterno. Cosa particolarmente notevole, la concentrazione degli ioni sodio era 50 mM all'interno e 460 mM all'esterno, mentre quella degli ioni potassio era 400 mM all'interno e 10 mM all'esterno. Inoltre il potenziale elettrico all'interno della membrana era di 70 mV più negativo di quello esterno. L'eccitazione di un'estremità della fibra causa un aumento locale della permeabilità della membrana agli ioni Na+ e il potenziale di membrana cambia segno, diventando di circa +50 mV: il cosiddetto ‛potenziale di azione'. Ne segue una propagazione dello stato di eccitazione lungo la membrana ed è quest'onda di alterazione della permeabilità che conduce il segnale lungo la fibra nervosa piuttosto che un qualche trasporto fisico di ioni in questa direzione longitudinale. Le proprietà delle membrane furono studiate da Hodgkin, Huxley e loro collaboratori non solo allo stato naturale, ma anche sostituendo il materiale interno con soluzioni saline di composizione nota. Si usarono specie ioniche marcate con traccianti radioattivi per seguire il movimento di ognuna di esse in determinate condizioni elettriche. Fu infilato un filo metallico lungo l'assone e si controllò il potenziale con una ‛pinza di voltaggio' inventata a questo scopo. Questo strumento fu il precursore del ‛potenziostato' usato quasi universalmente nella moderna elettrochimica. Anche la soglia di eccitabilità fu esaminata usando impulsi di corrente.
La spiegazione dell'attività nervosa fu tentata in termini di diffusione di ioni in un campo elettrico costante, usando un modello più adatto a una membrana spessa e tenendo conto della capacità elettrica di questa. Tale spiegazione è stata molto discussa ed elaborata, ma il meccanismo molecolare delle variazioni di permeabilità della membrana resta incerto. Un possibile meccanismo è l'allontanamento di protoni da specie presenti nella membrana sotto l'azione di un elevato campo elettrico locale, con conseguente variazione della configurazione di certe molecole (proteine).
La produzione di elettricità nelle anguille elettriche e in altri pesci elettrici è strettamente correlata con il meccanismo dell'attività nervosa. L'organo elettrico è composto da un ammasso di cellule note come ‛elettroplacche'. Un lato di ognuna di queste cellule è coperto di terminazioni nervose che, quando vengono eccitate, causano il cambiamento del potenziale della membrana dell'elettroplacca adiacente, fino alla depolarizzazione. L'altro lato non ha terminazioni nervose e pertanto resta inalterato. Perciò i potenziali di membrana attraverso le due superfici cellulari si sommano anziché annullarsi e la cellula produce una differenza di potenziale di circa 150 mV. Poiché possiede parecchie migliaia di elettroplacche, l'anguilla elettrica è in grado di produrre un voltaggio totale massimo di 600 V, che essa usa per stordire o uccidere altri pesci. Le raie hanno ammassi di elettroplacche più piccoli, che usano per rilevare la presenza di oggetti; esse hanno anche speciali recettori sensibili a piccolissime variazioni di campi elettrici.
I problemi che portarono allo studio dell'elettrochimica mantengono il loro interesse nel contesto della biologia moderna e molto resta da fare per raggiungere una completa comprensione dei complessi sistemi biologici. (Per un approfondimento dell'argomento trattato in questo capitolo si rinvia il lettore a Koryta, 1982).
Bibliografia
Bagotsky, V. S., Skundin, A. M., Chemical power sources, London 1980.
Baizer, M. M., Electrolytic reductive coupling. I. Acrylonitrile, in ‟Journal of the Electrochemical Society", 1964, CXI, pp. 215-228.
Bard, A. J., Encyclopedia of electrochemistry of the elements, 15 voll., New York 1974 ss.
Bard, A. J., Faulkner, L.R., Electrochemical methods, New York 1980.
Bard, A. J., Parsons, R., Jordan, J., (a cura di), Standard potentials in aqueous solutions, New York 1985.
Barker, G. C., Gardner, A. W., Osnovnye voprosy sovremennoj teoreticeskoj elektrochimii, Moskva 1965, pp. 34-42.
Beer, H.B., U.S.??? Patent, 3 632 498, 1972; E 711 385, 1973.
Bewick, A., Kunimatsu, K., Pons, B. S., Infrared spectroscopy of the elctrode-electrolyte interface, in ‟Electrochimica acta", 1980, XXV, pp. 465-468.
Bockris, J. O' M., Conway, B. E., Yeager, E., Comprehensive treatise of electrochemistry, 8 voll., New York 1980 ss.
Bond, A. M., Modernpolarographic methods in analytical chemistry, New York 1980.
Brattain, W. H., Garrett, C. G. B., Experiments on the interface between germanium and an electrolyte, in ‟Bell System technical journal", 1955, XXXIV, pp. 129-176.
Brdicka, R., Wiesner, K., Polarographische Bestimmung der Geschiwindigkeitskonstante für die Oxydation von Ferrohäm und anderen Ferrokomplexen durch H2O2, in ‟Naturwissenschaften", 1943, XXXI, p. 247.
Brodskij, A. M,. Gurevic, J. J., Levic, V. G., General threshold theory of electronic emission from the surface of a metal, in ‟Physica status solidi", 1970, XL, pp. 139-151.
Brodskij, A. M., Gurevic, J. J., Pleskov, J. V., Rotenberg, Z. A., Sovremennaja fotoelektrochimija. Fotoemissionye javlenija, Moskva 1971.
Dewald, J. F., Reaction of semiconductors with aqueous solutions, in Semiconductors (a cura di N. B. Hannay), New York 1959.
Donnan, F. G., Theorie der Membrangleichgewichte und Membranpotentiale bei Vorhandensein von nicht dialysierenden Electrolyten. Ein Beitrag zur physikalisch-chemischen Physiologie, in ‟Zeitschrift für Elektrochemie", 1911, XVII, pp. 571-581.
Dubpernell, G., Westbrook, J. H., Selected topics in the history of electrochemistry, Princeton n. j., 1978.
Fleischmann, M., Hendra, P. J., McQuillan, A. J., Raman spectra of pyridine adsorbed at a silver electrode, in ‟Chemical physics letters", 1974, XXVI, pp. 163-173.
Fontana, M. G., Greene, N. D., Corrosion engineering, New York 1967.
Frumkin, A. N.,??? Thesis, Odessa 1917.
Frumkin, A. N., Bemerkung zur Theorie der Wasserstoffüberspannung, in ‟Zeitschrift für hysikalische Chemie", 1932, A, CLX, pp. 116-118.
Frumkin, A. N., Dolin, P., Ershler, B. V., Kinetics of processes on the platinum electrode. Parts I, II and III, in ‟Acta physicochimica URSS", 1940, XIII, pp. 747-793.
Galvani, L., De viribus electricitatis in motu musculari commentarius, in ‟De Bononiensi Scientiarium et Artium Instituto atque Academia Commentarii", 1791, VII, pp. 363-418.
Gerischer, H., Semiconductor electrode reactions, in Advances in electrochemistry and electrochemical engineering (a cura di P. Delahay e C. W. Tobias), vol. I, Electrochemistry (a cura di P. Delahay), New York - London 1961, pp. 139-232.
Gibbs, J. W., On the equilibrium of heterogeneous substances, in ‟Transactions of the Connecticut Academy of Arts and Sciences", 1875, III, pp. 108-248.
Gouy, G., Sur la fonction électrocapillaire, in ‟Annales de chimie et de physique", 1903, s. 7, XXIX, pp. 145-241; 1906, s. 8, VIII, pp. 291-363; 1906, s. 8, IX, pp. 75-139; in ‟Annales de physique", 1917, VII, pp. 129 ss.
Grahame, D. C., The electrical double layer and the theory of electrocapillarity, in ‟Chemical review", 1947, XLI, pp. 441-501.
Gray, W. F. M., A shocking discovery, in ‟Journal of the Electrochemical Society" (New York) 1963, CX, pp. 210C-211C.
Grotthuss, C. J. D., Sur la décomposition de l'eau et des corps qu'elle tient en dissolution, à l'aide de l'électricité galvanique, in ‟Annales de chimie et de physique", 1806, LVIII, pp. 54-74.
Hammett, L. P., Lorch, A. E., The relation between the dissolution of metals in acids and the electrolytic evolution of hydrogen, in ‟Journal of the American Chemical Society", 1932, LIV, pp. 2128-2129.
Hansen, W. ., Kolb, D. M., Lynch, D. W. (a cura di), Electronic and molecular structure of electrode-electrolyte interfaces, Amsterdam 1983.
Hartley, H., Humphry Davy, London 1966.
Helmholtz, H. L. F. von, Studien über elektrische Grenzschichten, in ‟Annalen der Physik und Chemie", 1879, VII, pp. 337-382.
Helmholtz, H. L. F. von, Über galvanische Polarisation des Quecksilbers und darauf bezüglich neue Versuche des Herrn Arthur Koenig, in ‟Berliner akademische Monatsbericht", 1881, pp. 945-958.
Heyrovský, J., Electrolysis with a dropping mercury cathode. I. Deposition of alkali and alkaline earth metals, in ‟Philosophical magazine", 1923, XLV, pp. 303-315.
Hodgkin, A. L., Huxley, A. F., Potassium leakage from an active nerve fibre, in ‟Journal of physiology", 1947, CVI, pp. 341-367.
Koryta, J., Ions, electrodes and membranes, Chichester 1982.
Koryta, J., Stulik, K., Ions selective electrodes, Cambridge 1983.
Lange, E., Koenig, F. O., Elektrochemie der Phasengrenzen, in ‟Handbuch der Experimentalphysik", 1933, XII, 2, pp. 255-331.
Latimer, W., The oxidation states of the elements and their potentials in aqueous solutions, New York 1938, 19522.
Levic, V. G., Physicochemical hydrodynamics, Englewood Cliffs, N. J., 1962.
Lippmann, G., Relations entre les phénomènes électriques et capillaires, in ‟Annales de chimie et de physique", 1875, V, pp. 494-549.
Macdonald, D. D., Transient techniques in electrochemistry, New York 1977.
Mitchell, P., Chemiosmotic coupling and energy transduction, Bodmin, Cornwall, 1968.
Moore, W. J., Electrochemistry of nerves, in Special topics in electrochemistry (a cura di P. A. Rock), Amsterdam 1977, pp. 128-160.
Nernst, W., Die elektromotorische Wirksamkeit der Ionen, in ‟Zeitschrift für physikalische Chemie", 1889, IV, pp. 129-181.
Nernst, W., Theorie der Reaktiongeschwindigkeit in heterogenen Systemen, in ‟Zeitschrift für physikalische Chemie", 1904, XLVII, pp. 52-55.
Nicholson, W., Carlisle, A., Account of the new electrical or galvanic apparatus of Sig. A. Volta and experiments performed with the same, in ‟Journal of natural philosophical and chemical arts", 1800, IV, pp. 179-198.
Parsons, R., Equilibrium properties of electrified interphases, in Modern aspects of electrochemistry (a cura di J. O' M. Bockris e B. E. Conway), London 1954, pp. 103-179.
Pletcher, D., Industrial electrochemistry, London 1982.
Pourbaix, M., Atlas of electrochemical equilibria in aqueous solutions, Oxford 1966.
Ross, S., Faraday consults the scholars: the origin of the terms of electrochemistry, in ‟Notes and records of the Royal Society of London", 1961, XVI, pp. 187-220.
Rowlinson, J. S., Widom, B., Molecular theory of capillarity, Oxford 1982.
Tafel, J., Über die Polarisation bei kathodischer Wasserstoffentwicklung, in ‟Zeitschrift für physikalische Chemie", 1905, L, pp. 641-712.
Volta, A., A letter to Sir Joseph Banks, March 20th 1900. On electricity excited by the mere contact of conducting substances of different kinds, in ‟Philosophical transactions of the Royal Society of London", 1800, XC, 2, pp. 403-431.
Wagner, C., Traud, W., Über die Deutung von Korrosionsvorgängen durch Überlagerung von Elektrochemischen Teilvorgängen und über die Potentialbildung an Mischelektroden, in ‟Zeitschrift für Elektrochemie", 1938, XLIV, pp. 391-402.
Waterman, L., Preliminary report upon the excavations at Tel Umar, Iraq, Ann Arbor, Mich., 1931.
Williams, L. P., Michael Farday, London 1965.
Analisi chimica strumentale,
di Arnaldo Liberti e Guido Saini
SOMMARIO: 1. Introduzione. 2. Metodi elettroanalitici: a) potenziometria; b) elettrodi a membrana di vetro; c) elettrodi ione-selettivi; d) elettrodi a enzima; e) voltammetria, polarografia; f) polarografia a impulsi; g) tecniche di ridissoluzione (stripping) anodica; h) coulombometria. 3. Metodi basati sull'emissione e sull'assorbimento di radiazioni elettromagnetiche: a) fluorescenza di raggi X; b) emissione atomica; c) fluorescenza nel visibile e nell'ultravioletto; d) assorbimento atomico; e) assorbimento nel visibile e nell'ultravioletto; f) assorbimento nell'infrarosso; g) risonanza magnetica nucleare. 4. Altri metodi spettrometrici: a) spettrometrie di fotoelettroni; b) spettrometria di massa. 5. Metodi basati sulla distribuzione tra fasi: a) generalità sulla cromatografia; b) gascromatografia; c) cromatografia liquida (HPLC); d) cromatografia ionica. □ Bibliografia.
1. Introduzione
La chimica analitica si occupa dello studio e dello sviluppo dei metodi mediante i quali si possono individuare le specie chimiche presenti in un campione di materia e determinarne le quantità. Queste vengono di solito espresse come quantità relative, ossia come parti in peso di ogni specie per cento, per i costituenti maggiori o minori, e parti in peso per milione (ppm) o per miliardo (ppb) o per trilione (ppt), per i costituenti in tracce. Con l'espressione ‛specie chimiche' si possono intendere gli elementi chimici di cui è costituito il campione, oppure gli ioni presenti o i composti chimici che come tali esistono nel materiale in esame.
Fin dagli albori della chimica si è reso necessario studiare e mettere a punto metodi di analisi della materia. È significativo che la nascita della chimica come scienza venga fatta coincidere con la famosa esperienza compiuta da A.L. Lavoisier nel 1774, allo scopo di stabilire quali erano i principali componenti dell'aria. Lo sviluppo della chimica dalla sua origine è legato essenzialmente alle indagini analitiche eseguite allo scopo di caratterizzare la materia, individuarne la composizione e classificare ogni specie in funzione dei rapporti di combinazione secondo cui gli elementi sono tra loro legati.
Sino all'incirca agli anni trenta i procedimenti analitici erano fondamentalmente basati sull'impiego delle cosiddette tecniche gravimetriche e volumetriche; con le prime la determinazione di una data specie veniva eseguita attraverso la conversione di questa in un composto insolubile, del quale si misurava successivamente la massa. I metodi volumetrici erano basati sull'impiego di soluzioni di concentrazione nota di un reagente, che veniva aggiunto alla soluzione della specie in esame al fine di determinarne il contenuto mediante opportune reazioni (acido-base, ossidoriduzione, precipitazione o complessazione); facendo uso di un idoneo sistema indicatore per apprezzare il completamento della reazione utilizzata, dalla esatta misura del volume della soluzione impiegata come reagente era possibile risalire alla quantità della specie da determinare.
Le strumentazioni di base consistevano nella comune vetreria di laboratorio con qualche apparecchiatura più elaborata, generalmente di vetro, e in una bilancia con sensibilità pari a ± 0,1 mg, mediante la quale si eseguivano la misura della massa del campione e le altre pesate necessarie. Le metodiche volumetriche e gravimetriche vengono tuttora impiegate, ma l'evolversi della nostra società, con la creazione di un ingente numero di manufatti, il progresso tecnologico legato allo sviluppo di una varietà di prodotti, l'incrementato tenore di vita hanno comportato la necessità di adottare tecniche analitiche in grado di fornire le informazioni necessarie in una gamma assai ampia di casi per le più differenti finalità, quali la definizione delle specie chimiche che prendono parte a un processo, il controllo di qualità dei sistemi produttivi, per assicurare la costanza e la regolarità della produzione, la valutazione dell'ambiente, ecc. D'altra parte lo sviluppo di tecnologie analitiche (come le tecniche strumentali cromatografiche) ha avuto un'importanza fondamentale per il progresso di molti settori della chimica (chimica organica, biochimica, ecc.).
I compiti attuali della chimica analitica sono molteplici, in quanto si tratta di valutare non solo i costituenti principali di un dato sistema, e cioè i componenti maggiori - fine che poteva essere realizzato anche con i citati metodi di analisi convenzionali - ma anche i costituenti minori o in tracce in miscele particolarmente complesse, la cui presenza può avere influenza determinante. Mentre una quindicina di anni fa erano poche le sostanze che potevano essere determinate con sufficiente accuratezza in concentrazioni di poco inferiori a una parte per milione, oggi si è in condizione di eseguire determinazioni di molte sostanze presenti in parti per miliardo e, in casi molto favorevoli, in parti per trilione.
Facendo uso di tecniche particolarmente raffinate e in condizioni molto particolari, si sono rilevate concentrazioni dell'ordine di decine di atomi per cm3 (ad es. vapori di sodio elementare a temperatura ambiente. Si noti che, a pressione atmosferica, il numero di molecole presenti in 1 cm3 di un gas o vapore è dell'ordine di 1019). L'esigenza di poter rilevare elementi o composti in tracce si manifesta in molti campi: ad es. in quello dell'ambiente (acqua, aria, terreno, ambiente di lavoro), per poter seguire con la massima accuratezza i sintomi del degrado ambientale. Un altro esempio è quello della preparazione dei semiconduttori impiegati nella costruzione dei componenti elettronici. Il germanio e il silicio devono venire preparati allo stato di grandissima purezza prima di inquinarli di proposito con quantità esattamente predeterminate di altri elementi, in modo da conferire a essi la semiconduttività desiderata. Occorre quindi un accurato controllo della purezza sia delle materie prime impiegate sia dell'ambiente nel corso della preparazione industriale.
Per quanto riguarda gli alimenti, si possono avere inquinamenti da sostanze nocive sia nel corso della produzione (diserbanti, insetticidi, ormoni) sia in quello della conservazione o dei trattamenti successivi ai quali vengono sottoposti.
Altro importante compito della chimica analitica è quello di riuscire a separare e a determinare sostanze le cui strutture molecolari, e quindi anche le proprietà chimiche, sono assai simili. Per rendersi conto dell'importanza di questa attività è sufficiente ricordare che in un gruppo di composti con struttura molecolare molto simile può accadere di riscontrare che uno di essi è dotato di proprietà biologiche (ad es. tossicità) assai più degli altri, e quindi è di fondamentale importanza poter accertare la quantità di quel particolare composto.
I compiti indicati per la chimica analitica sono stati realizzati mediante l'impiego di procedimenti strumentali e cioè di apparecchiature che utilizzano principi fisici o chimico-fisici caratteristici del sistema in esame o della struttura delle specie che debbono essere determinate. In vari casi le apparecchiature possono fornire una risposta assai selettiva, così da non richiedere una preventiva separazione delle specie da analizzare, consentendo di pervenire alla sensibilità richiesta per le finalità indicate. Nel caso di miscele complesse, in cui può essere presente un gran numero di composti, si rende spesso necessario il loro frazionamento con il conseguente isolamento delle specie che interessano. L'obiettivo viene raggiunto mediante l'utilizzo di tecniche cromatografiche, che possono essere condotte secondo una varietà di procedimenti. Ovviamente i progressi compiuti dalla strumentazione nell'analisi chimica riflettono lo sviluppo dell'elettronica, senza il quale sarebbe stata impensabile la progettazione degli attuali strumenti. Tale apporto evidenzia un'altra caratteristica della moderna chimica analitica: l'utilizzo di mezzi strumentali in modo semiautomatico o automatico, al fine di eliminare l'intervento dell'operatore in molte attività e gli errori legati alla manualità, e ottenere il responso finale in forma grafica o digitale. Lo strumento può essere collegato con calcolatori che programmano lo svolgimento dell'analisi, elaborano i dati ed eventualmente confrontano la risposta dello strumento con i dati di sostanze note contenuti in una memoria, allo scopo di identificare le sostanze analizzate. Questo procedimento è quello generalmente eseguito per analisi di tipo industriale.
Le varie metodologie analitiche strumentali possono essere raggruppate nelle seguenti categorie: a) metodi elettroanalitici; b) metodi basati sull'emissione e sull'assorbimento di radiazioni; c) metodi basati sul principio della distribuzione tra fasi (cromatografia).
2. Metodi elettroanalitici
Si chiamano ‛metodi elettroanalitici' tutti i procedimenti di analisi in cui si perviene alla determinazione di una data specie attraverso la misura di una grandezza elettrica, utilizzando idonei dispositivi. I metodi elettroanalitici includono una varietà di tecniche; i più importanti sono: i metodi potenziometrici, fondati sulla misura della forza elettromotrice di una cella galvanica; i metodi conduttimetrici, nei quali si eseguono misure della resistenza elettrica di soluzioni di elettroliti; le tecniche voltammetriche, basate sul rilevamento della variazione dell'intensità di corrente in una cella elettrolitica al variare della differenza di potenziale applicata agli elettrodi; i metodi coulombometrici, con i quali la determinazione della quantità di una sostanza viene ricavata dalla misura del numero di coulomb consumati in un opportuno processo elettrolitico. Non verranno qui descritti i metodi conduttimetrici, di minore interesse perché meno selettivi degli altri.
a) Potenziometria
Nella potenziometria si crea una cella galvanica e dalla misura della sua forza elettromotrice si risale alla concentrazione di una specie presente nella soluzione in condizioni di equilibrio.
A titolo d'esempio viene illustrato brevemente il tipo di cella usato per la determinazione della concentrazione degli ioni idrogeno in una soluzione acquosa. Com'è noto, questa grandezza indica le caratteristiche di acidità (o di basicità) di una soluzione e viene misurata correntemente in chimica e in biologia. La concentrazione viene espressa in grammomolecole per litro.
Un parametro correlabile direttamente con la concentrazione degli ioni idrogeno [H+] è il pH, definito per semplicità (anche se in modo non del tutto corretto) come
pH = − log [H+].
La cella può essere costruita ponendo nella soluzione in esame una lamina di platino spugnoso sulla quale si fa gorgogliare idrogeno puro. A questa semicella ne viene collegata un'altra contenente una soluzione di cloruro di potassio in cui si trova immerso un filo d'argento ricoperto di uno strato del composto poco solubile AgCl (cloruro d'argento). Le due semicelle vengono collegate, ad es. mediante un setto poroso, come indicato nella fig. 1.
Delle due semicelle quella con il filo d'argento assume un potenziale costante se si mantiene costante la concentrazione del cloruro di potassio: essa quindi costituisce l'‛elettrodo di riferimento'. Il potenziale dell'altra semicella è una funzione logaritmica della concentrazione degli ioni idrogeno: l'elettrodo viene quindi chiamato ‛elettrodo indicatore'.
Nella cella avviene la reazione
2AgCl + H2 ⇄ 2Ag + 2H+ + 2Cl-
e la sua forza elettromotrice a 25 °C è data dall'espressione
E = E1 − 0,059 log [H+]/(pH2)1/2,
dove E1 è il potenziale noto dell'elettrodo di riferimento e pH2 la pressione parziale dell'idrogeno espressa in atmosfere. Se tale pressione viene portata a 1 atmosfera l'espressione diventa
E = E1 + 0,059pH.
b) Elettrodi a membrana di vetro
Il classico elettrodo di platino con gorgogliamento d'idrogeno, indicato come ‛elettrodo a idrogeno', è alquanto scomodo nel suo impiego e quindi si preferisce sostituirlo per la misura del pH con l'elettrodo a membrana di vetro, che ha la forma riportata nella fig. 2A. Nel suo interno vi è un elettrodo di riferimento argento-cloruro d'argento immerso in una soluzione con acidità costante contenente cloruri. La membrana di vetro, che costituisce il bulbo dell'elettrodo, è in contatto con la soluzione esterna di cui si vuole determinare la concentrazione in ioni idrogeno. L'elettrodo descritto assume, rispetto all'elettrodo di riferimento, un potenziale che è funzione logaritmica del rapporto tra le concentrazioni in ioni idrogeno delle soluzioni esterna e interna. Siccome la soluzione interna ha un'acidità fissa, ne consegue che l'elettrodo misura l'acidità della soluzione esterna.
L'elettrodo a membrana di vetro ha una resistenza elettrica di centinaia di megaohm e quindi per la misura della forza elettromotrice della cella di cui fa parte occorre fare uso di voltmetri elettronici dotati di amplificatori, che permettono di misurare agevolmente il pH e pertanto l'acidità delle soluzioni.
c) Elettrodi ione-selettivi
Sullo stesso principio dell'elettrodo a membrana sono costruiti altri elettrodi per determinare la concentrazione di vari ioni, come ad es. rame, argento, cadmio, piombo, e anioni come cloruri, bromuri, tiocianati, ecc. Tali elettrodi hanno uno schema simile a quello dell'elettrodo a membrana di vetro: la membrana di vetro è sostituita da una membrana sensibile ottenuta con vari procedimenti. Essa può essere una lamina sottile ricavata da un macrocristallo di un composto poco solubile dello ione considerato, oppure una membrana eterogenea ottenuta incorporando una polvere di un sale poco solubile dello ione considerato in una matrice formata da una materia plastica (v. fig. 2B). In un altro metodo si ricorre all'impregnazione di un diaframma poroso con uno scambiatore di ioni liquido, immiscibile con acqua (v. fig. 2C). La funzione dello scambiatore ionico è quella di scambiare con altri ioni la specie ionica che si deve determinare. L'elettrodo dispone di un serbatoio dello scambiatore, che alimenta di scambiatore il diaframma.
Questi elettrodi sono chiamati ‛ione-selettivi': in effetti non sono strettamente specifici nei confronti di un solo ione, in quanto la loro risposta viene influenzata dalla presenza di altri ioni. Anche con queste limitazioni essi risultano assai utili in campo analitico, perché permettono di eseguire misure rapide e attendibili della concentrazione di particolari ioni in un intervallo di concentrazione da qualche g/l a circa 0,1 mg/l. Inoltre, se la cella di misura viene collegata a un opportuno circuito elettrico, si può ottenere il rilevamento automatico e continuo della concentrazione di specie ioniche nelle acque. Altri elettrodi opportunamente modificati possono essere usati per determinare nell'aria gas come l'ammoniaca, il diossido di carbonio, il solfuro di idrogeno, ecc. Sono pure stati sviluppati elettrodi selettivi capaci di rilevare composti organici, e in particolare sostanze di interesse biologico.
d) Elettrodi a enzima
Ulteriori applicazioni della potenziometria diretta sono offerte dagli elettrodi a enzima, nei quali uno o più enzimi sono accoppiati a un opportuno elettrodo selettivo. L'enzima si trova immobilizzato sulla superficie dell'elettrodo: questo dispositivo viene impiegato per misurare un prodotto o un reagente che partecipa alla reazione enzimatica. Quando la sostanza diffonde nello strato di enzima, avviene la reazione enzimatica, che è specifica, e si forma un prodotto o viene consumato un reagente, che l'elettrodo selettivo è in grado di rivelare.
A titolo d'esempio si può citare l'elettrodo a urea, che si basa sull'impiego dell'enzima ureasi; l'enzima catalizza l'idrolisi dell'urea ad ammoniaca e diossido di carbonio. L'ureasi è immobilizzata su una membrana permeabile soltanto ai gas. L'urea diffonde sull'enzima, e l'ammoniaca formata attraversa la membrana e giunge su un elettrodo interno che ne rileva la concentrazione. L'accoppiamento dell'enzima con l'elettrodo selettivo contribuisce ad aumentare la selettività di questa tecnica.
Un'estensione dell'elettrodo a enzima è l'elettrodo a batteri, in cui una colonia viva di particolari batteri è immobilizzata sulla superficie di un elettrodo sensibile a un gas, che si forma nel corso di una reazione metabolica. Il gas attraversa lo strato di batteri e raggiunge l'elemento sensibile dell'elettrodo che funge da rivelatore. Un dispositivo di questo tipo può essere impiegato come sensore specifico per vari amminoacidi.
Un'ulteriore possibilità si ha con gli elettrodi a membrana accoppiati a tessuti viventi. Un sistema di questo tipo è l'elettrodo ad ammoniaca, sul quale si deposita un tessuto vivente (ad es. una sottile fetta di rene di maiale) che agisce come strato biocatalitico nella conversione di glutammina in ammoniaca e in altri prodotti. Elettrodi di questo tipo rivestono notevole importanza, perché consentono di analizzare rapidamente sostanze di grande interesse biologico e clinico. La determinazione di tali specie con i metodi classici di analisi risulta invece molto laboriosa e meno affidabile.
e) Voltammetria, polarografia
Se una soluzione contenente sostanze ossidabili o riducibili viene sottoposta a elettrolisi, il soluto può venire ridotto al catodo, oppure può venire ossidato all'anodo. Operando in opportune condizioni, l'intensità della corrente di elettrolisi che viene misurata è correlabile con la concentrazione del soluto e può quindi consentirne la determinazione. L'applicazione di questo principio ha dato origine alle ‛tecniche voltammetriche' di analisi. Tra le tecniche di questo tipo riveste particolare importanza la ‛polarografia'. Nella cella elettrolitica in cui si trova la soluzione da analizzare sono posti due elettrodi: un elettrodo di riferimento a potenziale costante e un elettrodo indicatore. Nella tecnica polarografica quest'ultimo è il cosiddetto elettrodo a goccia di mercurio, che consta di un tubo capillare di vetro collegato tramite un tubo flessibile con un serbatoio contenente mercurio (v. fig. 3). Il mercurio riempie il capillare e sgocciola dalla sua estremità inferiore, mentre il serbatoio contribuisce a mantenere costante il livello superiore, assicurando una velocità di efflusso costante. La superficie della goccia varia continuamente nel corso della sua vita, ma è sempre molto esigua, e l'elettrodo si comporta come un microelettrodo, facilmente polarizzabile. Esso è particolarmente adatto per lo studio di processi elettrolitici di riduzione; infatti, elettrolizzando una soluzione l'acqua, che è il solvente comunemente usato, non si riduce se non per valori del potenziale catodico molto negativi, data la sovratensione dell'idrogeno sul mercurio. Inoltre molti metalli che si formano per riduzione dai loro ioni si sciolgono nel mercurio formando amalgami. Lo sgocciolamento rinnova continuamente la superficie dell'elettrodo, che non risulta alterata dai prodotti di riduzione formatisi in precedenza.
Per realizzare condizioni di elettrolisi nelle quali si possa correlare la concentrazione della specie che si riduce sul catodo con l'intensità della corrente di elettrolisi occorre che il suo trasporto in soluzione avvenga in vicinanza del catodo unicamente tramite un processo di diffusione. Occorre quindi evitare sia il trasporto per convezione sia quello per migrazione (per effetto del campo esistente tra i due elettrodi). A questo si provvede mantenendo in riposo e a temperatura costante la soluzione e aggiungendo a essa una quantità sufficientemente grande di un elettrolita, i cui ioni siano difficilmente riducibili, in modo che nella massa della soluzione il trasporto di ioni per migrazione impegni prevalentemente gli ioni dell'elettrolita aggiunto. Nella cella, dalla quale è stato preventivamente eliminato l'ossigeno disciolto, si applica tra gli elettrodi una differenza di potenziale via via crescente: mentre il potenziale dell'elettrodo di riferimento si mantiene costante, il microelettrodo di mercurio assume un potenziale sempre più negativo. All'inizio nella cella passa una corrente debolissima, che poi, a partire da un certo valore del potenziale del microelettrodo, sale rapidamente, perché una delle specie presenti si riduce. L'intensità raggiunge infine un valore di soglia (v. fig. 4: la seghettatura è dovuta alla variazione della superficie della goccia di mercurio durante la sua vita). Il tracciato che si ottiene viene indicato come ‛onda polarografica'. Se si considera la riduzione di uno ione metallico come il cadmio, che ridotto a metallo forma un amalgama, il potenziale corrispondente alla metà dell'altezza dell'onda (potenziale di semionda o di semigradino E1/2) possiede un valore caratteristico, indipendente dalla concentrazione. L'altezza dell'onda (v. fig. 4) è invece proporzionale alla concentrazione dello ione metallico, qualora vengano mantenute costanti la velocità di sgocciolamento e il tipo e la concentrazione dell'elettrolita aggiunto. Se nella soluzione sono presenti varie specie riducibili, non necessariamente ioniche, ma solubili, e se i loro potenziali di semionda sono sufficientemente discosti l'uno dall'altro, si ottengono altrettante onde, dalla cui altezza è possibile risalire alle corrispondenti concentrazioni avendo in precedenza eseguito una taratura con soluzioni a concentrazione nota.
Il metodo polarografico permette di determinare specie inorganiche e organiche in concentrazione da circa 10-2 a circa 10-5 grammomolecole per litro.
f) Polarografia a impulsi
Per poter estendere a concentrazioni più basse l'applicazione del metodo polarografico occorre eliminare correnti interferenti quale quella di natura capacitiva che si origina nella formazione di un doppio strato di cariche elettriche all'atto della formazione e dell'accrescimento di ogni nuova goccia. Applicando impulsi di potenziale di breve durata e misurando l'intensità della corrente nel breve intervallo di tempo in cui la goccia ha raggiunto la massima superficie e il processo di carica è ormai terminato, diventa possibile portare la minima concentrazione determinabile con una tecnica differenziale a valori dell'ordine di circa 10-7 moli/l (v. fig. 5; siccome si opera con una tecnica differenziale il polarogramma è rappresentato da picchi la cui altezza è proporzionale alla concentrazione). Questa e altre tecniche sono state rese possibili dal ricorso a una strumentazione elettronica avanzata.
g) Tecniche di ridissoluzione (stripping) anodica
Un metodo elettroanalitico che può permettere la determinazione di concentrazioni dell'ordine di 10-10 moli/l di ioni metallici consiste nel concentrare su una superficie molto esigua di un catodo, ad es. di grafite, un metallo per riduzione da una soluzione molto diluita di un suo ione. Questo risultato si ottiene eseguendo l'elettrolisi della soluzione in esame per un tempo sufficientemente lungo. Dopo di ciò la polarità dell'elettrodo viene invertita, in modo da farlo funzionare come anodo. Variando il potenziale applicato all'elettrodo si raggiunge un valore per il quale il metallo si ossida e passa in soluzione, dando luogo a un picco di corrente dalla cui altezza si misura la concentrazione. Il metodo non richiede che tutto il metallo presente nella soluzione da analizzare venga depositato sull'elettrodo. Infatti è possibile ricavare la concentrazione per confronto con i risultati ottenuti operando in condizioni identiche con soluzioni a concentrazione nota del metallo da analizzare.
h) Coulombometria
La coulombometria è una tecnica elettroanalitica che si basa sull'impiego delle leggi di Faraday riguardanti il processo di elettrolisi. La quantità di una determinata specie chimica presente in una cella elettrolitica e riducibile al catodo od ossidabile all'anodo può essere dedotta dal consumo di elettricità nel corso dell'elettrolisi, a condizione che la quantità di elettricità che passa nella cella sia stata esclusivamente utilizzata per il processo di riduzione (o di ossidazione) preso in esame. Siccome, adottando opportuni accorgimenti, questa condizione può essere in molti casi realizzata, ne conseguono le molteplici possibilità di applicazione del metodo.
I procedimenti coulombometrici si classificano in due grandi categorie: Procedimenti a potenziale controllato e procedimenti a intensità di corrente costante; questi ultimi vengono indicati col termine di ‛titolazioni coulombometriche'.
La coulombometria a potenziale controllato consiste in un Processo elettrolitico, che si esegue a un elettrodo di lavoro - di platino o di mercurio - il cui Potenziale è mantenuto a un valore costante e predeterminato rispetto a un elettrodo di riferimento. Poiché in queste condizioni il solo processo elettrolitico che ha luogo riguarda la specie prescelta, che viene trasformata in modo praticamente completo, si risale alla quantità della sostanza in esame con l'ausilio di un coulombometro e cioè di un dispositivo che consente di misurare la quantità di elettricità impiegata nella reazione.
Le titolazioni coulombometriche sono invece essenzialmente delle titolazioni in cui i normali strumenti di impiego analitico - buretta e soluzione titolante - sono sostituiti da una sorgente di corrente costante e da un cronometro. La specie titolante è generata agli elettrodi; dalla conoscenza dei coulomb impiegati per il completamento della reazione, risultanti dal prodotto dell'intensità della corrente in ampere per il tempo in secondi, si risale alla concentrazione di una certa specie.
Questi procedimenti, che consentono di determinare una specie con grande accuratezza anche in soluzioni diluite, costituiscono la base di molti apparecchi per titolazioni automatiche.
3. Metodi basati sull'emissione e sull'assorbimento di radiazioni elettromagnetiche
I metodi di analisi basati sull'emissione e sull'assorbimento di radiazioni, genericamente indicati come ‛metodi spettrali', sono quelli di più diffuso impiego in chimica analitica e sono ricchi di informazioni sia sotto l'aspetto qualitativo sia sotto quello quantitativo le tecniche spettrali hanno come fondamento l'interazione delle radiazioni con sistemi atomici o molecolari; le radiazioni vengono emesse o assorbite quando hanno luogo transizioni tra livelli energetici stazionari. Data l'ampiezza dello spettro elettromagnetico si possono distinguere numerose tecniche spettrali, ciascuna delle quali copre un particolare intervallo di lunghezze d'onda.

Le apparecchiature impiegate presentano caratteristiche costruttive assai diverse, conseguenti sia alle caratteristiche specifiche dell'intervallo spettrale interessato sia alle prestazioni richieste. Uno schema dei metodi spettrali più importanti è riportato nella tab. I. Oltre ai suddetti metodi vanno ancora considerate le Spettrometrie di fotoelettroni e la Spettrometria di massa, nelle quali non vengono misurate radiazioni elettromagnetiche. Le due tecniche, riportate anch'esse nella tab. I, verranno trattate nel cap. 4.
a) Fluorescenza di raggi X
Se un fascio di raggi X di lunghezza d'onda opportuna viene inviato su una sostanza allo stato solido o liquido, una frazione del fascio incidente sarà assorbita dagli atomi presenti nel campione, con l'espulsione di elettroni appartenenti ai cosiddetti ‛gusci interni' (K, L).
Come indicato nella fig. 6, la lacuna creatasi viene immediatamente colmata dalla transizione di uno degli elettroni dei ‛gusci' più esterni. In questo processo si ha emissione di energia sotto forma di raggi X secondari (radiazione di fluorescenza).
Siccome le transizioni possibili sono relativamente poche, il fascio emesso può venire scomposto in una serie non numerosa di radiazioni con lunghezza d'onda caratteristica: ciascuna di esse ha lunghezza d'onda maggiore della radiazione eccitatrice. È noto che le frequenze delle radiazioni X emesse da ogni elemento obbediscono alla legge di Moseley e sono quindi correlabili con il numero atomico dell'elemento considerato. Lo spettro di emissione di raggi X di un elemento consente di individuarlo con una certa facilità. Se il materiale in esame è costituito da più elementi, sia liberi che combinati tra loro, l'individuazione delle frequenze nello spettro di raggi X emesso permette di risalire agli elementi presenti. La misura dell'intensità delle radiazioni emesse, confrontata con quella di un elemento di riferimento, aggiunto al campione in quantità nota, permette di ricavare indicazioni sulla loro concentrazione. In realtà, nelle determinazioni quantitative delle concentrazioni si manifestano interferenze di vario tipo che complicano l'elaborazione dei dati, ma, con l'uso di un calcolatore appositamente programmato, è possibile applicare correzioni ai dati sperimentali e giungere a ottenere valori attendibili delle concentrazioni.
Gli spettrometri con i quali vengono analizzate le radiazioni X emesse possono essere di due tipi diversi: a) a dispersione angolare; b) a dispersione di energia.
Negli spettrometri a dispersione angolare la separazione delle radiazioni con diversa lunghezza d'onda viene ottenuta come negli spettrometri classici: il fascio di raggi X generati dal campione viene inviato su una lamina di un cristallo, tagliata secondo un determinato asse cristallografico (v. fig. 7). Il fascio di raggi X raggiunge il cristallo che viene fatto ruotare. Quando l'angolo tra il fascio incidente e la superficie della lamina raggiunge un valore che soddisfa la legge di Bragg per la diffrazione
nλ = 2 δ senϑ
(n = ordine di diffrazione, λ = lunghezza d'onda di una delle componenti del fascio incidente, d = distanza tra i piani reticolari del cristallo in direzione perpendicolare alla superficie del cristallo, ϑ = angolo di diffrazione), il rivelatore (un contatore proporzionale o un contatore a scintillazione), che viene fatto ruotare su un goniometro di un angolo 2ϑ, riceve il fascio diffratto. Si registra in tal modo lo spettro di emissione del campione. Nella registrazione dello spettro occorre sostituire varie lamine per coprire un intervallo sufficientemente ampio di lunghezze d'onda.
Negli spettrometri a dispersione d'energia, per separare le diverse componenti spettrali di un fascio di raggi X si ricorre all'uso di un rivelatore che rileva l'energia dei fotoni incidenti e la trasmette come segnale elettrico a un analizzatore multicanale. L'analizzatore seleziona gli impulsi ricevuti dal rivelatore e li accumula in una serie di canali, ciascuno corrispondente a un certo intervallo di energie. L'intervallo di energie di ciascun canale può essere espresso anche come intervallo di lunghezze d'onda con l'impiego della relazione di Planck. Lo spettro delle energie ha quindi il suo equivalente nello spettro delle lunghezze d'onda (tenendo conto che la lunghezza d'onda è inversamente proporzionale all'energia della radiazione). Gli spettrometri a dispersione di energia rappresentano una concezione costruttiva assai più semplice ed economica di quella degli spettrometri a dispersione angolare, essendo la parte ottica sostituita da un dispositivo elettronico molto meno costoso: l'analizzatore multicanale. Negli spettrometri a dispersione d'energia il rivelatore è del tipo a silicio-litio, capace di rispondere selettivamente a fotoni dotati di energia diversa. Esso va mantenuto alla temperatura dell'azoto liquido (−195 °C).
La risoluzione degli spettrometri a dispersione di energia è generalmente minore di quella degli strumenti a dispersione angolare, ma per molti tipi di analisi le loro prestazioni sono ampiamente sufficienti.
Gli spettrometri a raggi X di modello più avanzato consentono l'analisi di tutti gli elementi con numero atomico superiore a quello del carbonio, mentre il rilevamento degli elementi con basso numero atomico è reso meno soddisfacente dalla scarsa sensibilità dei rivelatori ai raggi X emessi. I limiti di rivelabilità degli elementi con il metodo della fluorescenza di raggi X sono dell'ordine della parte per milione nei casi più favorevoli. Un vantaggio di questa tecnica nei confronti di altri metodi strumentali è dato dal fatto che essa non richiede la preventiva dissoluzione del campione; l'eliminazione di questo trattamento è particolarmente apprezzabile nell'analisi di rocce o di leghe metalliche molto resistenti all'attacco con reagenti chimici.
Con uno spettrometro a dispersione di energia di forma molto compatta montato su una sonda spaziale è stato possibile raccogliere preziose informazioni sulla composizione chimica delle rocce di pianeti del sistema solare. I raggi X primari erano prodotti con un nuclide radioattivo e il rivelatore utilizzato non richiedeva di essere mantenuto a bassa temperatura.
b) Emissione atomica
È noto che gli atomi isolati possono trovarsi sia allo stato fondamentale che in stati elettronici eccitati. Le transizioni da uno stato a un altro, che danno luogo a liberazione di energia, causano l'emissione di radiazioni con lunghezze d'onda ben definite, cui corrispondono righe diverse nello ‛spettro di emissione'. Il numero di righe presenti nello spettro aumenta fortemente con l'aumento del numero atomico. Ogni elemento emette uno spettro caratteristico nella regione dall'ultravioletto al visibile; pertanto se un campione di una sostanza viene portato allo stato di vapore atomico ed eccitato, lo spettro di emissione consente di individuare la maggior parte degli elementi presenti. L'identificazione si fa eseguendo la ricerca delle righe spettrali che per la loro intensità possono essere utilizzate per caratterizzare i singoli elementi. Il metodo è particolarmente adatto per riconoscere gli elementi metallici, le cui righe spettrali più caratteristiche si collocano nella regione spettrale superiore ai 200 nm, che può essere facilmente esplorata con gli spettrometri a emissione.
Negli spettrometri a emissione di tipo classico la sorgente è costituita da un generatore di scintille o da un arco elettrico entro il quale si immette il campione solido o in soluzione. In alternativa si può fare uso di una fiamma in cui il campione in soluzione viene introdotto in forma nebulizzata. Il fascio viene inviato su uno o più elementi disperdenti (prismi e/o reticoli) e focalizzato su una lastra fotografica che registra simultaneamente tutta la regione spettrale desiderata. In alternativa il dispositivo disperdente è un monocromatore con il quale possono essere isolate le singole righe spettrali. Nella sorgente il campione viene portato ad alta temperatura, volatilizzato e dissociato in atomi.
Nelle sorgenti che sono state menzionate la maggior parte degli atomi si trova allo stato fondamentale; solo una frazione degli atomi presenti viene eccitata ed è quindi in grado di emettere. L'intensità delle righe emesse è proporzionale alla quantità di atomi presenti, se si opera in condizioni di eccitazione perfettamente riproducibili. È quindi possibile utilizzare la spettrometria di emissione per la determinazione quantitativa di molti elementi. Siccome le condizioni di eccitazione non sono facilmente riproducibili, si confronta l'intensità di una riga spettrale dell'elemento da analizzare con quella di una riga di un elemento aggiunto al campione in proporzione nota. Quest'ultima riga serve come riferimento. Le due righe vanno scelte opportunamente, in modo che il rapporto tra le loro intensità sia praticamente indipendente dalle condizioni di eccitazione. Il confronto tra le intensità della riga dell'elemento del campione e della riga di riferimento viene fatto raccogliendone le radiazioni su tubi fotomoltiplicatori che le convertono in segnali elettrici. Lo spettrometro, precedentemente tarato con campioni di composizione nota, elabora i dati e fornisce il risultato.
Molti spettrometri per l'emissione atomica sono oggi dotati della cosiddetta ‛sorgente a plasma con accoppiamento induttivo' (spettrometri ICP). Si tratta di una torcia in cui viene immesso argo che viene ionizzato e sottoposto a un campo magnetico oscillante. Si genera in tal modo un plasma che raggiunge una temperatura intorno ai 10.000 °C. La soluzione del campione viene nebulizzata in una corrente d'argo che la convoglia nel plasma. In queste condizioni gli elementi presenti vengono portati allo stato atomico eccitato o allo stato ionico. Si realizza così una emissione più intensa di quella ottenibile con le sorgenti menzionate in precedenza, e quindi si raggiungono limiti di rivelabilità dell'ordine delle parti per miliardo.
c) Fluorescenza nel visibile e nell'ultravioletto
Molti composti, e anche varie specie ioniche inorganiche, se esposti a radiazioni ultraviolette, diventano capaci di emettere nella regione visibile. Nell'interazione tra le radiazioni ultraviolette e la materia si può avere la transizione delle molecole dallo stato fondamentale a stati elettronicamente e vibrazionalmente eccitati. Il ritorno allo stato fondamentale può avvenire in modi diversi, e in particolare mediante dissipazione termica, ossia per collisione delle molecole eccitate con altre molecole presenti, oppure con emissione di radiazioni luminose da parte delle molecole eccitate. Spesso l'energia associata agli stati vibrazionali viene dissipata mediante processi di rilassamento e solo l'energia elettronica viene emessa sotto forma di radiazioni. Ne consegue che la radiazione emessa ha lunghezza d'onda maggiore (ed energia minore) della radiazione eccitatrice.
La proprietà di emettere radiazioni di fluorescenza è caratteristica di certe molecole, in particolare di quelle che presentano uno scheletro rigido, come certi idrocarburi aromatici a nuclei condensati e molte altre sostanze. Ogni sostanza fluorescente emette le radiazioni in corrispondenza di certe regioni spettrali caratteristiche. Dalla misura dell'intensità della radiazione emessa da una soluzione di una sostanza fluorescente è possibile risalire alla sua concentrazione, essendovi una relazione lineare tra l'una e l'altra in un determinato intervallo di concentrazioni. Molte sostanze di notevole interesse biologico o farmacologico (ad es. alcaloidi come la chinina, vitamine come la riboflavina, ecc.) presentano una intensa fluorescenza, cosicché possono essere determinate con metodi fluorimetrici già a concentrazioni dell'ordine di parti per miliardo.
I fluorimetri impiegati in chimica analitica possono essere strumenti molto semplici, nei quali sia la radiazione eccitatrice che la radiazione di fluorescenza vengono selezionate con semplici filtri, oppure spettrofotofluorimetri dotati di un monocromatore per l'eccitazione e un altro per la radiazione emessa.
Questi ultimi possono essere costruiti in modo da consentire la registrazione sia degli spettri di eccitazione sia di quelli di fluorescenza, che, come si è detto, possono essere utilizzati per caratterizzare molti composti.
d) Assorbimento atomico
Se un vapore formato prevalentemente da atomi di un elemento allo stato fondamentale viene irradiato con un fascio monocromatico la cui energia corrisponda alla differenza di energia richiesta per l'eccitazione degli atomi, la radiazione viene assorbita. L'entità dell'assorbimento è proporzionale al numero di atomi presenti nello stato fondamentale. Se I0 è l'intensità del fascio monocromatico incidente e I l'intensità dopo che esso ha attraversato uno strato di vapore atomico di spessore S, è valida la relazione
A = log (I0/I) = KCS,
dove C è la concentrazione di atomi per unità di volume e K una costante di proporzionalità che può venire esplicitata. La grandezza A viene chiamata assorbanza. L'intensità della radiazione incidente viene modulata per distinguerla dalla radiazione emessa dagli atomi eccitati sempre presenti nel vapore atomico. Il rivelatore, che è un tubo fotomoltiplicatore, raccoglie la radiazione e la converte in una corrente. Solo la corrente modulata viene amplificata. Per produrre il vapore atomico si immette la soluzione del campione, nebulizzata, in una fiamma capace di portare il campione a una temperatura sufficientemente alta da dissociare in atomi i composti presenti. In alternativa si può impiegare un fornetto tubolare di grafite nel quale si introduce un piccolo volume di soluzione o anche il campione solido. Il riscaldamento viene effettuato facendo passare attraverso la grafite una corrente elettrica e programmando opportunamente l'incremento di temperatura. Si opera in atmosfera di argo, per evitare la formazione di ossidi difficilmente dissociabili. La radiazione monocromatica incidente proviene da una lampada spettrale capace di emettere lo spettro dell'elemento sottoposto ad analisi. Un monocromatore isola la riga spettrale desiderata.
La spettrometria di assorbimento atomico è particolarmente adatta per la determinazione dei metalli. I limiti di rivelabilità sono particolarmente bassi con la maggior parte dei metalli, perché nel vapore atomico presente nella fiamma o nel fornetto di grafite gli atomi si trovano prevalentemente nello stato fondamentale e sono quindi in grado di assorbire la radiazione incidente. I dati sono meglio riproducibili facendo uso della fiamma, mentre con il fornetto si ottengono limiti di rivelabilità più bassi, inferiori alla parte per miliardo e per molti elementi dell'ordine di parti per trilione.
e) Assorbimento nel visibile e nell'ultravioletto
Nella sezione dedicata alla fluorescenza nel visibile e nell'ultravioletto sono già stati esposti i processi di diseccitazione delle molecole. Le sostanze capaci di assorbire radiazioni nel visibile o nell'ultravioletto sono assai numerose. Alcune di esse si diseccitano dando luogo alla fluorescenza, mentre la maggior parte dissipa l'energia assorbita sotto forma di calore. Le misure di assorbimento vengono eseguite con uno spettrofotometro capace di operare nella regione spettrale compresa tra circa 195 e 900 nm. Gli strumenti di questo tipo sono generalmente costituiti da una sorgente, un monocromatore, un rivelatore e un dispositivo di registrazione e lettura, collegato con un amplificatore. La sorgente emette una radiazione policromatica. Il monocromatore isola le radiazioni comprese in un piccolo intervallo Δλ di lunghezze d'onda (spesso 0,2 nm) intorno a un certo valore λ. L'intervallo Δλ nell'intorno di λ viene detto ‛banda passante'. Negli strumenti a doppio raggio il pennello di radiazioni isolato dal monocromatore viene fatto passare alternativamente attraverso una vaschetta contenente il solvente e una vaschetta con la soluzione in esame. I due segnali vengono raccolti dal rivelatore, il quale li trasforma in segnali elettrici che vengono elaborati e presentati come trasmittanza T = (I/I0) - dove I è l'intensità della radiazione trasmessa dalla soluzione e I0 quella trasmessa dal solvente - oppure come assorbanza A = log (I0/I). Quest'ultima grandezza è correlata alla concentrazione dalla relazione di Lambert-Beer
A = εcs,
dove c è la concentrazione, solitamente espressa in grammomolecole per litro, ed s lo spessore dello strato di soluzione, espresso in cm. ε viene detta assorbività molare, e rappresenta il reciproco dello spessore di soluzione contenente i grammomolecola/l, che dà luogo a un'assorbanza unitaria. La relazione è applicabile generalmente in un intervallo di concentrazioni di circa due ordini di grandezza (ad es. da 0,1 a 60 parti per milione).
Gli spettrofotometri costruiti attualmente, previa taratura con soluzioni a concentrazione nota, elaborano i dati e li presentano espressi come concentrazione. Uno spettrofotometro fornisce anche la registrazione dello spettro di assorbimento (v. fig. 8). Il monocromatore è dotato di un motore che fa variare la lunghezza d'onda del fascio inviato sulle vaschette di misura, in modo da ottenere la trasmittanza o l'assorbanza al variare della lunghezza d'onda nell'intervallo spettrale desiderato.
f) Assorbimento nell'infrarosso
Mentre l'assorbimento della luce nel visibile e nell'ultravioletto è dovuto a transizioni elettroniche, le radiazioni infrarosse (IR), dotate di minore energia, possono causare transizioni negli stati vibrazionali delle molecole bi- e poliatomiche.
Le vibrazioni dalle quali sono animate le molecole possono venire scomposte in vari tipi di moti: di variazione della distanza tra due atomi (stiramento), di variazione dell'angolo fra tre atomi (deformazione), ecc. Pertanto se si espone un campione di un composto a un fascio di radiazioni IR di lunghezza d'onda opportuna, si può avere un assorbimento più o meno grande. In particolare si rileva che negli spettri di assorbimento compaiono bande che possono essere attribuite a transizioni vibrazionali dovute alla presenza di aggruppamenti atomici, quali −C=O, −O−H, −CH3, ecc.
L'intervallo di lunghezze d'onda in cui si collocano le suddette bande è generalmente poco influenzato dalla struttura della restante parte della molecola. Così, ad es., il gruppo −O−H dà luogo ad assorbimento da circa 2,77 a 2,70 μm. Uno stesso gruppo atomico può dare origine a più di una banda caratteristica. Ne consegue che dall'esame dello spettro IR di un composto è possibile desumere l'esistenza nella sua molecola di una serie di aggruppamenti atomici e quindi ricostruire, almeno in parte, la sua struttura molecolare. Si comprende quindi quale possa essere l'utilità di uno strumento quale lo spettrofotometro IR. La regione spettrale più importante per informazioni del tipo appena descritto è quella tra circa 6,6 e 14 μm.
Gli spettrofotometri IR esistenti nei laboratori chimici sono essenzialmente ispirati a due concetti costruttivi diversi. Nei modelli del tipo che chiameremo classico il fascio policromatico proveniente dalla sorgente viene disperso da un monocromatore che isola una stretta banda passante. Il pennello così selezionato giunge al rivelatore. Il campione da analizzare viene interposto tra la sorgente e il monocromatore. Gli strumenti sono del tipo a doppio raggio e lo spettro viene solitamente registrato come trasmittanza in funzione della lunghezza d'onda.
Gli spettrofotometri IR a trasformate di Fourier (detti più brevemente spettrofotometri IR-FT), di costruzione più recente, adottano uno schema costruttivo completamente diverso. Il fascio policromatico che ha attraversato il campione raggiunge un interferometro del tipo Michelson e dà luogo a un interferogramma che viene raccolto dal rivelatore ed elaborato da un calcolatore. L'interferogramma contiene tutte le informazioni relative alle radiazioni di diversa lunghezza d'onda prodotte dalla sorgente e assorbite in varia misura dal campione. Anche il fascio policromatico emesso dalla sorgente dà un interferogramma non modificato dall'assorbimento del campione. Il calcolatore elabora i dati facendo uso delle trasformate di Fourier e li presenta registrati come spettro di assorbimento. Gli spettrofotometri IR-FT offrono importanti vantaggi rispetto agli strumenti classici: siccome la radiazione incidente non viene scomposta in una serie di bande passanti, ciascuna di debole intensità, lo spettro ottenuto è maggiormente affidabile; inoltre, nel tempo richiesto da uno spettrofotometro del tipo classico per la registrazione dello spettro, lo strumento FT può accumulare un grande numero di dati relativi a molti interferogrammi. In questo modo i dati spuri dovuti agli errori casuali di misura vengono mediati, con il risultato che il rapporto segnale-fondo è assai migliore di quello ottenibile con uno strumento del tipo classico. Per questi e altri motivi gli spettrofotometri IR a trasformate di Fourier hanno ormai sostituito nei laboratori gli strumenti dell'altro tipo.
Il campione viene di solito esaminato allo stato puro; se si tratta di un solido, esso viene miscelato con bromuro di sodio (una sostanza trasparente all'IR nella regione spettrale già menzionata) e polverizzato finemente, in modo da mescolare intimamente le due sostanze. La polvere viene poi compressa, in modo da formare un dischetto di spessore molto esiguo.
In chimica analitica la spettrofotometria IR viene usata prevalentemente per l'individuazione di sostanze preventivamente separate da eventuali miscele; gli impieghi per determinazioni quantitative sono assai più limitati. La diffusione degli strumenti del tipo FT, che forniscono valori di trasmittanza assai più attendibili degli strumenti classici, fa prevedere un'estensione delle applicazioni all'analisi quantitativa.
g) Risonanza magnetica nucleare
Alcuni nuclidi, e tra di essi l'1H e il 13C, presentano la caratteristica per cui i loro nuclei possiedono un momento angolare di spin. Se un insieme di nuclei di 1H (ossia di protoni) venisse messo in un campo magnetico uniforme, essi potrebbero assumere due orientazioni di spin quasi ugualmente popolate a temperatura ambiente, dato che la differenza di energia tra i due stati è dell'ordine di 10-26 J, minore dell'energia termica a 25 °C (kT = 4,11 × 10-21 J).
Con l'impiego di un campo magnetico rotante (che può essere sostituito da una radiofrequenza opportuna) è possibile provocare la transizione di spin dal livello di energia inferiore a quello superiore. Le due popolazioni tendono quindi a eguagliarsi, ma esistono processi di rilassamento che tendono a riportare il sistema nelle condizioni iniziali.
Se, anziché considerare un insieme di nuclei di 1H, consideriamo un insieme di molecole contenenti atomi di 1H, il campo magnetico effettivo esercitato sui protoni risulta inferiore a quello applicato a causa dell'atmosfera elettronica che circonda i protoni, a sua volta influenzata dagli atomi degli altri elementi che costituiscono la molecola. In conseguenza di questi effetti la frequenza alla quale i protoni entrano in risonanza risulta diversa da quella calcolabile teoricamente; la differenza viene chiamata ‛spostamento chimico' (chemical shift). Se una molecola di un composto contiene vari atomi di 1H, ciascuno con diverso intorno chimico, siccome essi non sono equivalenti, la risonanza dei loro nuclei ha luogo a frequenze diverse.
Per misurare gli spostamenti chimici si usa uno spettrometro a risonanza magnetica nucleare. Esso è costituito da un magnete capace di creare un campo molto uniforme, variabile entro un piccolo intervallo intorno al valore richiesto per la risonanza, e dal campo magnetico rotante, oltre che da un dispositivo di rilevamento. Facendo variare il campo magnetico si possono registrare le frequenze alle quali i nuclei di idrogeno con diverso intorno chimico entrano in risonanza.
Lo studio teorico e il confronto empirico degli spettri RMN (o NMR) di un gran numero di composti ha permesso di stabilire relazioni tra la struttura molecolare del composto e la forma degli spettri. La spettrometria RMN permette quindi di ricavare preziose informazioni sulla struttura di un composto, preventivamente separato da altri eventualmente presenti. Le informazioni possono completare quelle che si ottengono con la spettrometria IR.
Nella fig. 9, A e B, è riportato lo spettro RMN protonico dell'etanolo CH3−CH2−OH rispettivamente a bassa e ad alta risoluzione. Nello spettro a bassa risoluzione i tre picchi possono essere assegnati agli aggruppamenti −OH, −CH2− e −CH3. Le aree dei picchi stanno tra loro come 1 : 2 : 3, ossia sono in relazione con i numeri di atomi d'idrogeno dei tre gruppi. Lo spettro ad alta risoluzione è più complesso perché ciascuno dei picchi risulta a sua volta scomposto in una serie di picchi, la cui presenza trova una razionale spiegazione se si tiene conto delle molteplici interazioni esistenti entro la molecola.
L'impiego della spettrometria di risonanza magnetica nucleare si è molto sviluppato nei laboratori chimici per l'ausilio che essa offre nello studio di nuovi composti, nell'identificazione di specie già note, nell'indagine su meccanismi di reazione. Esistono anche applicazioni della spettroscopia RMN all'analisi quantitativa (per es. per la determinazione rapida dell'acqua contenuta in un campione); si tratta comunque di applicazioni limitate.
Come si è detto, gli spettrometri RMN sono destinati a rilevare processi di risonanza che interessano due popolazioni assai poco diverse, e quindi assorbimenti di energia assai deboli. Si tratta pertanto di strumenti assai sofisticati. Se si passa dalla risonanza protonica, la più comunemente studiata, a quella del nuclide 13C, assai meno abbondante in natura del nuclide 12C, il cui nucleo non possiede momento di spin, il problema delle prestazioni richieste allo spettrometro diventa ancora più difficile. Per ottenere segnali affidabili, senza dover ricorrere all'arricchimento in 13C dei composti in studio, si deve fare uso di una tecnica basata sull'invio al campione, posto nel campo magnetico, di impulsi molto brevi di segnali a radiofrequenza dotati di notevole intensità. Si raccoglie un segnale in forma di un'onda via via smorzata. Si accumulano in un tempo relativamente breve moltissimi segnali di questo tipo, che vengono poi elaborati con l'uso delle trasformate di Fourier. Siccome l'accumulo dei dati permette di migliorare il rapporto tra il segnale e il fondo, diventa possibile registrare spettri affidabili delle sostanze in studio.
(Per maggiori particolari v. risonanza magnetica nucleare: Applicazioni chimiche, suppl.).
4. Altri metodi spettrometrici
a) Spettrometrie di fotoelettroni
Nel cap. 3, È a si è detto che l'emissione della radiazione di fluorescenza da parte di un atomo avviene in conseguenza dell'espulsione di un elettrone da un ‛guscio interno' dell'atomo considerato. Se si misura l'energia cinetica acquistata dall'elettrone espulso e l'energia del fotone incidente, si può ricavare l'energia di interazione tra il nucleo e l'elettrone considerato. Questo dato può permettere di identificare il tipo di elemento coinvolto nel processo. Ovviamente in un'esperienza di questo tipo possono venire espulsi elettroni di gusci diversi, e pertanto si otterrà una certa distribuzione delle energie cinetiche degli elettroni espulsi, ossia quello che può essere chiamato uno spettro di fotoelettroni. stato possibile mettere a punto strumenti per misurare l'energia degli elettroni espulsi: questa tecnica, che selve principalmente per individuare gli elementi presenti nel campione (ma che permette anche di ottenere indicazioni di grande utilità sugli elettroni impegnati nei legami di un particolare atomo con altri atomi), viene chiamata ESCA (Electron Spectroscopy for Chemical Analysis). L'emissione di elettroni può anche essere ottenuta con radiazioni meno energetiche dei raggi X, in modo da provocare l'espulsione di elettroni di valenza anziché di elettroni di gusci interni. Le radiazioni impiegate sono quelle della regione dell'ultravioletto lontano, con lunghezza d'onda inferiore a 180 nm. Siccome tali radiazioni vengono assorbite dall'aria, occorre eseguire le misure nel vuoto. Questa tecnica viene detta PES (PhotoElectron Spectroscopy). Entrambi i metodi sono particolarmente adatti per lo studio della superficie dei solidi, dato che la radiazione incidente, in particolare le radiazioni ultraviolette, penetra in uno strato di debole spessore. Siccome si presenta frequentemente la necessità di conoscere esattamente la composizione chimica del materiale esistente alla superficie di un solido, il metodo presenta notevole interesse dal punto di vista della chimica analitica. Spesso la composizione della superficie è diversa da quella della parte interna del solido, perché a contatto con gli agenti atmosferici essa si modifica. Lo studio dell'adesione di uno strato protettivo alla superficie di un solido può essere compiuto con questo metodo. Altre modificazioni superficiali che possono venire studiate con questa tecnica sono quelle che avvengono nei processi fotografici e in genere quelle che riguardano la riproduzione di immagini.
b) Spettrometria di massa
La spettrometria di massa è una tecnica analitica per la caratterizzazione di specie molecolari, basata sull'identificazione dei frammenti ionici che da esse si generano sotto l'azione di elettroni ad alta energia.
Nello spettrometro di massa le molecole del composto in esame, in quantità che può variare da pochi milligrammi a un nanogrammo, sono portate allo stato gassoso e sono sottoposte a bombardamento da parte di un fascio di elettroni, che dà luogo alla formazione di ioni positivi, ioni negativi e specie radicaliche. La semplice asportazione di un elettrone dalla molecola genera un radicale-ione, lo ‛ione molecolare', che possiede una massa eguale a quella della molecola. Si ha contemporaneamente la frammentazione della molecola con formazione di varie specie ioniche di cui viene misurato il rapporto massa/carica. Gli ioni possono essere rivelati con lo spettrometro se la loro vita media è superiore a un valore minimo, che è dell'ordine di 10-5 s. Se la loro stabilità è minore, essi possono subire sia ulteriori processi di frammentazione spontanea sia processi di associazione dovuti a collisione con radicali.
Dopo la frammentazione gli ioni positivi vengono accelerati sotto l'azione di un campo elettrico e separati in base al rapporto massa/carica con dispositivi che variano secondo il tipo di strumento impiegato.
Gli ioni, dopo il processo di separazione, raggiungono il rivelatore, che dà origine a una corrente ionica proporzionale al numero di cariche elettriche che essi portano.
Vi sono vari tipi di spettrometri (a focalizzazione semplice e doppia, cicloidali, a tempo di volo e a quadrupolo).
Il principio su cui è basata la separazione mediante uno spettrometro a focalizzazione semplice, schematizzato nella fig. 10, è il seguente. Il campione in esame, portato in fase gassosa, è immesso alla pressione di circa i torr nella camera di ionizzazione; in essa un filamento riscaldato a circa 2.000 °C genera un fascio di elettroni che, accelerati da un elettrodo positivo, attraversano una fenditura e bombardano le molecole presenti. Un campo elettrico accelera gli ioni positivi, i quali passano nella camera di separazione attraverso alcune fenditure che ne isolano uno stretto fascio. L'energia cinetica dei frammenti di massa m e carica e è uguale alla forza elettrica che agisce su di essi. Il campo magnetico, che agisce normalmente alla direzione del moto dei frammenti, determina la loro deflessione secondo traiettorie circolari in cui la forza centripeta dovuta al campo magnetico è bilanciata dalla forza centrifuga dovuta all'energia cinetica. Ne consegue che, per ioni che hanno la stessa carica, la massa è direttamente proporzionale al quadrato del raggio di curvatura. Per un dato valore di tensione di accelerazione, frammenti con una certa massa passeranno attraverso il tubo analizzatore e attraverso la fenditura e saranno raccolti su una placca collettrice. Si registrerà un flusso di ioni di questa massa, e cioè una corrente ionica che può essere amplificata e registrata. Facendo variare la tensione acceleratrice possono essere focalizzati ioni corrispondenti a frammenti di differente massa ed è in tale modo possibile ottenere il relativo spettro di massa.
Lo spettro di massa è in genere rappresentato come nella fig. 11, cioè con una serie di segmenti di altezza proporzionale alle abbondanze relative. In ascissa sono riportati i numeri di massa, e cioè i numeri interi più prossimi alla massa delle specie ioniche. Le altezze sono riferite a quelle del picco più intenso, che viene detto ‛picco base'.
L'identificazione di una data specie può essere eseguita mediante l'esame dello spettro di massa. Il peso molecolare può essere immediatamente determinato se è presente lo ione molecolare e cioè lo ione positivo risultante dalla cessione di un elettrone da parte della molecola neutra, senza frammentazione.
L'esperienza ottenuta esaminando gli spettri di massa di un gran numero di composti organici ha permesso di formulare una serie di regole di frammentazione delle molecole, cosicché dall'esame della serie di frammenti formati è possibile risalire alla struttura molecolare del composto in esame.
A titolo di esempio viene mostrato nella fig. 11 lo spettro di massa di un composto la cui massa molecolare è risultata 226 e al quale è stata attribuita la formula del 5-metilpentadecano in conseguenza dell'esame dei frammenti i cui valori sono riportati nella figura.
5. Metodi basati sulla distribuzione tra fasi
a) Generalità sulla cromatografia
Da lungo tempo il processo di estrazione, che si realizza dibattendo una soluzione con un solvente non miscibile con essa, è stato utilizzato per isolare un dato composto o un gruppo di composti dalla soluzione in esame. Tale processo si basa sul principio di ripartizione secondo cui una specie si distribuisce fra due fasi in modo da rendere costante il rapporto delle sue concentrazioni nelle due fasi. Pertanto per il soluto A che si distribuisce fra un solvenie organico e una fase acquosa si ha [A]0/[A]aq = KD, in cui [A]0 e [A]aq sono le concentrazioni nelle due fasi e KD è il coefficiente di ripartizione. Per effettuare l'isolamento di una data specie da una soluzione o da un sistema solido l'estrazione può essere eseguita in modo discontinuo, in modo continuo o in controcorrente; con quest'ultimo termine si indica il processo estrattivo in cui due solventi immiscibili vengono messi in contatto mentre fluiscono in direzione opposta.
Prendiamo in esame un processo in controcorrente discontinuo, in cui una quantità costante di solvente puro (fase stazionaria) viene messa in contatto con un ugual volume di un altro solvente (fase mobile); se questo solvente viene successivamente trasferito in recipienti contenenti la fase stazionaria e dibattuto con essa, si osserva che una qualunque specie si ripartisce nelle due fasi secondo una distribuzione gaussiana, governata dal valore di KD.
Il processo di ripartizione, e la conseguente distribuzione di una specie fra due solventi non miscibili, rappresenta uno dei numerosi metodi di distribuzione tra fasi diverse (come ad es. solido-liquido) ossia dei ‛metodi cromatografici'. Il termine ‛cromatografia' è stato introdotto dal botanico russo M. Tswett all'inizio di questo secolo per descrivere la separazione dei pigmenti delle foglie verdi ottenuta facendo percolare etere di petrolio su di una colonna impaccata con carbonato di calcio in polvere alla cui estremità superiore era posto un estratto di foglie. Con il procedere dell'esperimento, dalla banda colorata iniziale si formavano, lungo la colonna, bande separate di colore verde e aranciato dovute ai diversi costituenti del pigmento (clorofille e carotenoidi). In tutti i processi cromatografici si utilizza lo stesso principio e cioè la diversa velocità con cui i differenti componenti di una miscela migrano in una fase stazionaria sotto l'influenza di una fase mobile, la quale ha il compito di trascinare i componenti della miscela. La differente velocità è determinata dagli equilibri cui prendono parte le specie che debbono essere separate, e che dipendono dalla loro interazione con la fase stazionaria. Durante un processo cromatografico le molecole delle specie che debbono essere separate e analizzate passano dalla fase stazionaria a quella mobile e da questa nuovamente nella fase stazionaria, e tale trasferimento si ripete molte volte secondo i vari dispositivi sperimentali. Di conseguenza le molecole, allorché si trovano nella fase mobile, procedono nella direzione del flusso della fase mobile, mentre sono praticamente ferme durante il tempo in cui permangono nella fase stazionaria. La velocità di migrazione di ciascun soluto è determinata dal rapporto tra i tempi che esso trascorre nelle due fasi e quindi dal suo rapporto di distribuzione.
I processi che intervengono nel trasferimento da una fase mobile a una stazionaria sono quelli di ‛adsorbimento', ‛ripartizione', ‛scambio ionico' ed ‛esclusione'. Qualunque sia la natura dell'interazione fra i soluti di una miscela da analizzare e la fase stazionaria, il processo cromatografico che determina il loro frazionamento e la conseguente separazione dei costituenti è quello schematizzato nella fig. 12. Una miscela costituita da tre componenti, A, B e C, indicati rispettivamente con piccoli cerchi, quadrati e triangoli, è posta su di un supporto (I stadio); allorché la fase mobile la investe si ha una parziale separazione di questi costituenti (II stadio). Il frazionamento è più accentuato nel III stadio, mentre nel IV la separazione è pressoché completa, come risulta dal tracciato finale, che riporta la concentrazione delle tre specie nel sistema in cui è avvenuto il frazionamento.
La cromatografia costituisce il procedimento più efficace per la separazione dei costituenti di una miscela e per la determinazione di una qualunque specie; allorché questa è isolata, si può procedere alla sua identificazione e alla sua valutazione quantitativa.
Le tecniche cromatografiche si eseguono con apparecchiature e modalità differenti a seconda della natura della fase mobile impiegata, che può essere un gas o un liquido, e a seconda della natura della fase stazionaria, che può essere un liquido o un solido con particolari proprietà. I vari procedimenti sono schematizzati nella tab. II. Verranno ora presentate le tecniche di maggiore interesse.

b) Gascromatografia
L'analisi cromatografica che si realizza usando come fase mobile un gas viene denominata gascromatografia. La fase stazionaria può essere un solido o un liquido non volatile trattenuto da un supporto inerte. Nel primo caso si ha la cromatografia gas-solido (GSC), nel secondo la cromatografia gas-liquido (GLC). Gli schemi a blocchi e funzionale di un sistema cromatografico in fase gassosa sono riportati nella fig. 13. La fase mobile, detta anche gas di trasporto o gas vettore, costituita da un gas inerte quale elio, azoto o idrogeno, è fatta fluire con velocità costante attraverso una colonna cromatografica (il dispositivo in cui avviene il frazionamento della miscela in esame) mantenuta a temperatura costante per mezzo di una camera termostatica. Mediante un opportuno rivelatore - indicato con E nella fig. 13 - che fornisce di solito un responso differenziale misurando una proprietà del gas di eluizione prima e dopo il passaggio nella colonna, è possibile mettere in evidenza i singoli componenti della miscela in esame, che sono stati frazionati dalla colonna e che fuoriescono da essa in tempi successivi. Il responso del rivelatore è amplificato e inviato a un registratore da cui si ottiene il relativo tracciato, indicato come ‛gascromatogramma'.
Il campione da analizzare, costituito da sostanze gassose o volatili e termicamente stabili alla temperatura a cui si opera, viene iniettato a monte della colonna attraverso un setto di gomma mediante una siringa ipodermica. I gas e i vapori formatisi vengono trascinati dalla fase mobile, che fluisce lungo la colonna cromatografica. La separazione dei vari costituenti ha luogo in quanto essi si ripartiscono in modo differente tra la fase gassosa e la fase stazionaria.
La colonna cromatografica può essere di due tipi: impaccata o capillare. Quella del primo tipo è costituita da un tubo di vetro o di metallo, di 2-5 mm di diametro e di lunghezza variabile (in genere 1-10 m), che viene riempito con polvere di materiale solido inerte i cui granuli sono ricoperti da un sottile velo di liquido che costituisce la fase stazionaria e che è caratterizzato da una tensione di vapore molto bassa. Il materiale di riempimento si realizza mescolando la polvere con una soluzione del liquido desiderato disciolto in un solvente volatile e facendo poi evaporare il solvente. Una colonna capillare è costituita da un tubo di vetro o di silice fusa di diametro interno pari a 0,2-0,5 mm e di notevole lunghezza (20-100 m), sulla cui parete interna la fase stazionaria forma un velo liquido uniforme.
Entrambi i tipi di colonne forniscono prestazioni eccezionali, permettendo di separare miscele di grande complessità, sia naturali che artificiali. Tale fine è raggiunto sfruttando le forze intermolecolari fra un soluto e un solvente, quali quelle di dispersione, di induzione, di orientazione e le interazioni donatore-accettore comprendenti anche il legame a idrogeno.
Nell'analisi di specie non polari, come gli idrocarburi, le forze di dispersione costituiscono le uniche forze di interazione e conseguentemente i vari componenti fuoriescono dalla colonna in funzione della loro temperatura di ebollizione; per le specie polari, invece, le altre forze, che dipendono essenzialmente da polarizzabilità, potenziale di ionizzazione e momento dipolare, hanno un ruolo determinante e di conseguenza si possono avere fasi stazionarie dotate di selettività eccezionale.
L'efficienza di una colonna cromatografica è espressa in funzione del numero dei ‛piatti', valore che si ricava direttamente dal tracciato cromatografico: tale termine è derivato dalla terminologia impiegata per le colonne di distillazione; è possibile attualmente realizzare colonne con varie decine di migliaia di piatti. Il miglioramento tecnologico delle colonne è stato ottenuto con l'impiego di fasi liquide legate, in cui la fase stazionaria viene legata chimicamente al supporto mediante una opportuna reazione, e, per quanto riguarda le colonne capillari, con l'uso di fasi stazionarie immobilizzate ossia fissate alla colonna di vetro e di silice fusa mediante opportuni procedimenti. Ogni specie della miscela da separare viene identificata allorché fuoriesce dalla colonna percorsa da un flusso costante della fase mobile, utilizzando una varietà di rivelatori. La loro funzione è quella di fornire un'indicazione delle variazioni di composizione del gas vettore e tale fine viene raggiunto attraverso la misura di un parametro elettrico come la termoconducibilità, la ionizzazione, la termoionizzazione, la fotoionizzazione o la cattura di elettroni. Il responso del rivelatore è inviato a un registratore dove la fuoriuscita di una data specie dalla colonna cromatografica dà luogo a un picco. Esso viene tracciato in funzione del tempo che intercorre tra l'iniezione del campione e la fuoriuscita della specie ricercata; tale tempo viene denominato ‛tempo di ritenzione'. Il suo valore è specifico per una data specie in determinate condizioni di lavoro e consente di identificarla; poiché l'area del picco cromatografico (v. fig. 14) è proporzionale alla concentrazione della specie chimica, questa tecnica è ampiamente impiegata per la determinazione qualitativa e quantitativa di un dato composto.
La cromatografia consente di ricavare eccezionali informazioni sulla composizione di tutti i sistemi naturali o sintetici costituiti da specie gassose o vaporizzabili e, in virtù delle possibilità di frazionamento delle colonne cromatografiche e della sensibilità dei sistemi di rivelazione, grazie ai quali si possono rivelare quantità pari a 10-12 ÷ 10-13 g, è stato possibile risalire alla natura di sistemi complessi. A titolo di esempio è riportato nella fig. 14 il gascromatogramma di un olio essenziale di limone, in cui si evidenzia dal tracciato cromatografico che in questo sistema è presente un gran numero di costituenti, dell'ordine di qualche centinaio. Non v'è sistema naturale o artificiale sul quale la gascromatografia non abbia consentito di ricavare informazioni, e in particolare sui componenti minori, che possono influire in maniera determinante sulle caratteristiche del sistema.
La GC non può essere impiegata per composti non volatili o termicamente instabili, a meno che non si ricorra alla loro conversione in prodotti volatili. Può essere però impiegata anche per identificare composti polimerici attraverso i loro prodotti di pirolisi, e proprio a tale scopo viene impiegata nelle industrie di materie plastiche, della gomma e nella petrolchimica.
Uno dei problemi connessi all'impiego della GC è quello relativo all'identificazione di una specie che dà luogo a un picco cromatografico. Un primo approccio si ricava dalla valutazione del tempo di ritenzione, e cioè del tempo che un dato composto impiega per fuoriuscire dalla colonna, ma il procedimento più idoneo consiste nell'inviare l'effiuente dalla colonna cromatografica in uno spettrometro di massa (v. cap. 4, § b). Tale accoppiamento (gascromatografia-spettrometria di massa) costituisce un procedimento estremamente sensibile e selettivo per la determinazione di una data specie. Mediante la spettrometria di massa si identifica infatti una specie molecolare in base al modo in cui essa si frammenta quando è bombardata con elettroni di elevata energia, e, tramite l'esame degli ioni che vengono prodotti, si perviene alla sua identificazione.
Di eccezionale utilità è a questo riguardo l'impiego della frammentografia, e cioè della registrazione multipla di ioni specifici. Tale tecnica è di particolare importanza per identificare e determinare la composizione quantitativa di miscele complesse e per differenziare specie isomere. Nella fig. 15 è riportato il frammentogramma relativo alla determinazione di policlorodibenzodiossine PCDD. L'identificazione delle otto- (OcCDD), delle sette- (HpCDD), delle esa- (HxCDD), delle penta- (PeCCD) e delle tetra (TeCDD) è eseguita facendo la registrazione sui corrispondenti ioni molecolari e su altri due picchi isotopici più intensi, M+2 e M+4.
c) Cromatografia liquida (HPLC)
La cromatografia in fase gassosa, come si è detto, è impiegabile solo per le sostanze gassose o vaporizzabili; tuttavia una colonna cromatografica non può essere utilizzata a una temperatura superiore a circa 250 °C, perché la fase stazionaria si decompone.
Questa limitazione non esiste nella ‛cromatografia liquida', in cui si utilizza come fase mobile un liquido e si opera a temperatura ambiente. In queste condizioni qualsiasi specie può venire separata, identificata e determinata quantitativamente. Utilizzando come fase mobile un liquido, in luogo di un gas, e operando con una fase stazionaria supportata su particelle solide di dimensioni molto piccole, per esaltare la capacità di separazione, la velocità di flusso si abbassa notevolmente; per ovviare a questo inconveniente si è trovato opportuno esercitare a monte della colonna una pressione molto alta sulla fase mobile.
Utilizzando questi criteri è stata sviluppata la ‛cromatografia liquida ad alta pressione', detta anche più propriamente ‛cromatografia liquida ad alta risoluzione' (High Pressure - o Performance - Liquid Chromatography). Lo schema a blocchi di un sistema per HPLC è riportato nella flg. 16, insieme con lo schema funzionale. L'apparecchio è costituito da una pompa che convoglia alla colonna il liquido prescelto come fase mobile dopo che esso ha attraversato il dispositivo mediante il quale la sostanza in esame viene iniettata nella colonna. All'uscita della colonna è disposto un rivelatore collegato con il registratore.
La funzione della pompa è quella di assicurare un'elevata pressione di ingresso (3,5-35 MPa) e una portata controllata della fase mobile con un flusso costante (1-2 ml/min) attraverso la colonna in cui si trova la fase stazionaria. In genere la colonna ha un diametro interno pari a 2-4 mm e una lunghezza di 10-30 cm, ed è riempita di materiali omogenei costituiti da particelle di diametro molto piccolo (250 μm).
A monte della colonna vi è un dispositivo per l'introduzione del campione e all'uscita un rivelatore che segnala i vari componenti separati dalla colonna.
La registrazione della risposta del rivelatore in funzione del tempo dà luogo a un tracciato cromatografico, il ‛cr0matogramma liquido', che viene utilizzato per la valutazione qualitativa e quantitativa della sostanza analizzata. I dati del cromatogramma possono venire elaborati con opportuni sistemi computerizzati. Oltre alla fase stazionaria, anche la fase mobile svolge nella HPLC un ruolo attivo: infatti essa dà luogo a interazioni con i componenti della miscela da separare. Così, per variare l'entità di tali interazioni, la fase mobile, anziché consistere in un solo solvente, può essere una miscela di due solventi, e la composizione della miscela può essere mantenuta costante (procedimento isocratico) o fatta variare nel corso dell'eluizione. Questo secondo procedimento viene utilizzato quando i componenti di un campione possiedono caratteristiche di polarità molto diverse, per migliorare la separazione e per ridurre i tempi di eluizione.
I rivelatori impiegati appartengono a due grandi classi: quelli ottici e quelli elettrochimici. I rivelatori ottici ricevono un fascio di radiazioni che ha attraversato una vaschetta di dimensioni molto esigue in cui fluisce il liquido che esce dalla colonna. A seconda del rivelatore usato, le variazioni dell'intensità della luce (per effetto dell'assorbimento, o della fluorescenza, oppure dell'indice di rifrazione) prodotte dal passaggio dei costituenti del campione separati dalla colonna vengono trasformate in corrispondenti variazioni di tensione e registrate.
Il rivelatore ottico più usato è quello basato sull'assorbimento di raggi ultravioletti di lunghezza d'onda opportuna (di solito 254 nm), adatto alla rivelazione di molte sostanze organiche.
I rivelatori elettrochimici sono basati sulla misura di una grandezza elettrica della soluzione effluente; se le specie eluite sono cariche, si utilizza una microcella per la misura della conducibilità elettrica, mentre se le specie sono riducibili od ossidabili possono impiegarsi rivelatori voltammetrici.
La risposta di questi sensori è inviata a un registratore potenziometrico, al fine di ottenere il cromatogramma, o è elaborata da un calcolatore. Come nella gascromatografia, l'analisi qualitativa, intesa come identificazione del picco di ciascun componente, si esegue sulla base della misura dei tempi di eluizione, che vengono confrontati con quelli che si ottengono eluendo nelle stesse condizioni i singoli composti puri. L'analisi quantitativa è basata sulla misura delle aree dei picchi, previa appropriata calibrazione con miscele a contenuto noto.
Nella HPLC la separazione avviene attraverso le interazioni fra il campione, la fase mobile e la fase stazionaria, e, a seconda del materiale prescelto per la fase stazionaria, si può avere una cromatografla per adsorbimento, per ripartizione, per scambio ionico o per esclusione dimensionale. La fase stazionaria è di conseguenza costituita da solidi porosi che differiscono nella composizione chimica, nella struttura e nelle dimensioni delle particelle. Nella cromatografia per adsorbimento la fase stazionaria è in genere il gel di silice o l'allumina e i solventi più frequentemente usati sono, in ordine di polarità crescente, l'esano, l'isoottano, il cloroformio, l'acetonitrile, il metanolo e l'acqua. Nella cromatografia di ripartizione le fasi stazionarie sono costituite dalle cosiddette ‛fasi legate'; esse vengono preparate mediante reazioni chimiche cui prendono parte i gruppi idrossilici superflaali della silice e molecole lineari. La separazione di una miscela può essere eseguita mediante cromatografia a fase normale, intendendo con questo termine il procedimento in cui la polarità della fase mobile è minore di quella della fase stazionaria. Con alcune fasi legate (alchilsiliciche e feniliche) si può impiegare anche la tecnica della fase inversa, ossia eseguire l'eluizione con una fase mobile che ha una polarità maggiore di quella della fase stazionaria. Con questo procedimento è possibile analizzare una grande varietà di composti di interesse biologico, quali glucosidi, ormoni, amminoacidi; questa è pertanto la tecnica più diffusa.
Nella cromatografia di esclusione i costituenti di un campione vengono separati, in base alla dimensione delle loro molecole, su di un materiale costituito da un polimero poroso avente pori di determinate dimensioni. Poiché la fase stazionaria è costituita da un gel e la dimensione molecolare è il fattore determinante, questa metodica è definita anche cromatografia di permeazione su gel o di esclusione dimensionale. Se il campione contiene molecole di varie dimensioni, quelle che hanno dimensioni più grandi del diametro dei pori stessi passano velocemente attraverso la colonna. Le molecole più piccole vengono trattenute più a lungo dalle particelle, poiché possono penetrare entro tutti i pori. Il procedimento è schematizzato nella fig. 17: una miscela di molecole di dimensioni diverse, rappresentate sotto forma di cerchi (A), fluisce attraverso un materiale costituito da granuli porosi (B). Per effetto della permeazione selettiva la miscela si fraziona, come indicato in C.
Questa metodica, in base a un'opportuna scelta delle caratteristiche della fase stazionaria, può venire impiegata per l'analisi di specie sia ad alto sia a basso peso molecolare.
d) Cromatografia ionica
La cromatografia ionica (CI o IC) è una tecnica cromatografica per la separazione di ioni inorganici in funzione della loro affinità per una resina a scambio ionico. Le separazioni mediante CI sono dovute a differenze nell'equilibrio di distribuzione tra una fase mobile, che è una soluzione tampone, e una fase stazionaria, che è uno scambiatore ionico. La CI può essere considerata come una modificazione della cromatografia liquida ad alta risoluzione, in cui il processo di separazione è dovuto essenzialmente a un meccanismo di scambio ionico. Gli ioni del campione in esame competono con gli ioni della fase mobile nell'interazione con i gruppi scambiatori di carica opposta presenti nella fase stazionaria, che è impaccata nella colonna.
Lo schema a blocchi di un cromatografo ionico è riportato nella fig. 18. Mediante una pompa è fatta fluire nella colonna cromatografica - riempita con una fase stazionaria costituita da polimeri organici, ai quali sono stati fissati gruppi capaci di scambiare ioni, e che ha una piccola capacità di scambio - la soluzione tampone prescelta per l'eluizione. Se in una soluzione si debbono analizzare gli anioni, si adopera una colonna in cui lo ione scambiabile è lo ione idrogenocarbonato HCO3-. Allorché un campione della soluzione è iniettato in colonna si ha il frazionamento degli anioni presenti in funzione dell'affinità di questi per gli scambiatori ionici. Se l'affinità di un anione a interagire con lo scambiatore è grande, esso verrà trattenuto più a lungo nella colonna rispetto a quegli anioni che interagiscono in modo minore. Ne consegue che questi fuoriusciranno dalla colonna più rapidamente degli altri e si avrà in tal modo la separazione dei vari anioni.
Se in una soluzione si debbono analizzare i cationi si impiega una colonna in cui lo ione scambiabile è lo ione idrogeno. Poiché in entrambi i casi le specie separate sono ioni, e cioè specie conduttrici, il sistema di rivelazione è in genere costituito da una cella di conducibilità. La registrazione delle variazioni di conducibilità eseguita nel tempo, dovute alla eluizione delle varie specie ioniche dalla Colonna, consente di ricavare il relativo cromatogramma.
Quanto alla determinazione quantitativa, essa è basata sulla misura delle aree dei picchi che caratterizzano i singoli ioni nei cromatogrammi, previa taratura con soluzioni dei vari ioni con concentrazioni note.
Nella fig. 19 è mostrato il cromatogramma ionico relativo alla separazione di una miscela di anioni.
Bibliografia
Bauer, H. H., Christian, G. D., O'Reilly, J. E., Instrumental analysis, Boston 1978 (tr. it.: Analisi strumentale, Padova 1986).
Christian, G. D., Analytical chemistry, New York 19803 (tr. it.: Chimica analitica, Padova 1986).
Kolthoff, I. M., Elving, F. J. (a cura di), Treatise on analytical chemistry, 33 voll., New York 1979 ss.
Kolthoff, I. M., Sandell, E. B., Meehan, E. J., Bruckenstein, S., Quantitative chemical analysis, New York 19694 (tr. it.: Analisi chimica quantitativa, Padova 1980).
Poole, C. F., Schouette, S. A., Contemporary practice of chromatography, Amsterdam-New York 1984.
Saini, G., Liberti, A., Chimica analitica, Torino 1980.
Skoog, D. A., West, D. M., Principles of instrumental analysis, New York 1971.
Vogel, A. I., A textbook of quantitative inorganic analysis including elementary instrumental analysis, London 19723.
Wilson, C. L., Wilson, D. W., Comprehensive analytical chemistry, 4 voll., Amsterdam 1959 ss.