
Malattie da prioni e influenza aviaria
Malattie da prioni e influenza aviaria
Le malattie da prioni e l'influenza aviaria, entità profondamente diverse tra loro, sono esempi paradigmatici di malattie infettive emergenti. Questo loro carattere risiede nella intrinseca variabilità degli agenti che ne sono causa, nella mutevolezza della loro ecologia e nel frequente stabilirsi di cicli ed equilibri nuovi tra ambiente, specie ospiti e agenti infettivi. Nel caso dell'encefalopatia spongiforme bovina (BSE), le moderne tecnologie di riciclaggio e riutilizzo dei rifiuti animali e di distribuzione su scala mondiale dei mangimi hanno creato un'occasione unica per la diffusione del nuovo agente. Analogamente, l'allevamento avicolo intensivo, caratterizzato da elevatissime densità di animali per unità di superficie, cicli produttivi brevi e veloci che creano un continuo rinnovo del substrato di ospite disponibile e recettivo, così come le condizioni di promiscuità uomo-animale di certe aree del mondo più povere, possono creare occasioni eccezionali di diffusione per i virus dell'influenza aviaria.
La natura degli agenti responsabili di queste patologie e la difficoltà del loro controllo contribuiscono a caratterizzarle come emergenti. I prioni, agenti trasmissibili che, in base alla teoria più accreditata, sarebbero privi di acidi nucleici e costituiti esclusivamente dalla forma strutturalmente alterata di una proteina normalmente espressa dall'organismo colpito, continuano a rappresentare un ambito di ricerca e intervento enigmatico e ancora pieno di incertezze. Da qui scaturiscono le difficoltà nel definire misure di prevenzione e controllo calibrate e nel delineare scenari di rischio attendibili. Le più note tra le malattie da prioni sono la BSE, la malattia di Creutzfeldt-Jakob dell'uomo, la scrapie della pecora e la chronic wasting disease, unica malattia da prioni che colpisce animali allo stato selvatico e che appare attualmente in rapida diffusione tra i Cervidi dell'America Settentrionale.
Anche buona parte delle ragioni della popolarità dell'influenza aviaria come malattia emergente deriva dalle proprietà dei virus che ne sono responsabili. Le particolari caratteristiche del genoma a RNA segmentato dei virus influenzali sono all'origine delle loro enormi potenzialità in termini di variabilità genetica e di possibilità adattative. Tali caratteristiche ne hanno decretato il successo evolutivo e li hanno portati a occupare un posto di primo piano tra le malattie infettive che sono oggetto prioritario di interesse a livello mondiale.
Il 75% degli agenti patogeni definibili come 'emergenti' ha un'origine zoonosica, deriva cioè dalla trasmissione all'uomo di microrganismi propri delle specie animali; spesso, come nel caso dell'influenza aviaria, riconoscono nelle specie animali selvatiche il loro serbatoio naturale. In molti casi ‒ e tra questi anche la BSE e l'influenza aviaria ‒ la globalizzazione dei mercati di animali e prodotti unita alla crescita degli spostamenti delle persone su lunghe distanze, fa sì che i focolai di malattia corrano il rischio di trasformarsi in epidemie su scala mondiale.
Le malattie da prioni: caratteristiche e teorie eziologiche
Le malattie da prioni, o encefalopatie spongiformi trasmissibili (EST), rappresentano un gruppo di malattie neurodegenerative che colpiscono l'uomo e gli animali. Nonostante non possiedano alcun carattere clinico patologico proprio delle malattie infettive, le EST mostrano la sorprendente proprietà di essere trasmissibili. L'inoculazione in animali da laboratorio di materiale cerebrale proveniente da soggetti malati è infatti in grado di riprodurre sperimentalmente la malattia. La natura degli agenti delle EST è sin dalla fine degli anni Cinquanta del Novecento al centro di un acceso e appassionante dibattito scientifico. L'iniziale denominazione di 'virus lenti' o 'non convenzionali' ha perso progressivamente considerazione per il fallimento di tutti i tentativi di identificare un acido nucleico virale. Negli anni Sessanta venne dimostrata sperimentalmente la trasmissibilità di queste malattie, mentre gli agenti delle EST divennero una vera curiosità scientifica quando ne fu svelata l'estrema resistenza alle radiazioni ultraviolette, ionizzanti e ad altri trattamenti chimico-fisici efficaci nell'inattivazione degli altri microrganismi.
Nel 1981 venne dimostrata negli estratti cerebrali di animali malati, la presenza di strutture fibrillari che si scoprì in seguito essere composte da una glicoproteina che Stanley Prusiner chiamò proteina prionica o PrP (Proteinaceous infectious particle). La PrP rappresentò la base sulla quale Prusiner sviluppò la 'teoria prionica', una teoria rivoluzionaria che gli avrebbe valso nel 1997 il Premio Nobel per la medicina o fisiologia. Essa riconosceva l'esistenza di una nuova classe di agenti trasmissibili, i prioni, di natura esclusivamente proteica e privi di acidi nucleici. L'origine della PrP venne svelata nel 1985 dal gruppo di Charles Weissmann, il quale riconobbe che essa non era una componente o un prodotto dell'agente infettante, ma una proteina sintetizzata dallo stesso organismo ospite.
La funzione fisiologica della PrP è tuttora oscura; al contrario, il ruolo cruciale svolto da essa nella patogenesi delle EST è provato dalla completa resistenza alle EST mostrata dai topi transgenici mancanti del gene della PrP.
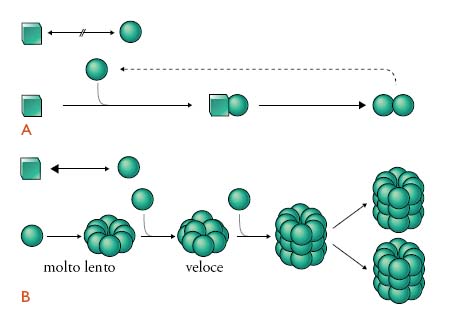
Nonostante le lacune tuttora esistenti, numerose evidenze scientifiche supportano la teoria di Prusiner; i prioni rappresenterebbero perciò una nuova classe di agenti infettivi, costituiti dalla forma patologica di una proteina cellulare normalmente espressa nell'organismo, denominata PrPC. Secondo il modello proposto da Prusiner, l'evento patogenetico centrale delle malattie da prioni è rappresentato dal cambiamento della struttura conformazionale della PrPC in una nuova isoforma patologica, denominata PrPSc (da PrP associata alla scrapie, malattia ovina prototipo delle EST). Questa avrebbe a sua volta, la capacità di 'imporre' o trasmettere alla PrPC la propria struttura conformazionale, replicando se stessa e innescando un processo di accumulo esponenziale nel sistema nervoso centrale (fig. 2). Questo processo esita in una malattia neurodegenerativa progressiva e fatale caratterizzata da vacuolizzazione dei neuroni e della sostanza grigia cerebrale (degenerazione spongiforme) e da accumulo di PrPSc nel tessuto nervoso in forma di aggregati e, talvolta, placche di amiloide. Indagini strutturali hanno mostrato nell'isoforma patologica un elevato contenuto in foglietti β rispetto alla PrPC, costituita prevalentemente da α-eliche. Per spiegare cosa dia l'avvio al processo patologico, Prusiner suggerisce un modello nel quale i prioni sono responsabili di malattie la cui origine è alternativamente spontanea (forme sporadiche), genetica o infettiva.
La malattia di Creutzfeldt-Jakob (MCJ) dell'uomo, che si manifesta con una incidenza di circa un caso per milione di abitanti per anno, sarebbe dovuta ‒ nella sua forma sporadica che comprende l'85-90% dei casi di EST umane ‒ alla conversione occasionale e spontanea della PrPC in PrPSc. Le forme genetiche di MCJ, così come le rare EST denominate sindrome di Gerstmann-Sträussler Sheinker e insonnia familiare fatale, sono invece legate a mutazioni del gene della PrP; in questi casi la conversione sarebbe facilitata dalla presenza di tali mutazioni, putativamente in grado di destabilizzare l'assetto conformazionale della proteina. La prima molecola di PrPSc sarebbe a quel punto capace di innescare il processo patologico. Nelle forme infettive, infine, la PrPSc deriverebbe da fonti esogene. Appartengono a questo gruppo il kuru, patologia oggi scomparsa e un tempo legata a rituali cannibalici praticati presso remote tribù della Papua Nuova Guinea, le forme iatrogene di MCJ, acquisite attraverso pratiche medico-chirurgiche realizzate con materiali o strumenti accidentalmente contaminati dall'agente della MCJ, e la variante della MCJ (vMCJ) che evidenze epidemiologiche e sperimentali attribuiscono al consumo di alimenti contaminati dall'agente della BSE. La vMCJ, descritta nel 1996, rappresenta la prima malattia da prioni per la quale viene dimostrata la capacità di trasmettersi da una specie animale all'uomo. Recentemente è stato dimostrato che una particolare forma di PrP ricombinante ottenuta da Escherichia coli, può essere convertita in strutture fibrillari ricche di foglietti β. Questo prodotto, inoculato in topi transgenici che esprimono quella particolare forma di PrP, è in grado di determinare una malattia con le caratteristiche delle EST, a sua volta ulteriormente trasmissibile nel topo. Questo esperimento, mirato alla produzione di prioni 'sintetici', pur richiedendo ulteriori verifiche sperimentali, potrebbe rappresentare la prova definitiva della validità della teoria prionica.
Nel caso delle EST animali, si ritiene che la maggior parte sia acquisita per via alimentare. La BSE ‒ patologia del bovino legata al consumo di mangimi contenenti farine animali infette ‒ si è in seguito trasmessa, sempre attraverso alimenti contaminati, anche a numerose specie di Bovidi (antilopi, bisonte, jak) e felini (ghepardo, tigre, leone, ocelot) selvatici allevati presso zoo inglesi. In concomitanza con l'epidemia di BSE, numerosi casi di malattia sono stati registrati anche nei gatti domestici. Anche la scrapie della pecora e della capra, la più nota e diffusa tra le EST degli animali, si ritiene venga trasmessa per via orale, forse attraverso la contaminazione dei pascoli. Alcune evidenze suggeririscono la possibilità che il contagio possa avvenire anche per via transplacentare. Analoghe modalità di trasmissione sono ipotizzate per la chronic wasting disease, l'unica EST descritta in animali che vivono allo stato selvatico, attualmente in forte diffusione tra i cervi dell'America settentrionale.
Non sono note le modalità di trasmissione della encefalopatia trasmissibile del visone, una rara EST che occasionalmente ha falcidiato allevamenti di questa specie. Anche in questo caso, si sospetta la trasmissione per via orale attraverso l'accidentale somministrazione di carni provenienti da ovini affetti da scrapie.
I ceppi di prioni
La comparsa dell'agente della BSE, responsabile di oltre 180.000 casi di malattia nella specie bovina, ha ricordato che, al pari di molti microrganismi, i prioni mostrano una propria variabilità spiegabile con l'esistenza di ceppi diversi, capaci di modificarsi e con differente patogenicità e spettro di ospite. Tradizionalmente, i ceppi di prioni si distinguono attraverso lo studio del quadro clinico-patologico indotto nei topi da laboratorio inoculati sperimentalmente. Ciascun ceppo mostra peculiarità in termini di durata del periodo di incubazione, localizzazione topografica cerebrale e severità della degenerazione e dell'accumulo di PrPSc. I diversi quadri sono caratteristici per ogni ceppo e si mantengono costanti nel corso di ripetuti passaggi nella stessa linea murina. Pur essendo empirico e indiretto, questo sistema dimostra in maniera incontestabile l'esistenza di una variabilità di questi agenti.
L'approccio della microbiologia tradizionale alla comprensione del fenomeno della variabilità di ceppo ‒ strettamente legato alla variabilità negli acidi nucleici che trasmettono le informazioni genetiche ‒ appare tuttavia inadeguato nel caso dei prioni, agenti apparentemente privi di tali molecole. Il meccanismo attraverso il quale i prioni, costituiti dalla sola PrPSc, codificano e perpetuano una informazione che li rende capaci di esprimere forme diverse di malattia ha rappresentato da sempre un forte argomento a sfavore della teoria prionica. Tale meccanismo non risiede in differenze della sequenza amminoacidica della PrP; nella medesima linea di topi inbred, portatrice perciò della stessa sequenza della PrP, possono infatti replicare differenti ceppi di prioni. Nel corso degli anni sono stati ottenuti importanti elementi a supporto del fatto che la specificità di ceppo sia codificata dalla struttura conformazionale della PrPSc e ‒ attraverso lo studio di proteine di lieviti con proprietà analoghe alla PrP ‒ che la variabilità dei ceppi possa essere legata alle modalità di aggregazione delle fibrille di PrPSc, ovvero alla struttura quaternaria di tale proteina.
La barriera di trasmissione e le infezioni subcliniche
La trasmissione delle EST da una specie all'altra incontra una più o meno evidente 'barriera di trasmissione', ovvero una difficoltà nel passare alla nuova specie. Le basi molecolari di tale barriera risiedono, in parte, nel livello di omologia della sequenza amminoacidica della PrP della specie che trasmette l'infezione e di quella che la contrae. Questo è provato dal fatto che in topi transgenici recanti il gene della PrP della specie che trasmette l'infezione la barriera interspecifica è, in molti casi, abolita e i tempi di incubazione risultano molto brevi. È questo il caso di topi portatori della PrP umana inoculati con MCJ; tuttavia, se gli stessi topi vengono inoculati con la vMCJ (quindi sempre nelle stesse condizioni di omologia di sequenza), i tempi di incubazione arrivano a essere addirittura più lunghi di quelli che si osservano nei topi wild type. È evidente pertanto che la barriera di trasmissione è un fenomeno cui partecipano almeno due componenti: la sequenza della PrP ed il ceppo di agente.
Il gene della PrP è altamente conservato nei Mammiferi. È stato suggerito che un limitato ma significativo numero di conformazioni della PrPSc sia possibile dal punto di vista termodinamico; queste costituirebbero il range di ceppi di prioni osservabile. Nell'ambito di una singola specie di mammifero, solo una parte di queste conformazioni sarebbe possibile. Qualora vi sia sostanziale analogia nelle conformazioni favorite della PrPSc di due specie, la trasmissione interspecifica verrebbe facilitata. Al contrario, lì dove non vi siano conformazioni preferenziali in comune, la trasmissione incontrerebbe difficoltà. È nota da tempo l'esistenza di ceppi di prioni che, pur patogeni per una certa specie, non producono malattia quando trasmessi a un'altra specie. Recenti studi hanno dimostrato che questo stato di apparente resistenza nasconde in realtà la replicazione e il lento e progressivo adattamento dell'agente al nuovo ospite, che funge perciò da portatore sano. È questa la ragione del timore che a fronte di 174 casi di vMCJ sinora registrati, il numero dei soggetti infetti senza segni di malattia possa essere molto più alto. Questo spiega anche l'attuale cautela adottata per prevenire un'eventuale ricircolazione intraspecifica dell'agente della BSE nella popolazione umana attraverso, per esempio, gli emoderivati o le pratiche chirurgiche.
Genetica delle malattie da prioni
Dalla scoperta del fondamentale ruolo della PrP nelle EST, molti studi si sono concentrati sul suo gene codificante. Tali studi hanno dimostrato, nella maggior parte delle specie animali, una notevole variabilità del gene della PrP. Nell'uomo, oltre alle numerose forme familiari nelle quali la malattia è legata a mutazioni del gene, è noto il ruolo del polimorfismo metionina/valina al codone 129. Sebbene oltre il 90% dei casi di MCJ sporadica si registrano in pazienti omozigoti per metionina o valina, tali genotipi sono presenti nella popolazione europea con una frequenza, rispettivamente, del 47 e del 7%. L'omozigosi per metionina al codone 129 ricorre inoltre in tutti i casi di vMCJ. Il ruolo della genetica nelle EST è altrettanto rilevante in campo animale. Nella specie ovina, in particolar modo, l'effetto delle diverse variazioni del gene della PrP nel condizionare la sensibilità e la resistenza alla scrapie è noto da tempo, tanto che l'UE ha recentemente varato un ambiziosissimo piano di controllo delle EST nella popolazione ovina europea basato sulla selezione genetica dei soggetti portatori del genotipo maggiormente resistente alla malattia. Il varo di una strategia tanto impegnativa e costosa indica l'attenzione che le autorità sanitarie europee, in seguito alla drammatica crisi della BSE, hanno posto al problema delle malattie da prioni negli animali e al loro rischio per l'uomo.
L'influenza aviaria: descrizione e classificazione
L'influenza aviaria è una malattia altamente contagiosa degli uccelli, descritta per la prima volta in Italia nel 1878 da Edoardo Perroncito. Da allora la malattia è stata segnalata in tutte le aree del mondo causando devastanti perdite economiche negli allevamenti avicoli (pollo, tacchino, anatra, oca, quaglia, fagiano); tali perdite sono dovute sia all'alta mortalità, sia alle drastiche misure di controllo che, al fine di prevenire l'ulteriore diffusione della malattia, prevedono la pronta distruzione di tutti gli animali presenti nell'area infetta. I virus influenzali sono virus a RNA segmentato appartenenti alla famiglia Orthomyxoviridae, classificati nei tipi A, B e C in base a differenze antigeniche della nucleoproteina e della proteina di matrice. Quando si parla di 'influenza umana' o di 'influenza aviaria' ci si riferisce alle malattie causate da virus influenzali che colpiscono prevalentemente l'uomo o gli uccelli. I virus di tipo A possono infettare l'uomo e varie specie animali (Uccelli, suini, cavalli, foche, Cetacei), ma riconoscono negli Uccelli selvatici il loro serbatoio naturale. I virus dell'influenza aviaria appartengono al tipo A. Essi sono ulteriormente classificati in sottotipi in base agli antigeni dell'emoagglutinina (H) e della neuroamminidasi (N) presenti sulla loro superficie. Esistono almeno sedici diversi sottotipi di virus influenzali di tipo A relativamente a differenze dell'emoagglutinina e nove relativamente alla neuroamminidasi.
I virus influenzali aviari possono essere responsabili di forme cliniche estremamente severe e a elevata mortalità (HPAI, Highly pathogenic avian influenza), così come di forme blande a bassa mortalità o di infezioni subcliniche (LPAI, Low pathogenic avian influenza). Tutti i virus aviari HPAI descritti sino a oggi appartengono ai sottotipi H5 e H7. Un ruolo chiave nella patogenicità dei virus influenzali aviari è svolto dall'emoagglutinina, glicoproteina determinante per la penetrazione e replicazione del virus nella cellula ospite. La possibilità di svolgere il suo ruolo dipende dall'attivazione di un suo precursore, operata attraverso il clivaggio della molecola in due subunità da parte delle proteasi cellulari. È stato osservato che nei ceppi avirulenti, tale clivaggio si verifica in un numero limitato di tipi cellulari; il virus è perciò in grado di colonizzare e replicarsi solo a livello del tratto respiratorio o intestinale, provocando quadri clinici modesti o infezioni subcliniche. Al contrario, il clivaggio dell'emoagglutinina dei ceppi virulenti è mediato da proteasi presenti in un elevato numero di tipi cellulari, dando ragione delle infezioni sistemiche e dei severi quadri clinici osservabili nei polli colpiti dai virus HPAI. L'attivazione proteolitica della emoagglutinina è condizionata da specifiche sequenze amminoacidiche, nel sito di clivaggio. Il confronto delle sequenze ha dimostrato, nei sottotipi H5 e H7 ad alta virulenza, la presenza di ripetizioni di amminoacidi basici nel sito di clivaggio dell'emoagglutinina; al contrario, il sito di clivaggio nei virus LPAI H5 e H7 mostra solitamente un singolo residuo basico. La diversa patogenicità dei virus influenzali aviari ‒ che rappresenta comunque un carattere poligenico ‒ sembrerebbe perciò essere particolarmente condizionata dal differente corredo enzimatico delle popolazioni cellulari dei vari organi e quindi dalla capacità degli stessi di fungere da attivatori del clivaggio dell'emoagglutinina.
Epidemiologia
L'epidemiologia dei virus influenzali aviari è legata agli Uccelli selvatici e, in particolare, a quelli acquatici (Anseriformi e Caradriformi). Queste specie sono considerate il serbatoio naturale dei virus influenzali, che albergano e replicano frequentemente in forma subclinica a livello intestinale. La fonte di infezione per i volatili domestici è difficilmente identificabile, tuttavia è ragionevole presumere che spesso derivi da contatto diretto o indiretto con volatili selvatici. La maggior parte dei virus che circolano tra gli Uccelli selvatici è poco o affatto virulento per le specie domestiche; è tuttavia dimostrato che virus LPAI H5 e H7, a seguito di cicli ripetuti di replicazione nelle specie aviarie domestiche possono dare origine ai sottotipi HPAI. Una volta giunta in un allevamento l'infezione si diffonde rapidamente attraverso le feci e i secreti orali e nasali. Lo spostamento di animali o la contaminazione indiretta di materiale, attrezzature e personale può facilmente trasferire l'infezione da un allevamento all'altro.
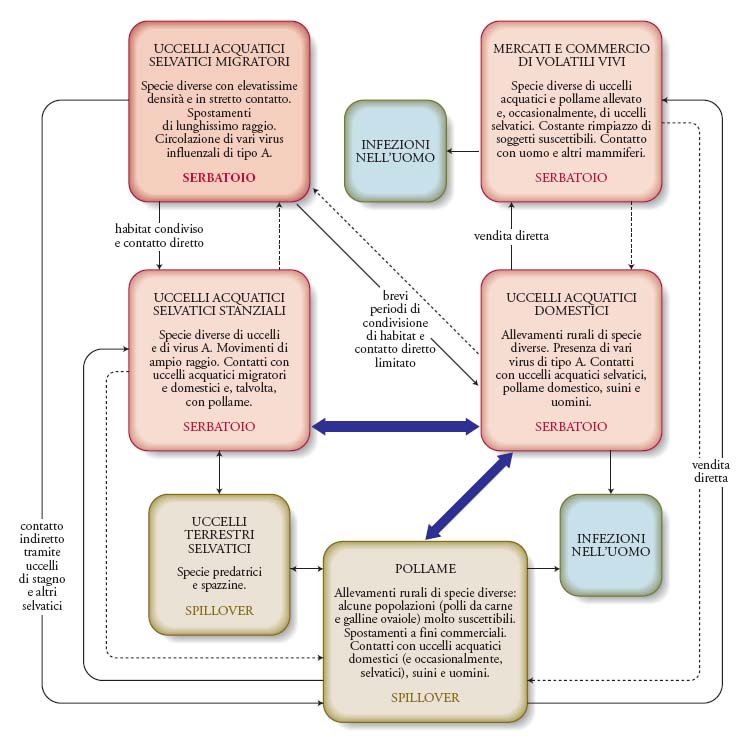
Sebbene questo rappresenti il modello di base dell'epidemiologia dei virus influenzali aviari, un'epidemia può essere compresa solo attraverso un approccio ecologico di ampia prospettiva, con specie che fungono, alternativamente, da serbatoi dell'infezione, da ospiti spillover (che replicano e trasmettono l'agente ma non sono in grado di mantenere un'epidemia per lungo tempo in assenza dell'ospite serbatoio) e da ospiti aberranti che, come nel caso dell'uomo, pur potendosi infettare non sono generalmente in grado di trasmettere l'infezione. Le ondate epidemiche osservate in Asia tra il 2003 e il 2005 a opera del virus A/H5N1 riconoscono un'epidemiologia estremamente complessa (fig. 3) nella quale il ruolo di serbatoio viene svolto da popolazioni di Uccelli selvatici o domestici in almeno quattro contesti ecologici differenti: quello silvestre, nel caso delle popolazioni di Uccelli acquatici migratori e stanziali, e quello antropico degli allevamenti rurali di Uccelli acquatici domestici (oche, anatre) e dei mercati di uccelli vivi. In tale ciclo, i polli costituiscono chiari esempi di ospiti spillover; l'uomo rappresenta invece soltanto un ospite aberrante, a rischio tuttavia di passare ‒ a causa di una eventuale modificazione del virus ‒ al ruolo di spillover, condizione favorevole all'avvio della trasmissione interumana e all'inizio di una fase pandemica.
I virus influenzali vanno incontro a frequenti modifiche, dando origine a nuovi ceppi o a nuovi sottotipi. Tali modifiche possono avvenire attraverso due modalità. La prima, definita come antigenic drift, avviene continuamente nel tempo ed è legata da singole mutazioni del corredo genetico virale, sotto la pressione selettiva operata dal sistema immunitario dell'ospite. La continua selezione e comparsa di nuovi ceppi virali spiega, nel caso dei virus influenzali umani, la limitata durata della protezione della vaccinazione antinfluenzale. La seconda modalità, denominata antigenic shift, si verifica più raramente ed è causa di modifiche sostanziali e improvvise del virus, spesso attraverso meccanismi di riassortimento genico tra virus di diversa origine che infettano lo stesso organismo. È il caso del suino, che può fungere da mixing vessel per la generazione di nuovi stipiti virali risultanti dal riassortimento umano-aviaro. Il risultato è la comparsa di virus nuovi, nei confronti dei quali le popolazioni esposte non possiedono alcuna memoria immunitaria. Se un nuovo sottotipo di influenza A infetta la popolazione umana si creano in questi casi le condizioni per lo sviluppo di epidemie su scala mondiale (pandemie).
Il rischio per l'uomo
I virus aviari generalmente non infettano l'uomo, tuttavia dal 1997 a oggi sono stati descritti oltre 150 casi di infezione nell'uomo:
1997, Cina (Hong Kong). Primo caso documentato di trasmissione diretta dagli Uccelli all'uomo di un virus aviario (A/H5N1). Diciotto persone che avevano avuto stretti contatti con polli in corso di un focolaio di HPAI furono ospedalizzate, sei di queste morirono.
1999, Cina (Hong Kong). Due casi di malattia risoltisi con la guarigione, causati da un virus aviario LPAI (A/H9N2) probabilmente contratto direttamente da pollame infetto.
2002, Virginia (USA). Evidenza sierologica di infezione (A/H7N2) in una persona durante una epidemia nei polli.
2003, Cina (Hong Kong). Due casi di HPAI (A/H5N1) tra i membri di una famiglia di Hong Kong di ritorno da un viaggio in Cina. Uno dei due casi ebbe sito fatale.
2003, Olanda. Ottantanove casi umani (A/H7N7), in prevalenza tra il personale professionalmente esposto, durante un'epidemia nel pollame. Sintomi da molto lievi (congiuntivite, lieve febbre) a gravi; un caso mortale in un veterinario. Per la prima volta viene documentata la probabile trasmissione interumana da tre lavoratori dell'industria avicola a loro familiari.
2003, Cina (Hong Kong). Un caso di malattia da LPAI (A/H9N2) risoltosi con la guarigione.
2003, New York (USA). Un caso di malattia (A/H7N2), risoltosi con la guarigione, in un paziente già affetto da altre patologie.
2004, Canada. Casi umani di congiuntivite associati ad un focolaio nel pollo (A/H7N3).
2004-2005, Asia. Dal gennaio 2003 sono stati segnalati oltre 130 casi umani (A/H5N1) di cui circa la metà mortali, in Cambogia, Cina, Thailandia, Vietnam, Indonesia, Turchia.
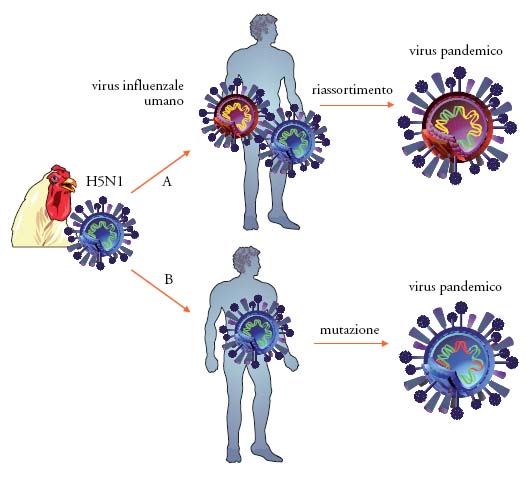
Tale casisitica indica con chiarezza che la maggior parte dei casi di infezione umana deriva dal contatto diretto con polli infetti o materiali contaminati dalle deiezioni, mentre non c'è alcuna evidenza di trasmissione attraverso il consumo di carni avicole o uova dopo la cottura. Si sa tuttavia ancora poco sui sottotipi e ceppi di influenza aviaria potenzialmente patogeni per l'uomo. Le maggiori preoccupazioni per l'uomo derivano tuttavia non dalla trasmissione del virus dalle specie aviarie, bensì dalla possibilità che virus influenzali aviari possano mutare o riassortirsi con virus influenzali umani dando origine a un nuovo stipite umano-aviario in grado di trasmettersi efficientemente da uomo a uomo e potenzialmente in grado di scatenare una nuova pandemia. Affascinanti studi di sequenziamento ed analisi filogenetica realizzati recentemente da Jeffriey K. Taubenberger hanno mostrato che virus pandemici possono generarsi non solo attraverso meccanismi di riassortimento, ma anche tramite mutazioni (fig. 4); il virus della pandemia 'spagnola' del 1918 (A/H1N1) non era infatti uno stipite riassortante, bensì un virus interamente aviario adattato all'uomo attraverso mutazioni.
La seconda pandemia registrata nel secolo scorso, denominata 'asiatica' si verificò nel 1957-1958. I morti stimati furono circa un milione; il virus reponsabile (A/H2N2) aveva origine da riassortimento tra un virus umano e uno aviario. Una terza pandemia, denominata Hong Kong si ebbe negli anni 1968-1969. Anche in questo caso il virus responsabile (A/H3N2), che causò circa un milione di morti, derivava da riassortimento umano-aviario. L'ultima epidemia su vasta scala è la 'russa' del 1977. L'identità del virus (A/H1N1) con quello che aveva circolato nell'uomo negli anni Cinquanta, fa ritenere che possa aver avuto origine da una accidentale fuga da qualche laboratorio. Non è possibile prevedere nè il momento preciso, nè l'impatto di una futura pandemia influenzale; la severità della malattia, la rapidità della sua diffusione e i gruppi a rischio nella popolazione sono variabili strettamente correlate al virus in causa e al suo assetto genetico.
Dal 2003 l'attenzione è rivolta nei confronti del virus A/H5N1 responsabile di epidemie nel pollame domestico e di episodi di mortalità tra gli Uccelli selvatici in molti Paesi asiatici (Cambogia, Cina, Hong Kong, Indonesia, Giappone, Laos, Corea del Sud, Thailandia, Vietnam, Kazakistan, Malesia, Mongolia). L'infezione, probabilmente seguendo le rotte migratorie degli Uccelli acquatici, si è diffusa nel corso del 2005 anche in alcuni Paesi europei (Russia, Ucraina, Romania, Turchia, Croazia). La perdurante circolazione di questo virus e i frequenti cambiamenti genetici e antigenici documentati preoccupano per la possibilità che tali modificazioni diano origine a un virus con capacità di trasmissione interumana, condizione indispensabile per il verificarsi di una pandemia. Per far fronte a tale rischio, la rete di sorveglianza nei confronti dell'influenza è attiva a livello mondiale e sono stati predisposti protocolli di intervento nei quali si integrano misure di profilassi e controllo in ambito umano e veterinario. I farmaci antivirali rappresentano la prima difesa in caso di pandemia e in mancanza di una immediata disponibilità del vaccino diretto contro il virus pandemico. Attualmente non è ancora disponibile un vaccino, per uso umano, contro il virus H5N1. Tuttavia i laboratori facenti parte della rete globale di sorveglianza dell'influenza, hanno preparato un virus prototipo che costituisce la base per l'allestimento del vaccino specifico.
Bibliografia
Agrimi 2003: Agrimi, Umberto e altri, Animal transmissible spongiform encephalopathies and genetics, "Veterinary research communications", 27, 2003, pp. 31-38.
Alper 1966: Alper, Tikvah - Haig, D.A. - Clarke, Michael C., The exceptionally small size of the scrapie agent, "Biochemichal and biophysical research communications", 22, 1966, pp. 278-284.
Alper 1967: Alper, Tikvah e altri, Does the agent of scrapie replicate without nucleic acid?, "Nature", 214, 1967, pp. 764-766.
Bessen, Marsh 1992: Bessen, Richard A. - Marsh, Richard F., Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent, "Journal of virology", 66, 1992, pp. 2096-2101.
Bruce 1993: Bruce, Moira E., Scrapie strain variation and mutation, "British medical bulletin", 49, 1993, pp. 822-838.
Bruce 1997: Bruce, Moira E., Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent, "Nature", 389, 1997, pp. 498-501.
Büeler 1992: Büeler, Hansruedi e altri, Mice devoid of PrP are resistant to scrapie, "Cell", 73, 1992, pp. 1339-1347.
Capua, Marangon 2000: Capua, Ilaria - Marangon, Stefano, The avian influenza epidemic in Italy (1999-2000): a review, "Avian pathology", 29, 2000, pp. 289-294.
Collinge 2001: Collinge, John, Prion diseases of humans and animals: their causes and molecular basis, "Annual review of neuroscience", 24, 2001, pp. 519-550.
Cunningham 2005: Cunningham, Andrew, A walk on the wild side: emerging wildlife diseases, "British medical journal", 331, 2005, pp. 1214-1215.
Diringer 1983: Diringer, Heino e altri, Scrapie infectivity, fibrils and low molecular weight protein, "Nature", 306, 1983, pp. 476-478.
Hill 1997: Hill, Andrew F. e altri, The same prion strain causes vCJD and BSE, "Nature", 389, 1997, pp. 448-450.
Horimoto, Kawaoka 2001: Horimoto, Taisuke - Kawaoka, Yoshihiro, Pandemic threat posed by avian influenza A viruses, "Clinical microbiology reviews", 14, 2001, pp. 129-149.
Legname 2004: Legname, Giuseppe e altri, Synthetic mammalian prions, "Science", 305, 2004, pp. 673-676.
Merz 1981: Merz, Patricia A. e altri, Abnormal fibrils from scrapie-infected brain, "Acta neuropathologica", 54, 1981, pp. 63-74.
Morris, Jackson 2005: Morris, Roger S. - Jackson, Ron, Epidemiology of H5N1 avian influenza in Asia and implications for regional control. A contracted report for the Food and Agriculture Organization of the United Nations covering the period january 2003 to february 2005, Palmerston North, EpiCentre, Massey University (New Zealand), 2005.
Oesch 1985: Oesch, Bruno e altri, A cellular gene encodes scrapie PrP 27-30 protein, "Cell", 40, 1985, pp. 735-746.
Perroncito 1878: Perroncito, Edoardo, Epizoozia tifoide nei gallinacei, "Annali Accademia Agricoltura Torino", 21, 1878, pp. 87-126.
Prusiner 1982: Prusiner, Stanley B., Novel proteinaceous infectious particles cause scrapie, "Science", 216, 1982, pp. 136-144.
Prusiner 1983: Prusiner, Stanley B., Scrapie prions aggregate to form amyloid-like birefringent rods, "Cell", 35, pp. 349-358.
Prusiner 1999: Prusiner, Stanley B., An introduction to prion biology and diseases, in: Prion biology and diseases, edited by Stanley B. Prusiner, Cold Spring Harbor (N.Y.), Cold Spring Harbor Laboratory Press, 1999, pp. 1-66.
Race 2002: Race, Richard e altri, Subclinical scrapie infection in a resistant species: persistence, replication and adaptation of infectivity during four passages, "Journal of infectious diseases", 186, 2002, pp. 166-170.
Russell, Webster 2005: Russell, Charles J. - Webster, Robert G., The genesis of a pandemic influenza virus, "Cell", 123, 2005, pp. 368-371.
Soldevila 2006: Soldevila, Marta e altri, The prion protein gene in humans revisited: lesson from a worldwide resequencing study, "Genome research", 16, 2006, pp. 231-239 (article published online ahead of print at http://www.genome.org/cgi/doi/10.1101/gr.4345506).
Taubenberger 2005: Taubenberger, Jeffrey K. e altri, Characterization of the 1918 influenza virus polymerase genes, "Nature", 437, 2005, pp. 889-893.
Tanaka 2004: Tanaka, Motosumi e altri, Conformational variations in an infectious protein determine prion strain differences, "Nature", 428, 2004, pp. 323-328.
Weissmann 2004: Weissmann, Charles, The state of prion, "Nature reviews. Microbiology", 2, 2004, pp. 861-871.
Will 1996: Will, Robert G. e altri, A new variant of Creutzfeldt-Jakob disease in the UK, "Lancet", 347, 1996, pp. 921-925.