
Stereochimica
Stereochimica
La stereochimica può essere definita come lo studio della disposizione spaziale degli atomi all'interno di molecole o di aggregati molecolari e dei fenomeni che dipendono dalle diversità fra le disposizioni stesse. Secondo questa definizione la stereochimica dovrebbe abbracciare l'intero campo della chimica, dal momento che i fenomeni chimici sono legati in definitiva a tali diversità e alle differenze di energia a esse associate. In questo senso la stereochimica non rappresenta tanto un ramo della chimica, quanto, piuttosto, un modo di affrontarla. Comunque, in senso più stretto, il termine stereochimica è spesso usato per definire lo studio e l'analisi della stereoisomeria, ossia di quel fenomeno a causa del quale a una data formula di struttura può corrispondere più di un composto.
Composti differenti, seppur costituiti dagli stessi elementi combinati nelle stesse proporzioni, sono noti come isomeri. Il primo esempio noto risale al 1824, anno in cui venne osservato come il cianato d'argento [1a] e il fulminato d'argento [1b] avessero differenti proprietà pur fornendo gli stessi risultati all'analisi elementare. In una nota editoriale a piè di pagina in un lavoro di Friedrich Wöhler, Joseph Louis Gay-Lussac suggerì l'ipotesi che i due composti contenessero gli stessi atomi, ma combinati differentemente.
Ag+(N≡C−O)−
Ag+(C≡N−O)−
La formula di struttura di un composto definisce la costituzione, o meglio, i reciproci legami tra gli atomi di una molecola. Le formule di struttura di varie migliaia di composti vennero determinate, durante il XIX sec., unicamente per mezzo di metodi chimici; solo molto più tardi furono disponibili metodi fisici per definire la configurazione spaziale delle molecole. Durante questo periodo vennero riconosciuti anche alcuni tipi di isomeria. Gli isomeri di struttura corrispondono a differenti formule di struttura: essi sono spesso classificati in tre sottospecie. L'etere dimetilico [2a] e l'etanolo [2b] sono isomeri funzionali, contengono, cioè, gruppi funzionali diversi:
CH3-O-CH3
CH3-CH2-OH
L'1-cloropropano [3a] e il 2-cloropropano [3b] vengono definiti isomeri di posizione, differendo nella posizione del sostituente cloro; anche l'n-butano [3c] e l'isobutano [3d] potrebbero esser considerati isomeri di posizione,
Cl-CH2-CH2-CH3
CH3-CH-CH3
∣Cl
CH3-CH2-CH2-CH3
formula
ma sono più spesso definiti isomeri di catena, presentan-do una diversa disposizione degli atomi di carbonio costituenti. Comunque, casi di isomeria vennero osserva-ti anche quando gli isomeri avevano la stessa formula di struttura.
Definizioni
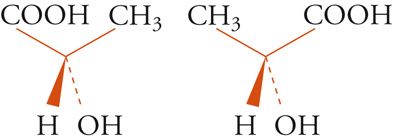
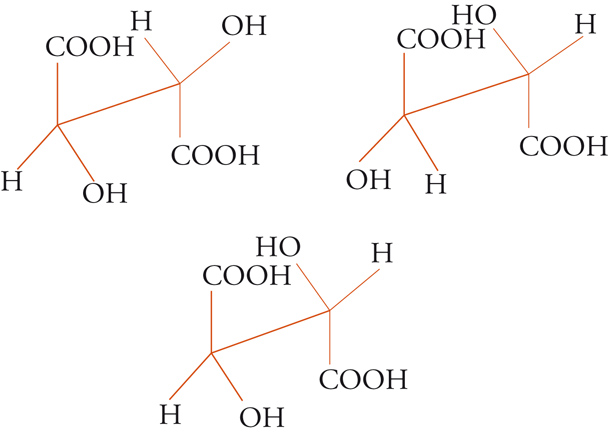
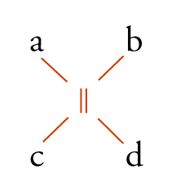
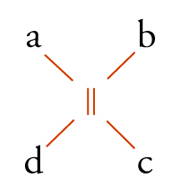
Con lo sviluppo del concetto di valenza direzionale, l'apparente ambiguità di composti isomeri con la stessa formula di struttura poté essere spiegata ammettendo che tali formule dovessero tener conto non solo dei reciproci legami degli atomi nella molecola, ma anche della loro disposizione spaziale. Si può dire che questa spiegazione, cui giunsero indipendentemente e quasi contemporaneamente, nel 1874, Jacobus H. van't Hoff e Joseph Achille Le Bel, segni l'inizio della stereochimica. Era stato osservato che alcune coppie di stereoisomeri ruotavano il piano della luce polarizzata in opposte direzioni, pur essendo altrimenti indistinguibili. Louis Pasteur, con le sue ricerche sull'acido tartarico, concluse che le molecole di acido tartarico destrogiro e levogiro erano dissimmetriche, cioè immagini speculari non sovrapponibili l'una all'altra. Altre coppie di isomeri ottici vennero trovate a partire dal 1874. Van't Hoff e Le Bel osservarono come tutti gli esempi noti avessero una caratteristica comune: la presenza di un atomo di carbonio con quattro sostituenti diversi, definito da van't Hoff atomo di carbonio asimmetrico. Infatti, ponendo ai vertici di un tetraedro, al cui centro si trovi un atomo di carbonio, quattro sostituenti diversi, si otterrebbero due sole strutture non sovrapponibili, tra loro correlate come un oggetto con la sua immagine speculare. Secondo questa descrizione, i due isomeri ottici dell'acido lattico corrispondono alle due strutture mostrate nella fig. 2, immagini speculari non sovrapponibili l'una all'altra. Si dice che le due strutture possiedono la stessa costituzione, ma configurazione opposta. Per l'acido tartarico, che contiene due atomi di carbonio asimmetrici, sono possibili tre forme (fig. 3) non interconvertibili per rotazione attorno al legame centrale. Due di queste forme corrispondono ai due acidi otticamente attivi, la terza a un acido otticamente inattivo, detto mesotartarico, avente proprietà chimiche e fisiche lievemente differenti. In ciascuna delle due forme otticamente attive i due atomi di carbonio asimmetrici presentano la stessa configurazione: sono correlati tra loro da un asse binario di rotazione. Nella forma meso i due atomi di carbonio sono correlati attraverso un centro di simmetria (asse improprio di rotazione) o, dopo rotazione attorno al legame centrale, da un piano di simmetria: la loro configurazione risulta opposta. Tale molecola può essere sovrapposta alla sua immagine speculare e ciò spiega la mancanza di attività ottica. Questo tipo di stereoisomeria, in cui coppie di isomeri corrispondono a immagini speculari non sovrapponibili, non richiede necessariamente la presenza di un atomo di carbonio asimmetrico (né, addirittura, la presenza di atomi di carbonio nella molecola). Queste coppie di isomeri sono ora denominate, di solito, enantiomeri. Stereoisomeri che non corrispondono a coppie di enantiomeri sono solitamente chiamati diastereoisomeri, sebbene per distinguere i vari casi che possono verificarsi siano stati anche usati alcuni altri termini. Così gli acidi maleico [4a] e fumarico [4b]
[4a] formula[4
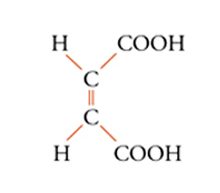
b] formula
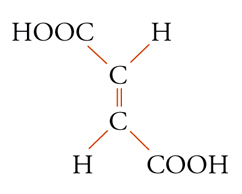
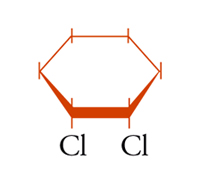
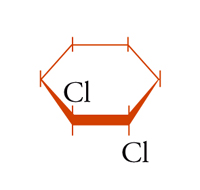
possono essere definiti isomeri geometrici. Anche in molecole cicliche possono aversi casi di isomeria geometrica o cis-trans: per esempio, nel cis- e trans-1,2-diclorocicloesano [5a] e [5b], dove l'isomero trans corrisponde effettivamente a una coppia di enantiomeri.
[5a] formula[5
b] formula
Pasteur, osservando come molti composti naturali, prodotti da organismi viventi, fossero otticamente attivi, concluse che gli organismi sono caratterizzati da una qualche intrinseca asimmetria, che conduce alla produzione di una sola delle due possibili molecole antipodi, laddove le sintesi di laboratorio dello stesso composto conducono a un prodotto otticamente inattivo consistente in una miscela equimolecolare di entrambi gli antipodi, ossia a una miscela racemica. Egli osservò che, in presenza di microrganismi, l'acido tartarico otticamente inattivo diviene gradualmente levogiro; le molecole destrogire si trasformano in acido piruvico, mentre quelle levogire restano inalterate. Metodi chimici per risolvere miscele racemiche vennero sviluppati a partire dalla fine del XIX secolo. Per esempio, l'addizione di una base otticamente attiva a una miscela di acidi racemici fornisce due sali diastereoisomeri, che possono essere separati per cristallizzazione frazionata. Per acidificazione di una soluzione di uno dei prodotti così separati e successiva ricristallizzazione si recupera poi uno degli enantiomeri allo stato puro.
È necessario aggiungere che la maggior parte degli elementi si presenta in più di una forma (per es., il carbonio come diamante e come grafite), le varie forme di un elemento sono solitamente dette forme allotropiche.
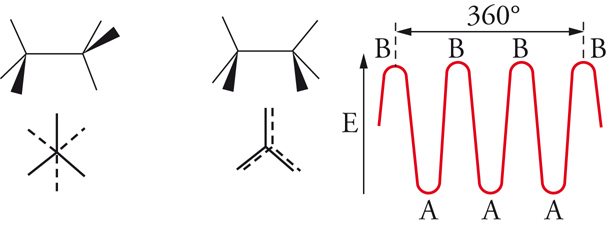
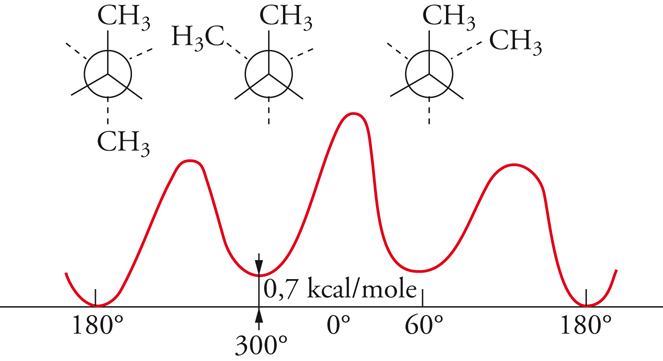
All'incirca fino al 1935 si riteneva che la rotazione intorno a legami chimici semplici fosse più o meno libera. Erano infatti state isolate molte coppie stabili di isomeri geometrici corrispondenti a una disposizione cis o trans dei sostituenti intorno a doppi legami, mentre nessun esempio, che implicasse rotazione intorno a un legame semplice, era noto. Nel 1936, Kenneth S. Pitzer mostrò che la rotazione attorno al legame semplice carbonio-carbonio dell'etano non era completamente libera, ma era associata a una barriera di energia di circa 3 kcal/mole. Il minimo di energia si presenta quando le due terne di atomi di idrogeno alle opposte estremità del legame assumono una posizione sfalsata, il massimo quando esse sono in posizione eclissata (fig. 4). Per l'etano, la barriera rotazionale ha una simmetria ternaria, mentre, per le rotazioni intorno al legame centrale del butano, le posizioni corrispondenti a rotazioni di 120° non sono più equivalenti (fig. 5). È noto che la struttura con i gruppi metilici terminali ruotati di 180° l'uno rispetto all'altro ha un'energia potenziale di circa 0,7 kcal/mole minore rispetto a quella nella quale i gruppi metilici sono ruotati di 60°. Le varie forme che una molecola può assumere per rotazione intorno ai legami sono dette conformazioni. Gli isomeri conformazionali, corrispondenti ai vari minimi di energia, possono essere rivelati con metodi spettroscopici, specialmente di risonanza magnetica nucleare e di assorbimento infrarosso, ma le barriere energetiche per le interconversioni di tali isomeri sono di solito così basse che già a temperatura ambiente si raggiunge rapidamente l'equilibrio. Anche nel caso degli isomeri conformazionali possiamo parlare di enantiomeri o di diastereoisomeri, proprio come nel caso degli stereoisomeri configurazionali, ma in verità la distinzione tra isomeri conformazionali e configurazionali non è ben definita. Talora questa distinzione è basata su criteri energetici, talvolta su modelli strutturali, entrambe le definizioni implicano una certa ambiguità. Sembra più opportuno considerare tutti i casi di isomeria formalmente derivabili per rotazione attorno a legami (inclusi i doppi legami, come nel caso dell'isomeria cis-trans) come esempi di isomeria conformazionale, qualunque possa essere il meccanismo di interconversione.
Aspetti teorici
Un oggetto non sovrapponibile alla sua immagine speculare è detto chirale, un semplice esempio è la mano. Gli oggetti sovrapponibili alle loro immagini speculari, per esempio un cubo, sono definite achirali. Supponendo che le molecole siano rigidi ordinamenti di atomi puntiformi, possiamo distinguere le molecole chirali da quelle achirali in base a proprietà di simmetria. La condizione necessaria e sufficiente per l'achiralità è la presenza di un elemento improprio di simmetria (elemento di simmetria di seconda specie), cioè un piano di riflessione, un centro d'inversione, assi alternanti n-ari di rotazione. La presenza di elementi di simmetria di prima specie (assi n-ari di rotazione) è compatibile con la chiralità, cosicché non è corretto identificare la chiralità con l'asimmetria. La parola dissimmetrico è spesso usata per indicare l'assenza di elementi impropri di simmetria, essa è perciò sinonimo di chirale.
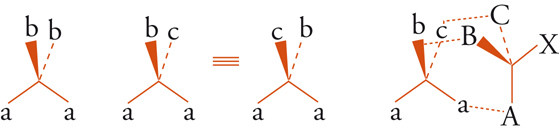
Esaminiamo ora alcune relazioni intramolecolari. Consideriamo dapprima una molecola tetraedrica Caabb con simmetria C2v. La rotazione intorno a un asse binario orienta la molecola in modo tale che risulta indistinguibile rispetto all'orientazione iniziale (fig. 6). Consideriamo ora una molecola tetraedrica Caabc con simmetria Cs: i due sostituenti a, a sono ora correlati per riflessione attraverso il piano di specularità, ma sono distinguibili con un mezzo di riconoscimento chirale, come suggerito nella fig. 6. Gli enzimi, responsabili dei processi chimici che avvengono in sistemi viventi, sono macromolecole chirali e perciò possono distinguere due siffatti sostituenti, detti enantiotopici e, talvolta, prochirali. Sono noti numerosi esempi biologici in cui i due gruppi enantiotopici subiscono una diversa modificazione chimica: per esempio, nell'ossidazione enzimatica dell'etanolo ad acetaldeide, solo uno dei due atomi di idrogeno enantiotopici del gruppo metilenico è rimosso in maniera specifica.
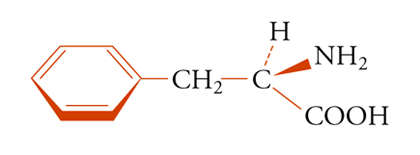
Sostituenti, che siano correlati da un asse puro di rotazione, sono indistinguibili con ogni mezzo e sono spesso definiti omotopici. Se almeno uno dei due sostituenti b o c della molecola tetraedrica Caabc (fig. 6) è chirale, la figura non ha più simmetria Cs e i due sostituenti a, a non sono più correlati da operazioni di simmetria. Questi gruppi possono esser chiamati diastereotopici. Un esempio è fornito dai due protoni metilenici della fenil-alanina [6]:
[6] formula
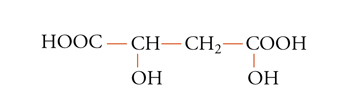
I due sostituenti a, a sono pure diastereotopici se entrambi gli altri due sostituenti sono chirali ed enantiomorfi. Un esempio è fornito dai protoni metilenici della forma meso dell'acido 2,4-diidrossiglutarico [7]:
[7] formula
Fino al 1950 non si sapeva con certezza quale delle due strutture enantiomorfe possibili, per ogni molecola otticamente attiva, corrispondesse all'isomero destrogiro e quale a quello levogiro. Fu comunque possibile correlare tra loro, con metodi chimici, le configurazioni di molti composti otticamente attivi, cioè stabilire le loro configurazioni relative a qualche composto di riferimento. A questo scopo alla gliceraldeide venne arbitrariamente assegnata la configurazione [8a] rappresentata dalla formula [8b] di proiezione secondo la convenzione di Fischer:
[8a] formula[8
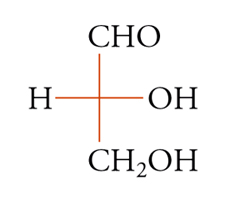
b] formula
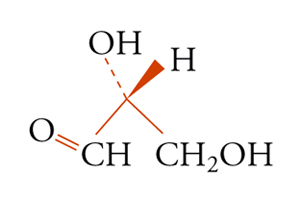
Era noto, per esempio, che tutti gli amminoacidi naturali presenti nelle molecole proteiche presentavano la stessa configurazione (L) all'atomo di carbonio asimmetrico e potevano esser rappresentati secondo la [9] riferendosi alla formula arbitraria per la gliceraldeide [8b]:
[9] formula
Un metodo generale per correlare la chiralità molecolare con quella macroscopica è basato su determinazioni di diffrazione di raggi X e venne proposto intorno al 1950 da Johannes M. Bijvoet. La disposizione spaziale degli atomi che ne risultò fu essenzialmente quella mostrata nella fig. 3 per l'acido destrogiro, disposizione che si accordava con le correlazioni chimiche basate sulla struttura della gliceraldeide. Risultò perciò corretta la convenzione di Fischer.
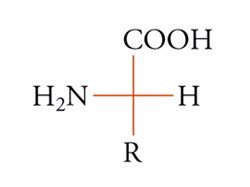
La configurazione assoluta di una molecola chirale può essere sempre descritta fornendo le coordinate spaziali (xi, yi, zi) degli atomi in un dato sistema di coordinate destrorso. Se per una qualsiasi ragione i vettori base X′, Y′, Z′ vengono sostituiti dai nuovi vettori X′, Y′, Z′, il sistema di nuove coordinate deve essere anch'esso destrorso, in questo caso il determinante della trasformazione lineare è positivo. La configurazione assoluta può anche essere descritta attraverso una rappresentazione grafica, come nella convenzione di Fischer, comunque nessuno dei due metodi citati è sempre praticabile. È quindi utile poter specificare la configurazione assoluta per mezzo di una notazione. Delle diverse che sono state proposte, la più usata è quella di Cahn, Ingold e Prelog. Nella sua formulazione più semplice, essa specifica la configurazione assoluta di un tetraedro chirale (sistema tridimensionale) e di conseguenza quella di quattro gruppi diversi qualsiasi disposti attorno a un atomo o a più generici elementi di asimmetria (centro, asse, piano). I quattro gruppi vengono posti in un dato ordine, secondo un insieme di regole arbitrarie. A seconda che i primi tre gruppi si susseguano in senso orario o antiorario sul piano che li contiene (dal punto di vista di un osservatore posto dalla parte del quarto gruppo), si applica il simbolo R o S (rectus o sinister). Inoltre per un centro pseudoasimmetrico, si applicano i simboli r o s all'atomo o ad altro elemento di asimmetria, o a ognuno di essi se ve ne sono diversi. Le principali regole per ordinare i gruppi sono le seguenti: i gruppi sono ordinati in prima analisi secondo il numero atomico decrescente degli atomi legati all'elemento di asimmetria, oppure, se non vi è distinzione, secondo quello degli atomi immediatamente seguenti e così via, procedendo fino a trovare atomi aventi numeri atomici diversi. I doppietti elettronici vengono considerati come sostituenti e viene loro assegnata la più bassa priorità. Se due gruppi differiscono solo isotopicamente, vengono ordinati secondo massa isotopica decrescente e se differiscono solo stereochimicamente l'ordine è cis prima di trans e R prima di S. Secondo queste regole, i gruppi attorno all'atomo di carbonio asimmetrico dell'amminoacido naturale L-alanina [10] vengono ordinati secondo la sequenza NH2〈COOH〈CH3〈H, cosicché la configurazione risulta S. Sebbene tutti gli α-amminoacidi delle
[10] formula
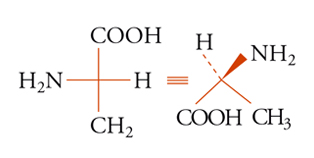
proteine abbiano la configurazione mostrata nella [9], a seconda della precedenza del gruppo R sul gruppo COOH, essi saranno R o S. In maggioranza risultano S, ma, per esempio, la L-cisteina (R=CH2SH) è R. L'acido tartarico destrogiro (fig. 3) è R, R.
Le olefine del tipo abC=Ccd sono achirali, ma i due possibili stereoisomeri [11a] e [11b] possono essere designati come seqcis (o Z, zusammen) e seqtrans (o E, entgegen), a seconda che i gruppi di più alto ordine alle due estremità del doppio legame siano in cis o in trans fra loro.
[11a] formula[11
b] formula3.
Stereochimica e trasformazioni chimiche
Sinora abbiamo considerato principalmente l'analisi e la descrizione di vari tipi di stereoisomeria (compresa l'isomeria conformazionale). Le nostre descrizioni sono state essenzialmente statiche, abbiamo cioè tralasciato del tutto il fatto che la chimica riguarda fenomeni dinamici. Gli aspetti stereochimici della reattività chimica sono della massima importanza per molte ragioni. Per esempio, è particolarmente importante per i composti di interesse biologico, quali i farmaci, per i quali si è constatato che, in genere, solo uno dei molti possibili stereoisomeri mostra la voluta attività biologica. Inoltre sicuramente gli studi stereochimici hanno contribuito notevolmente alla chiarificazione dei meccanismi di reazione chimica. Il miglior esempio è probabilmente la reazione di sostituzione nucleofila
[12] X− + CR1R2R3Y → Y− + CR1R2R3X
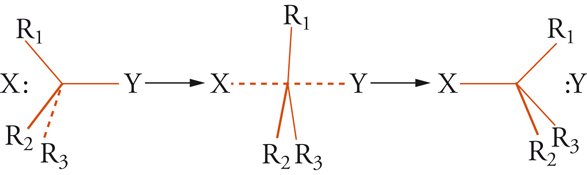
dove X− e Y− sono anioni elettrondonatori (nucleofili). Per questo tipo di reazione possono essere supposti due tipi limite di meccanismo denominati rispettivamente come SN1 e SN2. Nel primo meccanismo, il sostituente Y è rimosso come Y− fornendo uno ione carbonio planare [CR1R2R3]+, il quale può reagire con X− formando XCR1R2R3, oppure con Y− formando il composto di partenza. Nel secondo tipo di meccanismo il nucleofilo X− si avvicina al centro del triangolo R1R2R3. Nello stadio intermedio le distanze X…C e C…Y sono all'incirca uguali e la piramide CR1R2R3 diventa planare. La reazione ha fine quando si forma il legame X-C e si allontana Y− (fig. 7).
I due meccanismi limite mostrano una diversa cinetica, ma possono anche esser distinti, in linea di principio, attraverso considerazioni stereochimiche. Se il composto di partenza CR1R2R3Y fosse otticamente attivo, il meccanismo SN2 dovrebbe condurre a un prodotto otticamente attivo con inversione di configurazione assoluta, mentre il meccanismo SN1 fornirebbe prodotti racemici. Nella realtà, le reazioni di sostituzione nucleofila seguono l'uno o l'altro meccanismo a seconda della natura del sostituente Y, di R1, R2, R3 e del solvente usato nella reazione.
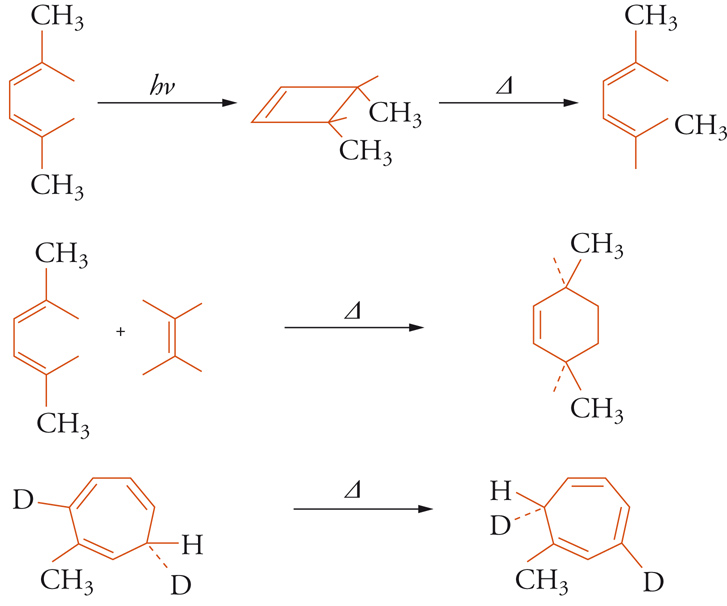
A partire dal 1965 Robert B. Woodward e Roald Hoffmann, in una serie di lavori classici, mostrarono come un'ampia gamma di reazioni stereospecifiche potesse essere raggruppata sotto il nome di reazioni pericicliche, interpretabili secondo la teoria degli orbitali molecolari. In tale gruppo rientrano le reazioni elettrocicliche, di cicloaddizione e sigmatropiche, di ciascuna delle quali è mostrato nella fig. 8 un esempio.
Ognuna delle reazioni mostrate nella fig. 8 potrebbe condurre, in linea di principio, a due stereoisomeri. Uno dei due possibili stereoisomeri si forma solo quando l'energia di attivazione necessaria è fornita per riscaldamento (reazione termica), mentre l'altro si forma se l'energia è fornita dalla luce (reazione fotochimica). La regola, che decide il decorso stereochimico della reazione, è basata sul numero, pari o dispari, di legami rotti e formati. Per esempio, la chiusura dell'anello del 2,5-dimetil-esa-2,4-diene (fig. 7) per via fotochimica potrebbe condurre ai due isomeri ottici del 3,4-dimetilciclobutene, il cis, che presenta i gruppi metilici dalla stessa parte del piano dell'anello, e il trans, con i gruppi metilici da parti opposte. In realtà si forma però solo l'isomero cis. Se si suppone che il decorso della reazione sia concertato, ossia che la rottura e la formazione dei legami avvengano contemporaneamente, le rotazioni attorno ai doppi legami, richieste per porre entrambi i gruppi metilici dalla stessa parte del piano, devono avvenire in direzione opposta, l'una in senso orario, l'altra in senso antiorario. Questo tipo di rotazioni concertate viene detto disrotatorio. Evidentemente l'apertura termica del cis-dimetilciclobutene segue un andamento opposto: non appena i legami dell'anello si aprono, i due gruppi metilici ruotano nello stesso senso, si ha cioè un processo conrotatorio. Per le corrispondenti reazioni dei trieni coniugati e dei cicloesadieni le regole sono invertite, ossia la reazione termica è disrotatoria e quella fotochimica conrotatoria. Dicotomie stereochimiche simili esistono per le altre classi di reazioni mostrate nella fig. 7. Woodward e Hoffmann riconobbero le caratteristiche comuni di tali reazioni e mostrarono come potessero essere inquadrate secondo un principio, quello della conservazione della simmetria dell'orbitale, che è stato confermato come una delle più importanti generalizzazioni della chimica organica, un principio che ha stimolato una grande quantità di lavoro, sia sperimentale sia teorico.
Stereochimica dei polimeri
I polimeri sono macromolecole costituite da una regolare ripetizione di unità più piccole. Così le proteine sono polimeri costituiti da α-amminoacidi, gli acidi nucleici polimeri di nucleotidi e la cellulosa è un polimero di unità di glucosio. In questi importanti tipi di polimeri biologici le subunità sono tutte omochirali.Tutti gli α-amminoacidi, unità strutturali delle proteine, appartengono alla serie L (come raffigurato in [10] per l'alanina). Gli amminoacidi della serie D infatti rappresentano una sorta di stranezza in biochimica, sebbene essi siano comunque presenti in certi composti aventi proprietà antibiotiche e prodotti da microrganismi. Se subunità identicamente chirali sono legate assieme in modo regolare, il tipo più generale di periodicità che ne risulta è una combinazione di una traslazione e di una rotazione, ossia, in altri termini, una disposizione a elica di subunità; invero, il tipo principale di regolarità osservato in natura nelle strutture di proteine, acidi nucleici e polisaccaridi è quello elicoidale.
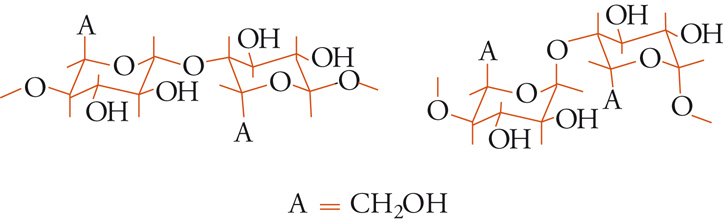
Le due principali classi di polimeri polisaccaridici naturali, amido e cellulosa, sono costituite in prevalenza da subunità di D-glucosio, sebbene siano anche presenti piccole quantità di altri zuccheri (D-mannosio, D e L-galattosio, D-xilosio, L-arabinosio), acidi uronici e amminozuccheri. La diversità tra amido e cellulosa è puramente stereochimica, in quanto riguarda una variazione di configurazione. Nell'amido le subunità sono unite (fig. 9) mediante un legame α-glucosidico, nella cellulosa mediante un legame β-glucosidico.
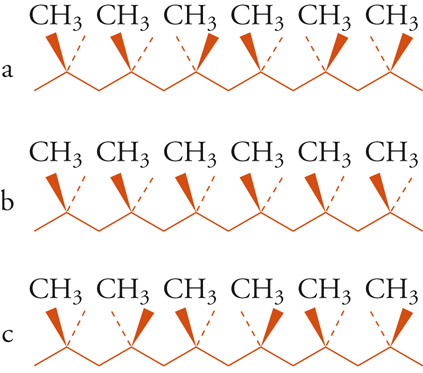
Differenze stereochimiche possono anche influenzare in maniera sorprendente le proprietà di polimeri sintetici, ciò può avere importanza dal punto di vista commerciale. Il polipropilene preparato mediante polimerizzazione catalizzata da acidi presenta una sequenza di configurazioni più o meno casuale (fig. 10) ed è un materiale cedevole, gommoso, di nessuna particolare utilità. Nel 1955 Giulio Natta e i suoi collaboratori scoprirono che polimeri preparati per mezzo di certi catalizzatori organometallici presentano strutture stereoregolari, illustrate nella fig. 10. Nel polipropilene isotattico tutti i gruppi metilici stanno dalla stessa parte del piano individuato dalla catena principale, mentre in quello sindiotattico i gruppi metilici si trovano alternativamente da una parte e dall'altra del piano. Nella struttura isotattica i gruppi successivi sono correlati da traslazione, in quella sindiotattica da un asse binario di rotazione. Si deve notare che gli atomi di idrogeno dei gruppi metilenici sono diastereotopici nella struttura isotattica e omotopici in quella sindiotattica. Il polipropilene isotattico è un materiale fibroso che può essere filato e tessuto, a differenza del polimero atattico, cioè non stereoregolare.
Considerazioni finali
Pasteur per primo riconobbe che i microrganismi devono possedere qualche tipo di profonda asimmetria che permette loro di distinguere tra enantiomeri e si convinse che una proprietà dei sistemi viventi era appunto la capacità di produrre o metabolizzare uno degli enantiomeri. I polimeri naturali di tutti i sistemi viventi sono omochirali: le proteine sono costituite esclusivamente da L-amminoacidi, gli acidi nucleici da D-ribo(o -desossiribo-) -nucleotidi, ecc. Una volta che il processo di selettività sia stato instaurato, si capisce come possa essere perpetuato. Tuttavia, è molto difficile capire come tale selettività sia sorta in un mondo prebiotico. Come scrisse Pasteur: "Forze simmetriche non viventi non possono produrre asimmetria agendo su molecole o atomi simmetrici".
Il fatto che tutto ciò che vive sulla Terra sia omochirale suggerisce l'idea che l'attuale omochiralità sia sorta attraverso un evento di piccolissima probabilità ma di grande efficacia nel favorire lo sviluppo preferenziale di un tipo di polimero omochirale rispetto al corrispondente polimero stereoirregolare. Tale evento può essersi verificato anche una sola volta durante il lungo periodo di storia prebiologica terrestre. Se le cose stanno così, cercare una spiegazione dell'origine dell'omochiralità nei sistemi viventi appare una perdita di tempo: più verosimile essa sembrerà, meno probabilità avrà di essere esatta!
La stereochimica ha un futuro? La stereochimica in senso stretto, come studio e analisi di stereoisomeri, ha avuto un passato importante, ma si può dire senza esagerazione che, con i metodi fisici moderni per la determinazione della struttura, i problemi che rimangono ancora irrisolti riguardino principalmente definizioni e classificazione.
Il crescente numero di strutture dettagliate a disposizione per le varie molecole, grazie all'applicazione di metodi fisici di analisi (diffrazione di raggi X, di neutroni, di elettroni, spettroscopia a microonde), probabilmente permetterà di scoprire molti generi di correlazione tra i diversi tipi di parametri geometrici. Queste correlazioni forniscono informazioni sulle caratteristiche della superficie energetica multidimensionale (superficie di Born-Oppenheimer) che possono essere trasferite tra classi di molecole strettamente affini. Informazioni dello stesso tipo si possono ottenere, in linea di principio, mediante calcoli quantomeccanici, sarà quindi interessante vedere in quale misura i due tipi di approccio al problema risulteranno complementari. Con lo sviluppo di campi di forza semiempirici, è possibile calcolare, con tecniche di minimizzazione dell'energia, strutture di equilibrio e insieme proprietà termochimiche e vibrazionali per molti tipi di molecole. Questo può essere già fatto per gli idrocarburi, con un'accuratezza paragonabile a quella dei migliori metodi sperimentali, ma con minor dispendio di energie e di tempo. Una sfida per il futuro può essere l'applicazione di questi metodi per il calcolo delle conformazioni stabili di una proteina a partire dalla conoscenza della sua sequenza di amminoacidi.
Attualmente molto poco è noto sui modi secondo i quali gli atomi si riassestano durante le reazioni chimiche. Non mancano modelli qualitativi che riflettono l'essenza di questi riassestamenti, ma i particolari rimangono oscuri a causa della loro inaccessibilità agli studi sperimentali e delle difficoltà di ordine pratico connesse con l'esecuzione di calcoli quantomeccanici su modelli di sistemi tanto complessi. Un possibile approccio a questo problema sta nella correlazione tra parametri strutturali osservati in una serie di molecole affini o tra altre subunità che si trovino congelate in un edificio cristallino e pertanto siano sperimentalmente accessibili. Per esempio, i dati strutturali che si riferiscono alle interazioni intramolecolari N…C=O, in alcune strutture cristalline, mostrano come, man mano che la distanza N…C diminuisce, la funzione carbonilica diventi sempre meno planare e la distanza C-O aumenti. Queste variazioni ricordano la sequenza di variazioni che si pensa avvengano in una reazione di addizione nucleofila al carbonile (SN) e i dati possono perciò essere presi come base sperimentale per tracciare il percorso della reazione. È troppo presto, comunque, per stabilire se questo tipo di approccio possa essere di applicazione generale.
Interessanti risvolti potrebbero venire dallo studio degli aspetti stereochimici degli stati intermedi di una reazione, dall'analisi strutturale di enzimi cristallini interagenti con substrati non naturali che imitino alcune proprietà di quelli naturali. I risultati sperimentali finora ottenuti inducono a ritenere che negli enzimi proteolitici (quelli che catalizzano l'idrolisi dei legami peptidici) la disposizione degli atomi vicini al sito attivo (ma non altrove) sia molto simile per tutti gli enzimi. Si possono in tal modo riconoscere, e forse anche riprodurre in composti sintetici, i requisiti strutturali necessari per un'attività catalitica ottimale. Sicuramente lo sviluppo di linee di ricerca in questo campo beneficerebbero di una approfondita conoscenza degli aspetti stereochimici delle reazioni chimiche coinvolte.
Bibliografia
Bijvoet 1951: Bijvoet, Johannes e altri, Determination of the absolute configuration of optically active compounds by means of X-rays, "Nature", 168, 1951, pp. 271-272.
Bovey 1969: Bovey, Frank A., Polymer conformation and configuration, New York, Academic Press, 1969.
Cahn 1956: Cahn, Robert S. - Ingold, Christopher K. - Pre-log, Vladimir, The specification of asymmetric configuration in organic chemistry, "Experientia", 12, 1956, pp. 81-94.
Cahn 1964: Cahn, Robert S., An introduction to the sequence rule, "Journal of chemical education", 41, 1964, pp. 116-125.
Cahn 1966: Cahn, Robert S. - Ingold, Christopher K. - Pre-log, Vladimir, Specification of molecular chirality, "Angewandte Chemie", 5, 1966, pp. 385-415.
Chiordoglu 1971: Chiordoglu, Grégoire, Conformational analysis, New York, Academic Press, 1971.
Dunitz 1975: Dunitz, Jack D., Chemical reaction paths, "Philosophical transactions of the Royal Society B", 272, 1975, pp. 99-108.
Dunitz, Waser 1972: Dunitz, Jack D. - Waser, Jürg, Geometric constraints in six- and eight-membered rings, "Journal of the American Chemical Society", 94, 1972, pp. 5645-5650.
Dunitz, Waser 1972: Dunitz, Jack D. - Waser, Jürg, The planarity of the equilateral isogonal pentagon, "Elemente der Mathematik", 27, 1972, pp. 25-32.
Eliel 1962: Eliel, Ernest L., Stereochemistry of carbon compounds, New York, McGraw-Hill, 1962.
Eliel 1965: Eliel, Ernest L. e altri, Conformational analysis, New York-London, Interscience, 1965.
Gillespie 1972: Gillespie, Roland J., Molecular geometry, London-New York, Van Nostrand Reinhold, 1972.
International Union of Pure and Applied Chemistry 1970: IUPAC tentative rules for the nomenclature of or-ganic chemistry. Section E. Fundamental stereochemistry, "Journal of organic chemistry", 35, 1970, pp. 2849-2867.
Kemp, Pitzer 1936: Kemp, J.D. - Pitzer, Kenneth S., Hindered rotation of the methyl groups in ethane, "Journal of chem-ical physics", 4, 1936, p. 749.
Kim 1973: Kim, Seong Hwan e altri, Three-dimensional structure of yeast phenylalanine transfer RNA: folding of the poly-nucleotide chain, "Science", 179, 1973, pp. 285-288.
Kim 1974: Kim, Seong Hwan e altri, Three-dimensional ter-tiary structure of yeast phenylalanine transfer RNA, "Science", 185, 1974, pp. 435-440.
Kuhn 1972: Kuhn, Hans, Self-organization of molecular systems and evolution of the genetic apparatus, "Angewandte Chemie", 11, 1972, pp. 798-820.
Le Bel 1874: Le Bel, Joseph-Achille, Sur les relations qui existent entre les formules atomiques des corps organiques et le pouvoir rotatoire de leurs dissolutions, "Bulletin de la Société Chimique de Paris", 22, 1874, pp. 337-347.
Lewis 1916: Lewis, Gilbert N., The atom and the molecule, "Journal of the American Chemical Society", 38, 1916, pp. 762-785.
Lowe 1973: Lowe, John P., The barrier to internal rotation in ethane, "Science", 179, 1973, pp. 527-532.
Natta 1955: Natta, Giulio e altri, Crystalline high polymers of α-olefins, "Journal of the American Chemical Society", 77, 1955, pp. 1708-1710.
Mazzanti, Moraglio 1955: Mazzanti, Giorgio - Moraglio, Giovanni, Crystalline high polymers of α-olefins, "Journal of the American Chemical Society", 77, 1955, pp. 1708-1710.
Pasteur 1848: Pasteur, Louis, Mémoire sur la relation qui peut exister entre la forme cristalline et la composition chimique, et sur la cause de la polarisation rotatoire, "Comptes rendus hebdomadaires des séances de l'Académie des Sciences", 26, 1848, pp. 535-539.
Prelog, Wieland 1944: Prelog, Vladimir - Wieland, Peter, Über die Spaltung der Tröger'schen Base in optischenAntipoden. Ein Beitrag zur Stereochemie des dreiwertigen Stickstoffs, "Helvetica chimica acta", 27, 1944, pp. 1127-1134.
Ruch 1972: Ruch, Ernst, Algebraic aspects of the chirality phenomenon in chemistry, "Accounts of chemical research", 5, 1972, pp. 49-56.
Tanford 1967: Tanford, Charles, Physical chemistry of macromolecules, New York, Wiley, 1967.
Van't Hoff 1875: van't Hoff, Jacobus H., La chimie dans l'es-pace, Rotterdam, Bazendijk, 1875.
Walsh 1953: Walsh, Arthur D., The electronic orbitals, shapes and spectra of polyatomic molecules, "Journal of the Chemical Society", 1953, pp. 2260-2329.
Watson, Crick 1953: Watson, James D. - Crick, Francis H.C., Molecular structure of nucleic acids. A structure for deoxy-ribose nucleic acid, "Nature", 171, 1953, pp. 737-738.
Werner 1905: Werner, Alfred, Neuere Anschauungen auf dem Gebiete der anorganischen Chemie, Braunschweig, Vieweg, 1905.
Wöhler 1824: Wöhler, Friedrich, Recherches analytiques sur l'acide cyanique, "Annales de chimie et de physique", 27, 1824, pp. 196-200.
Wöhler 1828: Wöhler, Friedrich, Über künstliche Bildung des Harnstoffes, ‟Annalen der Physik und Chemie", 12, 1828, pp. 253-256.
Woodward, Hoffmann 1970: Woodward, Robert B. - Hoffmann, Roald, The conservation of orbital symmetry, Weinheim, Verlag Chemie/Academic Press, 1970.