
Imprinting genomico
Imprinting genomico
Durante lo sviluppo dei Mammiferi i genomi dei genitori sono funzionalmente non equivalenti, in quanto alcuni loci omologhi subiscono l'effetto di modificazioni epigenetiche specifiche per la linea germinale, quando i cromosomi si separano nelle linee maschili e femminili. Questo processo conferisce a una sottoclasse di loci omologhi, definiti 'geni imprinted', un'espressione dipendente dal genitore d'origine. Alcuni di questi geni sono espressi se ereditati dal padre, altri se ereditati dalla madre. L'altro allele parentale viene, infatti, mantenuto in uno stato in parte represso mediante la metilazione del DNA, una delle più importanti modificazioni epigenetiche ereditabili. Alcuni geni sottoposti a imprinting sono raggruppati in particolari domini cromosomici e controllati da un elemento regolativo, definito 'centro di imprinting'. Tali geni svolgono un ruolo significativo nello sviluppo: un errato imprinting o un'espressione non appropriata di questi geni sono alla base di diverse malattie genetiche umane.
Lo sviluppo per partenogenesi è apparentemente impossibile nei Mammiferi. Numerosi esperimenti volti alla ricostruzione dello zigote per la produzione di embrioni androgenetici (due genomi paterni) e partenogenetici/ginogenetici (due genomi materni) hanno dimostrato che, per uno sviluppo corretto, è necessaria la presenza di entrambi i genomi. Poiché entrambi i genomi parentali contribuiscono con geni simili, l'incapacità di questi embrioni geneticamente bilanciati di svilupparsi deve essere attribuita a differenze epigenetiche tra i genomi parentali. I genomi parentali risultano non equivalenti per la sottoclasse di geni imprinted, la cui espressione dipende dalla loro origine parentale. Molti tipi differenti di geni imprinted condividono caratteristiche strutturali che conferiscono loro tale comportamento.
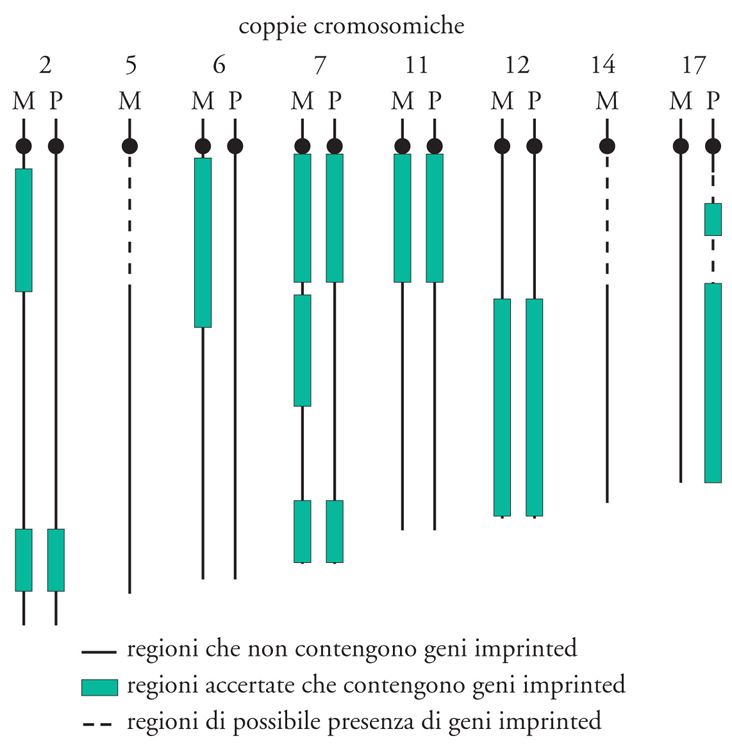
Solo una piccola parte dei geni è regolata da imprinting genomico. Benché il loro numero non sia noto, è stato stimato che nel genoma di topo siano circa 200. Le regioni del cromosoma che contengono alcuni importanti geni imprinted sono state identificate mediante il test di non complementazione. Utilizzando specifiche traslocazioni e riordinamenti (rearrangement) cromosomici, è possibile produrre embrioni caratterizzati da duplicazioni di regioni paterne (PatDp) o materne (MatDp) con reciproche deficienze. Questi embrioni sono bilanciati geneticamente e, nella maggior parte dei casi, in essi non si rivelano effetti fenotipici apprezzabili. Solo le regioni cromosomiche contenenti geni imprinted, identificate sui cromosomi 2, 6, 7, 11, 12, 17, determinano effetti fenotipici in seguito all'acquisto o alla perdita di funzione per uno di questi geni. È importante sottolineare che questo tipo di esperimenti ha consentito di identificare le 'regioni imprinted' in modo grossolano, in quanto non tutti i geni ivi contenuti in realtà sono tali. (fig. 1).
Identificazione di geni imprinted
Il primo gene imprinted fu identificato per caso, quando il gene Igf2 fu distrutto mediante ricombinazione omologa: topi che avevano ereditato la mutazione dal padre mostravano un ritardo nella crescita, quando invece la ereditavano dalla madre avevano una taglia normale. Anche Mash2 fu identificato come gene imprinted in seguito a studi analoghi: questo gene è importante per lo sviluppo del trofoblasto e quando viene ereditato in forma mutata per via materna causa morte embrionale precoce, mentre lo sviluppo dell'embrione che lo eredita per via paterna procede normalmente. L'identificazione del gene imprinted Igf2r è avvenuta invece mediante studi di genetica classica: la mutazione per delezione Tme è letale nell'embrione quando ereditata per via materna, mentre l'eredità paterna non produce alcun effetto. Anomalie genetiche umane, in particolare la sindrome di Prader-Willi e Angelman, sono state utilizzate per identificare il gene imprinted SNRPN e, a questo, ha fatto immediato seguito la caratterizzazione di altri geni imprinted adiacenti e dei loro omologhi di topo. Sembra che i geni imprinted siano raggruppati in domini; questa proprietà ha consentito l'identificazione di altri geni imprinted, H19 e p57Kip2, e Mas, rispettivamente localizzati in prossimità di Igf2 e Igf2r.
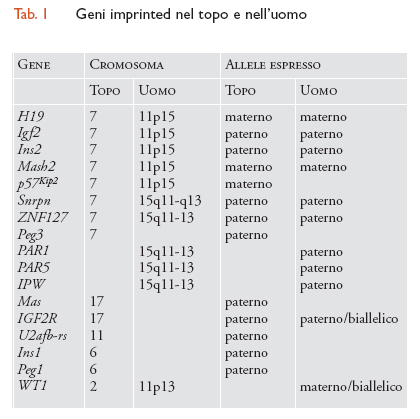
Più recentemente sono state condotte analisi sistematiche all'interno di genomi che hanno consentito l'identificazione di altri geni imprinted. Esistono potenzialmente due modi per identificare tali geni, il primo dei quali sfrutta la differente metilazione dei geni imprinted nei genomi parentali. Il differente stato di metilazione dei genomi materni e paterni è stato infatti utilizzato per elaborare il metodo di 'analisi del genoma mediante siti di restrizione di riferimento' (RLGS, Restriction landmark genome scanning). Il principale vantaggio di questo sistema consiste nella possibilità di utilizzare virtualmente qualsiasi tessuto, sia dell'embrione sia dell'adulto, poiché la metilazione dovuta all'imprinting è conservata anche laddove i geni imprinted non sono espressi. Il secondo approccio consiste nell'isolamento del gene espresso. Per l'isolamento di geni imprinted viene utilizzato il metodo di ibridazione per sottrazione, unitamente a procedure di selezione differenziale. Per la sottrazione è stata messa a punto una metodica basata su un'amplificazione di cDNA (DNA complementare) e successiva ibridazione per sottrazione. In seguito ad analisi differenziale di cDNA sottratti, assieme ai cDNA provenienti da uova fecondate e da embrioni di controllo, sono stati identificati almeno quattro geni. Due di questi, Peg2 e Peg4, si rivelarono come i geni imprinted già identificati Igf2 e Snrpn mentre Peg1/Mest e Peg3 si rivelarono nuovi geni imprinted. Altri geni imprinted saranno probabilmente identificati con questa tecnica e dovrebbe essere possibile isolare anche geni sottoposti a imprinting materno (MEGS, Maternally imprinted genes), utilizzando embrioni androgenetici e normali (tab. 1).
Il ruolo svolto dai geni imprinted durante lo sviluppo
La maggior parte dei primi esperimenti per comprendere il ruolo svolto dai geni imprinted fu condotta su embrioni androgenetici e ginogenetici, fornendo molte informazioni sugli effetti cumulativi di tali geni. Con l'identificazione di un numero crescente di geni di questo tipo sarà possibile esaminare come essi, individualmente, determinino lo sviluppo dell'organismo. Gli embrioni androgenetici e ginogenetici mostrano una mortalità embrionale precoce, sopravvivendo fino allo stadio di 6-8 somiti i primi e 25 somiti i secondi, con fenotipi opposti nella formazione dei tessuti extraembrionali ed embrionali. Ulteriori studi sullo sviluppo sono stati effettuati mediante ricostituzione della blastocisti: è infatti possibile far sviluppare embrioni partenogenetici o ginogenetici dopo fusione con tessuto trofoectodermico normale. Tuttavia, neanche le blastocisti ricostituite sono in grado di terminare lo sviluppo, e ciò sta a indicare che questo blocco ha cause molto più complesse. Sono stati condotti numerosi esperimenti per studiare il destino di cellule uniparentali in embrioni chimerici con cellule normali. In questi esperimenti è stato accertato che la presenza di cellule normali consente il 'recupero' delle cellule uniparentali, permettendone uno sviluppo più avanzato di quanto non farebbero da sole. In secondo luogo, la distribuzione delle cellule uniparentali e i conseguenti effetti fenotipici sugli embrioni chimerici forniscono uno strumento per determinare le caratteristiche di tali cellule e il ruolo potenziale svolto dai geni imprinted nello sviluppo.
Generalmente, le cellule uniparentali si distribuiscono in modo casuale fino all'undicesimo giorno e mezzo di gestazione, per poi raggiungere posizioni specifiche per cellule androgenetiche e ginogenetiche. Il primo effetto evidente osservato nelle chimere riguarda la crescita fetale: la presenza di cellule partenogenetiche/ginogenetiche (PG/GG) causa un ritardo della crescita, mentre quella di cellule androgenetiche un'accelerazione. Le chimere contenenti cellule PG si sviluppano, anche dopo la nascita e nell'età adulta, pur rimanendo individui piccoli, ma, se il contributo delle cellule uniparentali è molto alto, pur rimanendo individui piccoli, si verifica mortalità embrionale. I feti chimerici contenenti cellule androgenetiche (AG) sono più grandi dei controlli. Le chimere di questo tipo mostrano effetti fenotipici maggiori specialmente riguardo all'asse scheletrico, probabilmente a causa di una maggiore proliferazione delle cellule staminali. Le cellule PG mostrano, al contrario, una diminuzione della proliferazione delle cellule staminali, che si osserva anche nella massa cellulare interna degli embrioni prima dell'impianto.
L'effetto fenotipico più eclatante prodotto dalle cellule AG nelle chimere consiste nell'iperproliferazione di cartilagine, caratterizzata da iperplasia e ipertrofia dei condrociti, specialmente nelle costole ma anche nelle vertebre. Si assiste altresì a ingrandimento e fusione delle dita e alla presenza di dita supplementari. In ogni caso, le cellule AG rimangono predominanti tra le cellule staminali presenti in vari tessuti. Le cellule PG sono invece quasi completamente assenti nei tessuti mesodermici. Diversi studi hanno dimostrato che queste vengono eliminate dalla muscolatura scheletrica delle chimere adulte, anche se tali cellule sono presenti nei miotomi e in altri compartimenti somitici di chimere di 10 e 11 giorni e non si osservano differenze per quanto riguarda il contributo alla formazione dei somiti e del tubo neurale, all'interno del quale persistono. Inoltre, embrioni PG esprimono vari geni di regolazione della miogenesi, a dimostrazione che il programma di differenziamento muscolare non è difettivo. Però, nonostante la loro determinazione al differenziamento miogenico, le cellule PG vengono escluse dalla muscolatura scheletrica a partire dal tredicesimo giorno e quelle che rimangono vengono accumulate nel tessuto connettivo intramuscolare. Le ragioni del mancato sviluppo di cellule PG nel tessuto muscolare non sono state ancora chiarite e i geni imprinted responsabili non sono stati identificati; è comunque possibile che geni imprinted siano coinvolti nello sviluppo dei mioblasti somitici attraverso la linea miogenica.
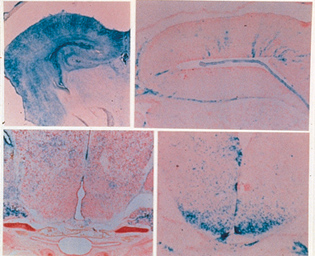
Nel cervello i geni imprinted possono svolgere un ruolo centrale nello sviluppo e nel comportamento. Mentre le cellule PG sono ben rappresentate nel cervello, quelle AG ne sono quasi completamente escluse. Analisi ulteriori hanno però dimostrato una distribuzione molto più specifica delle cellule uniparentali nel cervello: le cellule PG sono presenti nella parte anteriore del cervello, mentre sono quasi completamente assenti nell'ipotalamo. Al contrario, le cellule AG sono molto rappresentate nell'ipotalamo ma assenti nella corteccia. Studi sul comportamento hanno dimostrato che individui chimerici con cellule PG sono aggressivi, con un significativo aumento dell'aggressività in maschi che presentano un alto contributo di cellule PG nel cervello, mentre nelle femmine non si osserva alcuna variazione. La latenza di attacco nei confronti di un maschio estraneo da parte di un maschio chimerico è ridotta da 4 o 5 min a circa 16 sec. L'effetto sull'aggressività è particolarmente interessante, poiché in un'unica chimera umana contenente cellule PG, l'individuo mostrava comportamento aggressivo intermittente. Conseguenze comportamentali dell'imprinting sono state osservate anche nelle sindromi di Prader-Willi e Angelman, in uomini e topi con disomia, rispettivamente, dei cromosomi 7 e 2. Nel loro insieme questi dati sottolineano l'importanza dei geni imprinted nella crescita, sviluppo e funzionalità del cervello (fig. 2).
Il ruolo di singoli geni imprinted
L'identificazione di geni imprinted rende possibile studiarne il ruolo sia singolarmente sia di concerto all'interno di gruppi predeterminati. Questi studi possono essere messi in relazione con i diversi fenotipi di chimere con cellule uniparentali AG e PG. Per esempio, una delle principali differenze fenotipiche tra embrioni AG e PG risiede nello sviluppo dei tessuti extraembrionali, generalmente ridotti negli embrioni di tipo PG: il primo gene imprinted che determina esclusivamente lo sviluppo del trofoblasto è stato identificato e denominato Mash2. I geni imprinted noti per essere coinvolti direttamente nel controllo della crescita del feto sono Igf2 e Igf2r. Igf2 è espresso solo dall'allele paterno ed è necessario per una normale crescita fetale, come dimostrato da esperimenti di mutazione sitospecifica. Igf2r, localizzato sul cromosoma 17, viene espresso dall'allele materno. Vi sono indicazioni che il ruolo di IGF2R non è da ricercare nella trasduzione del segnale provocato da IGF2 ma, piuttosto, nella degradazione di IGF2 di localizzazione extracellulare, attraverso un processo di endocitosi mediato dal recettore specifico. Quindi, i topi mutati mancanti di IGF2R risultano, alla nascita, di dimensioni del 30% maggiori rispetto ai controlli: tale effetto sulla crescita può essere normalizzato attraverso la combinazione delle mutazioni di Igf2r e Igf2.
Il gene p57Kip2 è un potente inibitore di numerosi complessi ciclina/Cdk, essendo in grado di formare legami molto forti. Il suo omologo umano, localizzato sul cromosoma 11pI5-5, è implicato nella sindrome di Beckwith-Wiedmann, causa di crescita eccessiva, che è alla base del tumore di Wilm e di altre forme come il rabdomiosarcoma. Studi compiuti su numerose forme di tumore e l'osservazione di una diminuzione della proliferazione delle cellule di tipo PG fanno ritenere che questo gene si esprima esclusivamente attraverso l'allele materno. Il complesso dei risultati esposti offre un quadro sufficientemente chiaro degli effetti cumulativi determinati dai geni imprinted nel corso dello sviluppo; chiaramente, le interazioni tra geni imprinted devono essere estremamente complesse e, spesso, nelle cellule di tipo uniparentale si può verificare una regolazione non corretta di questi geni. Solo attraverso l'integrazione di questi studi con altri su MatDp e PatDp relativi a specifiche regioni cromosomali si potrà tracciare un quadro dettagliato e accurato delle interazioni esistenti tra geni imprinted. Inoltre, con il progredire dell'identificazione di altri geni imprinted, sarà possibile stabilirne il ruolo sia singolarmente sia in gruppo, attraverso l'uso di animali transgenici in cui è possibile causare la perdita e l'acquisizione di determinate funzioni. Sarà, infine, possibile reintrodurre singoli geni imprinted all'interno di linee cellulari embrionali AG e PG e determinare in che modo ciò alteri le loro proprietà.
Meccanismi di imprinting
L'imprinting genetico è un processo che si articola in diverse fasi durante le quali i loci imprinted vanno incontro a una serie di modificazioni che iniziano proprio nella linea germinale, quando i cromosomi parentali segregano nei gameti. Nelle cellule germinali primordiali avviene un primo evento che consiste nella perdita di tutti gli imprinting ereditati, così da rendere impossibile la distinzione dei cromosomi omologhi parentali. In questa fase, dunque, i cromosomi parentali sono probabilmente del tutto equivalenti. Ciò può essere osservato a livello molecolare con la perdita di metilazione dei siti imprinted. A uno stadio più avanzato della gametogenesi, si assiste alla metilazione di loci imprinted e di altri ed è questo il momento in cui i geni imprinted possono subire metilazioni diverse nella spermatogenesi e nell'ovogenesi. Evidenze sperimentali mostrano come gli alleli parentali dei geni imprinted possano continuare a subire modificazioni perfino dopo la fecondazione, risultando infine, nei tessuti embrionali, diversamente metilati. La modificazione del DNA mediante metilazione dei dinucleotidi CpG ha proprietà che permettono di spiegare l'imprinting: infatti, la metilazione del DNA influenza l'espressione genica, è ereditaria ed è reversibile. Il dinucleotide CpG è presente nel genoma dei Mammiferi in misura molto inferiore rispetto alla frequenza attesa (25%), ma con un'alta percentuale di metilazione; circa il 2% dei dinucleotidi CpG si trova raggruppato in tratti del genoma di circa 1 kb di lunghezza, in cui la frequenza risulta corrispondente all'attesa; la maggior parte delle 'isole CpG' (così sono denominate queste regioni ricche di CpG) è localizzata all'estremità 5′ dei geni e non è comunque metilata, indipendentemente dall'attività di trascrizione del gene.
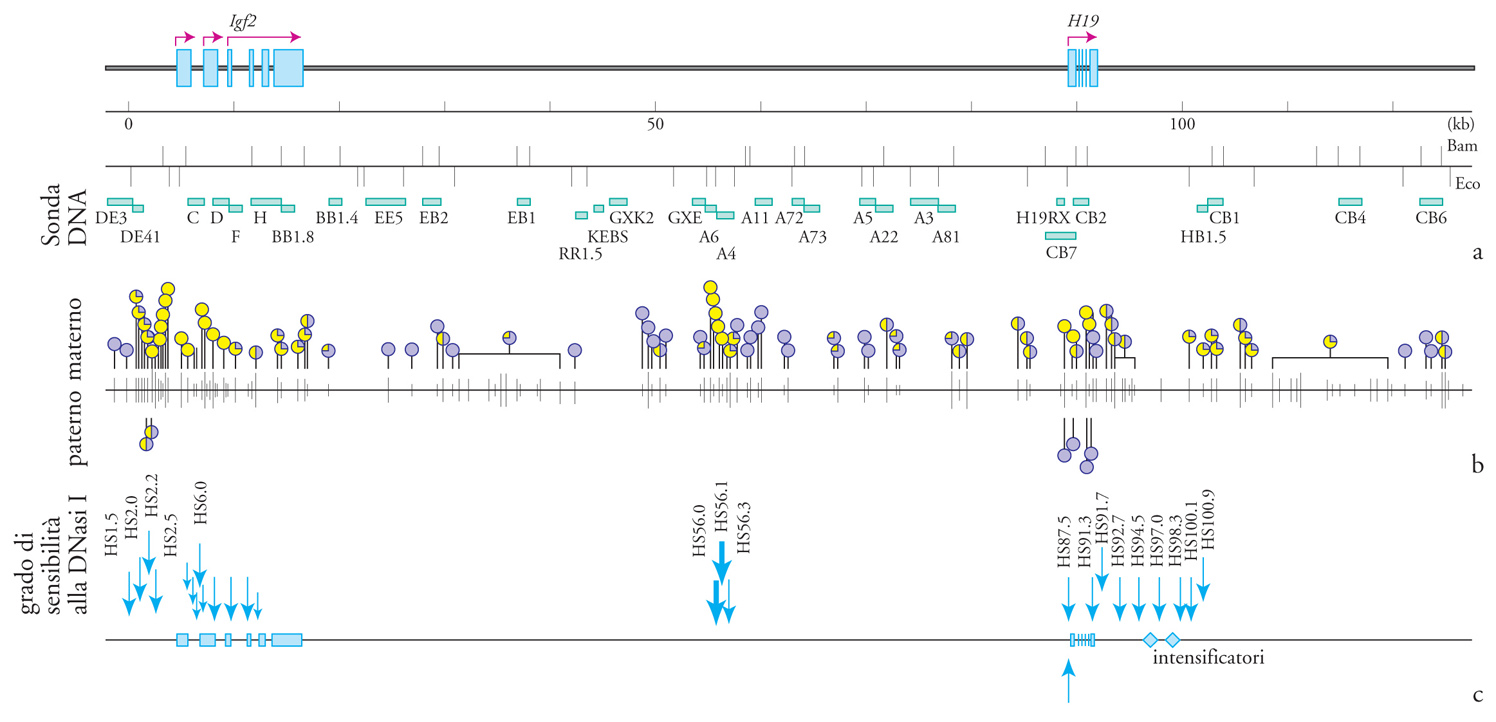
Le isole ricche di CpG sono di solito non metilate, tranne quando sono associate a geni imprinted, nel qual caso viene metilata quella di uno degli alleli parentali. Nel caso del gene H19, l'allele paterno inattivo è altamente metilato, al contrario di quello materno attivo che risulta non metilato nella regione del promotore e del primo esone. In questo secondo caso si nota anche una diversa sensibilità all'attacco della DNasi I, che indica una conformazione cromatinica più rilassata (fig. 3). Il gene Igf2, situato 90 kb a monte di H19, mostra un livello di metilazione differenziale più fine, in particolare nella regione del promotore 1 e in quella che si estende dalle vicinanze del quinto esone fino all'inizio del sesto. L'allele paterno di Igf2 presenta un livello di metilazione leggermente maggiore di quello materno ma, al contrario di H19, entrambi gli alleli risultano attivi e sensibili in uguale misura all'attacco della DNasi I. Il gene Igf2r contiene due regioni che mostrano un diverso livello di metilazione tra i due alleli parentali.
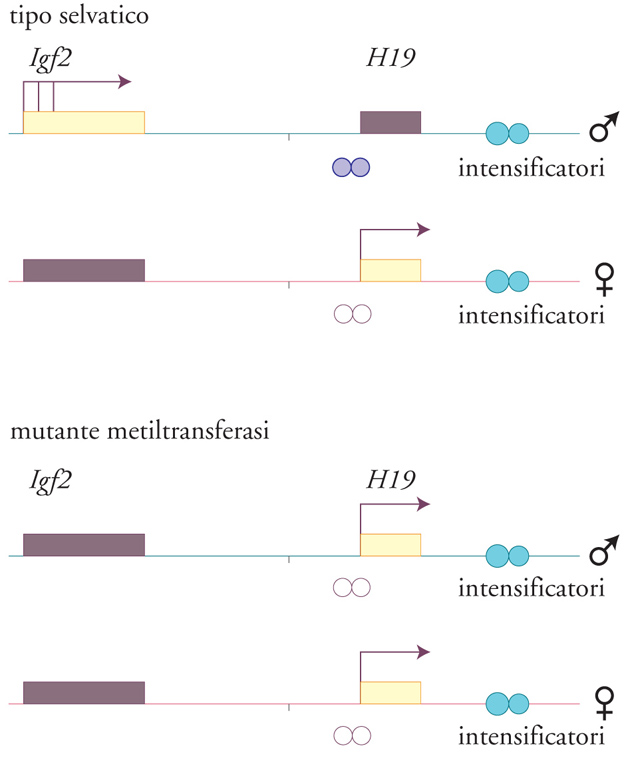
Un certo numero di regioni metilate nei geni di derivazione materna e paterna è in stretta associazione con geni trascrizionalmente attivi o silenti. Comunque è lecito concludere che, a eccezione del caso di Igf2, in cui l'isola CpG presente al 5′ risulta non metilata in entrambi gli alleli, esiste una stretta correlazione tra il fenomeno della metilazione delle regioni regolative dei geni imprinted e la loro inattività trascrizionale. L'importanza di questo aspetto nel mantenimento dell'imprinting è stata dimostrata da un esperimento di delezione del gene che codifica la metiltransferasi di topo attraverso la tecnica di inattivazione genica mirata (gene targeting) in linee cellulari embrionali. La mancanza di questo enzima provocava la morte degli embrioni prima del decimo giorno di gestazione; inoltre, negli embrioni mutanti, le due copie del gene H19 risultavano attive mentre entrambe le copie di Igf2 e Igf2r erano silenti, a dimostrazione dell'effetto negativo, dal punto di vista della trascrizione, che la metilazione ha sul gene H19 e dell'effetto positivo che essa ha sui geni Igf2 e Igf2r (fig. 4).
Caratteristiche strutturali dei geni imprinted e loro organizzazione in domini
Alcune isole a CpG presenti in geni imprinted contengono moduli di sequenze ripetute di lunghezze variabili: per esempio, la regione 2 del gene Igf2r, con metilazione differenziata, contiene una serie di moduli ripetuti, nella stessa direzione, variabili in dimensioni dalle 25 alle 75 coppie di basi, che sono presenti anche in altri geni. Nella regione a monte del gene H19 è inoltre presente un elemento ripetuto di sei coppie di basi, molto vicino ai dinucleotidi CpG metilati in maniera differenziale; analoghe sequenze ripetute sono state osservate in prossimità di regioni di metilazione differenziale del gene p57Kip2. Tuttavia, non sembra esserci tra esse omologia di sequenza, segno di un probabile coinvolgimento delle unità ripetute in modificazioni locali della struttura cromatinica, in conseguenza dello stato di metilazione della regione e di altre potenziali modificazioni epigenetiche del DNA e della cromatina, quali per esempio l'eterocromatina, che influenzerebbero l'accessibilità del DNA a fattori trascrizionali essenziali.
Alcuni dei geni imprinted, come H19, IPW, PAR1 e PAR5, non possiedono alcuna fase di lettura aperta (open reading frame): è stato perciò ipotizzato che il loro prodotto funzionale possa essere l'RNA corrispondente, come accade per il gene, Xist, legato all'inattivazione del cromosoma X. Ciò ha portato a ipotizzare che le molecole di RNA di alcuni geni imprinted come H19 potrebbero avere un simile ruolo di silenziatori di geni in cis in domini imprinted. Malgrado ciò, il prodotto del gene H19 è principalmente localizzato nel citoplasma, come anche quello del gene IPW. Il ruolo di queste molecole di RNA resta così ancora dubbio. Con il progredire dell'identificazione di nuovi geni imprinted, si delinea sempre più l'ipotesi secondo cui questo tipo di geni potrebbe essere organizzato in specifici domini che ne contengono un certo numero. Questi domini hanno la caratteristica principale di essere asincroni dal punto di vista della replicazione, essendo sempre la regione paterna la prima a essere replicata. In questo modo un piano replicativo differenziale a carico dei due alleli parentali fornisce un punto di riferimento per un dominio genomico di grandi dimensioni, permettendo all'apparato trascrizionale cellulare e agli apparati responsabili delle modificazioni epigenetiche del DNA di distinguerli e di controllarli in modo appropriato.
Nella porzione distale del cromosoma 7 del topo sono stati sinora identificati cinque geni imprinted, quattro dei quali giacciono in una regione che si estende per circa 300 kb. H19 e Igf2 sono separati da un tratto di DNA che misura 70 kb e si trovano nello stesso orientamento dal punto di vista trascrizionale. Ins2 è situato circa 20 kb a monte di Igf2, con il medesimo orientamento trascrizionale, ed è a espressione paterna nel sacco vitellino ma dimostra di possedere espressione biallelica nel pancreas. Mash2 è posto ancora più a monte e la sua espressione, esclusivamente materna, è ristretta allo spongiotrofoblasto. Il gene di più recente identificazione in questa regione è il p57Kip2. Dati ottenuti per l'uomo suggeriscono che possa esserci un elemento di controllo di Igf2 a monte dell'estremità 5′, che influenza la metilazione dei geni imprinted presenti nella regione, come H19 e Igf2. La loro simile espressione ha portato a suggerire che i due geni condividano gli stessi elementi di regolazione per i quali potrebbero competere; così, l'allele materno non metilato di H19 potrebbe interagire con gli intensificatori (enhancer) inibendo in tal modo l'espressione di Igf2 dello stesso cromosoma materno. Invece, quando il gene H19 è metilato, come nel cromosoma paterno, esso non interagisce con gli intensificatori, che restano liberi di attivare l'espressione di Igf2. Sino a ora gli unici intensificatori identificati in questa regione sono situati a valle del gene H19 e la loro azione è ristretta ai tessuti di origine endodermica, indicando implicitamente la necessità della presenza di altri intensificatori per l'espressione di geni del miotomo.
In ulteriori esperimenti si operò la delezione di un tratto corrispondente al gene H19 e di un segmento a monte delle dimensioni di 10 kb mediante il metodo dell'inattivazione genica mirata (gene targeting). Il risultato fu quello di indurre l'attivazione della copia materna del gene Igf2, normalmente silente, e di provocare un'espressione biallelica di Igf2. Inoltre, questa manipolazione fece assumere alla regione a monte del gene un profilo di metilazione di tipo paterno. Questi risultati dimostrano che il gene H19, l'RNA che ne deriva e la regione a monte che lo precede giocano un ruolo determinante nel controllo dell'imprinting, per lo meno del gene Igf2. Esperimenti di delezione della zona a valle di H19 hanno inoltre mostrato come questi elementi siano usati sia da H19 sia da Igf2, come risulta da un calo dell'espressione di entrambi i geni nei tessuti endodermici, rispettivamente dopo trasmissione materna e paterna. Al contrario, in questo esperimento non si sono registrati effetti sull'espressione di Mash2, suggerendo che l'espressione di questo gene può non essere correlata con quella degli altri due geni. L'imprinting di H19 e Igf2 sembra quindi essere collegato e regolato in alcuni tessuti da un elemento di controllo comune.
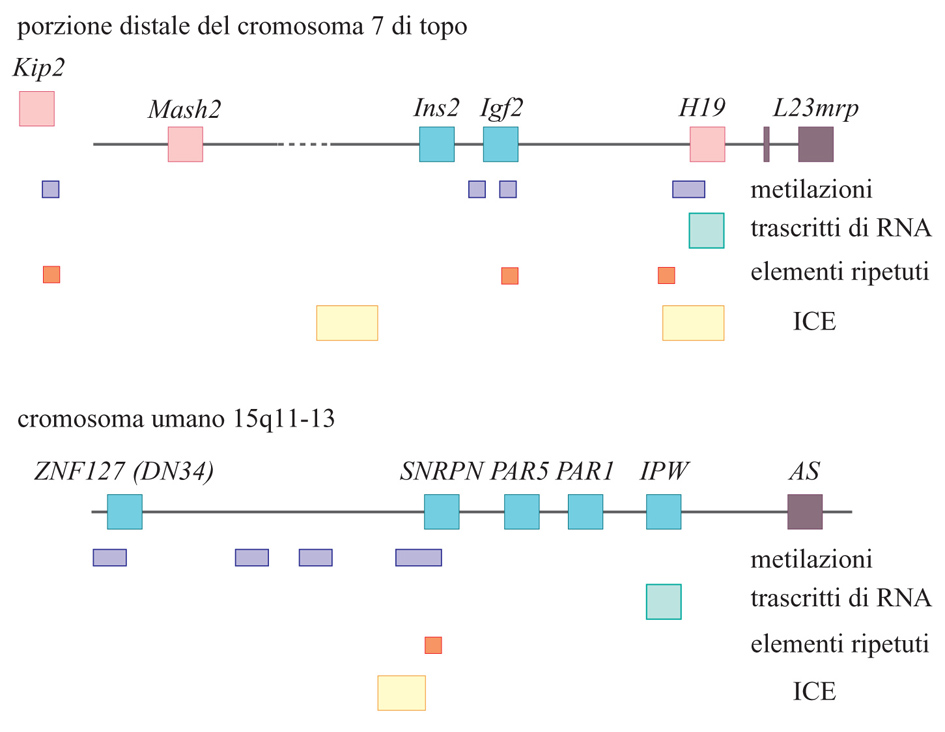
L'altro dominio imprinted nella parte centrale del cromosoma 7 del topo risulta sintenico rispetto al 15q11-13 dell'uomo, che contiene regioni coinvolte nello sviluppo di disordini genetici noti come 'sindrome di Angelman' (AS) e 'sindrome di Prader-Willi' (PWS). Sembra che del gene PWS sia espresso solo l'allele paterno, mentre dell'AS solo quello materno; in questa regione sono noti cinque geni imprinted che comprendono, all'interno di una regione di 1,5 Mb (megabasi), SNRPN, IPW, ZNF127, PAR1 e PAR5; di tutti questi geni sono espressi esclusivamente gli alleli paterni. Un dato interessante è rappresentato dal fatto che in pazienti affetti da PWS è andata perduta l'espressione di SNPRN, PAR1 e PAR5, in seguito a trasmissione paterna di piccole delezioni nella regione a monte di SNRPN, che contiene l'isola di CpG a metilazione differenziale. Tutti questi risultati hanno portato a supporre la possibile esistenza di un elemento genetico di controllo dell'imprinting (ICE, Imprinting control element) che regoli l'espressione di geni confinanti nel cromosoma paterno. Alcune analisi hanno anche dimostrato un'influenza sul tempo di replicazione della regione di 500 o 650 kb intorno a SNRPN. Successivi esperimenti di microdelezioni operate in questa regione hanno dimostrato che esiste un'associazione con l'AS, anche se si pensa che il gene o i geni putativi AS mappino centinaia di chilobasi più lontano. È quindi possibile che, all'interno del dominio imprinted AS/PWS, ci sia un singolo elemento di controllo (ICE) che possa regolare l'epigenotipo (la conformazione della cromatina) parentale di questa regione dei cromosomi (fig. 5).
L'imprinting genomico può quindi influenzare sia l'espressione di singoli geni sia quella di ampie regioni contenenti raggruppamenti di geni. Probabilmente, in entrambi i casi, questo avviene con l'ausilio di fattori specifici. Il nucleo è caratterizzato da un elevato livello di organizzazione, con strutture specializzate che consentono la compartimentazione di differenti funzioni specifiche, in grado di consentire una regolazione differente di regioni cromosomiche quasi identiche. La metilazione del DNA rappresenta forse il primo segno connotativo dell'avvenuto imprinting, ma potrebbero intervenire anche altre modificazioni, quali l'acetilazione degli istoni, l'ubiquitinazione, la replicazione differenziale, l'utilizzo di regioni di ancoraggio all'impalcatura del nucleo da parte di elementi di delimitazione o, infine, l'esistenza di fattori specifici per le cellule della linea germinale non ancora identificati. L'imprinting genomico è un fenomeno che conferisce una serie di differenze funzionali ai due genomi parentali. Numerosi geni imprinted sono stati trovati o sono in fase di identificazione e il loro ruolo nello sviluppo e nelle malattie viene progressivamente chiarito. Tra questi vi sono i geni per fattori di crescita, recettori, fattori di trascrizione, enzimi e alcuni geni particolari che codificano molecole di RNA, la cui funzione non è a tutt'oggi nota.
Attualmente sono in corso molti studi volti a individuare ulteriori geni imprinted e a chiarire il loro ruolo individuale e le interazioni con altri geni di questo tipo e di tipo non imprinted. I geni imprinted mostrano caratteristiche comuni e spesso sembrano essere organizzati in raggruppamenti all'interno di domini, anch'essi dotati di peculiarità, come quelli dei domini di origine paterna, che sono in grado di replicarsi in anticipo rispetto agli altri. C'è, inoltre, una serie di evidenze a favore dell'esistenza di un unico elemento regolativo in grado di controllare l'imprinting di un'intera regione, probabilmente in connessione con elementi gene-specifici. Nel meccanismo dell'imprinting il fenomeno della metilazione del DNA rappresenta una componente fondamentale, essendo utilizzato tanto nel mantenimento dell'imprint parentale, quanto nel controllo accurato dell'espressione degli alleli imprinted. Il fenomeno dell'imprinting si manifesta con un processo ciclico che inizia nella linea germinale, con l'annullamento di tutti gli imprint parentali, a cui fa seguito l'attribuzione dei nuovi imprint durante la gametogenesi. Non è ancora chiaro come questi eventi precoci portino al preciso imprinting degli alleli parentali. Mentre continuano le ricerche sul ruolo e sull'importanza di questo enigmatico fenomeno nel campo della biologia dello sviluppo e delle malattie genetiche, non è ancora concluso il dibattito sul suo reale significato dal punto di vista biologico ed evolutivo.
bibliografia
Barlow 1991: Barlow, Denise P. e altri, The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus, "Nature", 349, 1991, pp. 84-87.
Bartolomei 1993: Bartolomei, Marisa S. e altri, Epigenetic mechanisms underlying the imprinting of the mouse H19 gene, "Genes and development", 7, 1993, pp. 1663-1673.
Barton 1991: Barton, Sheila C. e altri, Influence of paternally imprinted genes on development, "Development", 113, 1991, pp. 679-688.
Brannan 1990: Brannan, Camilynn I. e altri, The product of the H19 gene may function as an RNA, "Molecular and cellular biology", 10, 1990, pp. 28-36.
Buiting 1995: Buiting, Karin S. e altri, Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15, "Nature genetics", 9, 1995, pp. 395-400.
Cattanach 1995: Cattanach, Bruce M. - Barr, J. - Jones, J., Use of chromosome rearrangements for investigations into imprinting in the mouse, in: Genomic imprinting. Causes and consequences, edited by Rolf Ohlsson, Kerstin Hall, Martin Ritzen, Cambridge, Cambridge University Press, 1995, pp. 327-341.
De Chiara 1991: De Chiara, Thomas M. - Robertson, Elizabeth I. - Efstratiadis, Argiris, Parental imprinting of the mouse insulin-like growth factor II gene, "Cell", 64, 1991, pp. 849-859.
Efstratiadis 1994: Efstratiadis, Argiris, Parental imprinting of autosomal mammalian genes, "Current opinion in genetics and development", 4, 1994, pp. 265-280.
Ferguson-Smith 1993: Ferguson-Smith, Anne C. e altri, Parental-origin-specific epigenetic modification of the mouse H19 gene, "Nature", 362, 1993, pp. 751-755.
Guillemot 1995: Guillemot, François e altri, Genomic imprinting of Mash2, a mouse gene required for trophoblast development, "Nature genetics", 9, 1995, pp. 235-242.
Hatada, Mukai 1995: Hatada, Izuho - Mukai, Tsunehiro, Genomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouse, "Nature genetics", 11, 1995, pp. 204-206.
Hayashizaki 1994: Hayashizaki, Yoshihide e altri, Identification of an imprinted U2af binding protein related sequence on mouse chromosome 11 using the RLGS method, "Nature genetics", 6, 1994, pp. 33-40.
Kitsberg 1993: Kitsberg, Daniel e altri, Allele-specific replication timing of imprinted gene regions, "Nature", 364, 1993, pp. 459-463.
Leighton 1995: Leighton, Philip A. e altri, Disruption of imprinting caused by deletion of the H19 gene region in mice, "Nature", 375, 1995, pp. 34-39.
Li 1993: Li, En - Beard, Caroline - Jaenisch, Rudolf, Role for DNA methylation in genomic imprinting, "Nature", 366, 1993, pp. 362-365.
Reik 1989: Reik, Wolf, Genomic imprinting and genetic disorders in man, "Trends in genetics", 5, 1989, pp. 331-336.
Solter 1988: Solter, Davor, Differential imprinting and expression of maternal and paternal genomes, "Annual review of genetics", 22, 1988, pp. 127-146.
Strain 1995: Strain, Lisa e altri, A human parthenogenetic chimaera, "Nature genetics", 11, 1995, pp. 164-169.
Surani 1986: Surani, M. Azim, Evidences and consequences of differences between maternal and paternal genomes during embryogenesis in the mouse, in: Experimental approaches to mammalian embryonic development, edited by Janet Rossant, Roger A. Pedersen, Cambridge, Cambridge University Press, 1986, pp. 401-435.