
Fotochimica
Fotochimica
Una suddivisione importante presente nella chimica si basa sulla distinzione tra reazioni che hanno luogo tra atomi e molecole nel loro stato elettronico fondamentale e quelle in cui sono coinvolti stati elettronici eccitati. Nello stato fondamentale, tutti gli elettroni occupano gli orbitali molecolari aventi i livelli di energia più bassi possibili, mentre ogni altra distribuzione rappresenta uno stato elettronico eccitato. In generale, l'energia richiesta per la transizione dallo stato fondamentale a quello eccitato, cioè per la promozione di un elettrone a un livello inizialmente non occupato, è compresa nell'intervallo di 42÷190 kcal/mol. Essa, quindi, è dello stesso ordine di grandezza (oppure superiore) dell'energia richiesta per molte reazioni tra molecole che si trovano nello stato fondamentale e che coinvolgono la rottura di legami. Normalmente, tale eccitazione energetica è la conseguenza dell'assorbimento di un fotone, la cui lunghezza d'onda si trova nella regione visibile o ultravioletta dello spettro elettromagnetico (700÷150 nm).
La molecola eccitata si trova necessariamente in uno stato di minore stabilità rispetto al corrispondente stato fondamentale e possiede perciò una tendenza intrinseca sia a liberare nuovamente l'eccesso di energia, sia a utilizzarla nelle reazioni chimiche. I processi puramente fisici di liberazione dell'energia eccedente riportano la molecola nel suo stato fondamentale originario, mentre lo stato eccitato può anche subire trasformazioni chimiche che sono appropriatamente chiamate reazioni fotochimiche, per il fatto che hanno inizio in seguito all'assorbimento della luce. La fotochimica, pertanto, riguarda in primo luogo tali eventi chimici, che coinvolgono atomi e molecole in sistemi organici, inorganici e biologici. La chimica delle radiazioni si occupa, invece, degli effetti di radiazioni a più alto contenuto energetico, normalmente sotto i 100 nm, inclusi i raggi X, i raggi γ e i bombardamenti elettronici; tali radiazioni producono specie ionizzate oltre che stati neutri eccitati. Per tradizione, questo argomento è considerato vicino alla fotochimica, ma distinto da essa.
Le teorie fondamentali della chimica quantistica, dell'ottica e della spettroscopia atomica furono sviluppate nei loro aspetti teorici a partire proprio dallo studio degli stati eccitati. Tali sviluppi riguardavano l'introduzione della spettroscopia e della fotocinetica molecolare, nonché l'uso accresciuto del formalismo della teoria dei gruppi in connessione con la teoria degli orbitali molecolari, elaborata soprattutto da Robert S. Mulliken. Queste tecniche hanno fornito non soltanto una maggiore capacità d'osservazione della natura e della dinamica degli stati eccitati, ma hanno anche permesso, gradualmente, di rimuovere la barriera tra la chimica dello stato fondamentale e quella degli stati eccitati. I chimico-fisici hanno studiato in maniera molto ampia e approfondita la fotochimica degli atomi e delle molecole semplici in fase gassosa. Da allora sono state sviluppate svariate tecniche per realizzare potenti sorgenti di luce ultravioletta, per ottenere fasci monocromatici e per l'analisi spettroscopica e chimica dei prodotti ottenuti. L'uso di questi metodi per la rilevazione e la misurazione quantitativa di processi fotofisici e fotochimici in fase gassosa relativamente semplici contribuìalla formazione di molti concetti base della fotochimicamoderna.
Un interesse molto più diffuso, seguito da una rapida espansione in tutte le sue multidisciplinari direzioni, fu determinato principalmente dall'acquisizione della conoscenza delle strutture molecolari per via spettroscopica. Tali risultati spinsero i chimici organici a irradiare diverse specie chimiche, inclusi i prodotti naturali e i loro derivati, individuando un ampio insieme di gruppi cromofori, spesso complessi, inseriti in differenti combinazioni strutturali. Sono state così scoperte molte trasformazioni fotochimiche fondamentali. Un altro importante sviluppo parallelo riguardò l'introduzione e il perfezionamento di metodi per la rilevazione, come pure per l'analisi cinetica e strutturale, di stati eccitati e di specie transitorie aventi vita breve. I più recenti progressi nella strumentazione includono miglioramenti della risoluzione temporale e della rilevazione fotoelettrica, l'impiego di tecniche a bassa temperatura, l'accumulo di informazioni e deconvoluzioni per mezzo di calcolatori in combinazione con sorgenti monocromatiche di alta potenza e precisione, nonché di rivelatori di emissione, assorbimento e risonanza magnetica.
Processi fotofisici primari
L'energia E di un fotone dipende dalla sua frequenza ν o dalla sua lunghezza d'onda λ secondo la relazione:
[1] formula
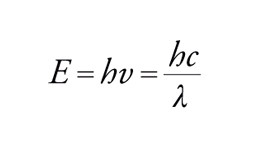
dove h è la costante di Planck, uguale a 6,63∙10−27 erg∙s. Johannes Stark e Albert Einstein stabilirono, con la legge sull'equivalenza fotochimica, che nel processo fotochimico primario ogni quanto assorbito attiva una sola molecola. Da questo principio è nato il concetto di resa quantica, che esprime il numero di molecole che hanno emesso radiazioni per fluorescenza o fosforescenza, hanno reagito oppure sono state formate, per ogni quanto di luce assorbito. Presupposto fondamentale per la misura quantitativa della luce assorbita da una data sostanza è la registrazione del suo spettro di assorbimento nella regione delle lunghezze d'onda usate per l'irraggiamento. L'assorbimento molare ε (indicato anche come coefficiente di estinzione molare), che è costante per ogni sostanza per una data lunghezza d'onda e proporzionale alla probabilità della transizione della molecola dallo stato fondamentale allo stato eccitato, si può determinare dall'equazione:
[2] formula
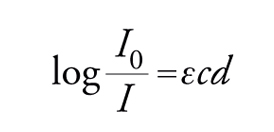
detta legge di Lambert e Beer, dove I0 e I sono le intensità della luce incidente e trasmessa, rispettivamente, c è la concentrazione della soluzione in moli/litro, e d è lo spessore della cella in cm.

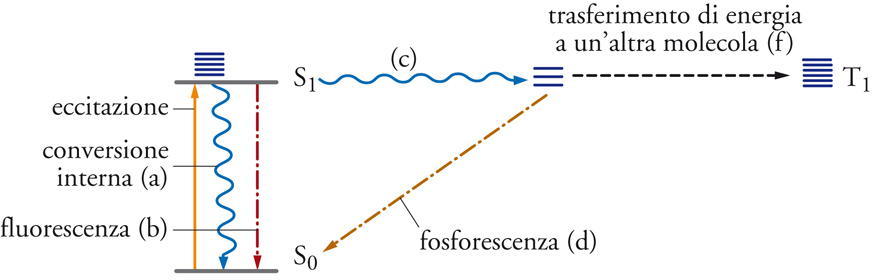
Dopo l'assorbimento della luce da parte di una molecola e il suo passaggio in uno stato eccitato, esistono diversi meccanismi possibili per la liberazione dell'energia elettronica di eccitazione con ritorno della molecola allo stato fondamentale di partenza senza nessun cambiamento permanente. Molte di queste vie per la dissipazione dell'energia sono costituite da processi fotofisici monomolecolari. Alcuni servono alla ripartizione dell'energia di eccitazione tra i vari livelli di energia molecolare e altri intervengono anche quando l'eccitazione dà luogo, alla fine, a una trasformazione chimica. La tab. 1 elenca i più comuni tra questi processi fisici di disattivazione, per ciascuno dei quali sono dati gli intervalli approssimativi dei valori delle costanti di velocità tipiche delle molecole organiche. Tali processi sono schematizzati nel diagramma contenuto nella fig. 1, detto diagramma di Jablonski. La maggior parte delle molecole possiede un numero pari di elettroni, i quali, nello stato fondamentale, occupano orbitali con spin opposti appaiati (stato fondamentale di singoletto, S0). Negli stati elettronici eccitati, i due elettroni non accoppiati sono in orbitali differenti e possono avere ancora la stessa molteplicità di spin (stati eccitati di singoletto, S1, S2, ecc.) o spin disaccoppiati (stati di tripletto, T1, T2, ecc.). Tutti questi stati corrispondono a specie distinte con differenti strutture e reattività. Lo stato di tripletto è invariabilmente di energia più bassa rispetto al corrispondente stato di singoletto. Questa differenza nasce dal fatto che elettroni non accoppiati con spin paralleli non possono occupare simultaneamente lo stesso spazio (principio di Pauli). Nello stato di tripletto, la repulsione tra elettroni risulta perciò ridotta.
L'assorbimento di un fotone è soggetto a regole di selezione che favoriscono la conservazione dello spin. Di conseguenza, i più comuni processi di assorbimento che risultano permessi per quanto riguarda i momenti di spin portano dallo stato di singoletto fondamentale S0 al primo stato eccitato di singoletto o a uno più alto, a seconda del valore dell'energia e della lunghezza d'onda della luce incidente. Anche la transizione T1→T2 è permessa per quanto riguarda lo spin, ma ovviamente è osservabile solo a condizione che la popolazione del più basso stato di tripletto sia sufficientemente elevata. Essa può realizzarsi nel caso che si verifichino l'una, l'altra o entrambe le seguenti circostanze: un tempo di vita molto lungo per lo stato eccitato e un'intensità molto alta della luce, come avviene negli esperimenti con una radiazione continua fornita da un laser o nella fotolisi a lampo o a flash (flash photolysis), nella quale si generano impulsi di radiazione di 10−5 s. Poiché i vari stati elettronici hanno sovente conformazioni diverse, in generale l'assorbimento popola inizialmente un livello vibrazionale a elevata energia dello stato eccitato, ed è seguito da un rilassamento vibrazionale con una distribuzione dell'energia in accordo con la statistica di Boltzmann all'interno dello stesso stato.
La trasformazione non radiativa di un qualsiasi stato eccitato, termicamente in equilibrio, in un altro stato elettronico a energia più bassa comporta una transizione tra i livelli degli stati energetici vibrazionali-rotazionali isoenergetici, seguita da un rilassamento vibrazionale. Queste transizioni sono classificate come conversioni interne, quando i due stati hanno la stessa molteplicità di spin, e come passaggi dallo stato di singoletto a quello di tripletto (intersystem crossing), quando la transizione è accoppiata a una variazione di spin e lega due stati di differente molteplicità. Lo stato fondamentale originale della molecola, quindi, può essere ristabilito mediante lo spopolamento puramente fisico di uno dei suoi stati eccitati. Si manifesta una correlazione inversa tra la velocità delle transizioni non irraggianti e la differenza di energia tra i livelli fondamentali degli stati elettronici interessati. Infatti, poiché gli stati eccitati sono raggruppati con elevata densità, le conversioni interne avvengono rapidamente. Benché l'intersystem crossing sia un processo non permesso per quanto riguarda lo spin, la velocità della transizione S1→T1 può tuttavia essere molto alta, poiché la separazione di energia è a sua volta piccola.
La luminescenza è provocata dall'emissione spontanea di un fotone. La fluorescenza, invece, è una transizione molto rapida che avviene con emissione di radiazione fra due stati della stessa molteplicità, solitamente S1→S0, mentre la fosforescenza è l'analoga transizione più lenta T1→S0, che non è permessa per quanto riguarda lo spin. Il trasferimento di energia e le reazioni chimiche, come per esempio la formazione di un nuovo prodotto stabile, possono avvenire sia monomolecolarmente sia bimolecolarmente. In molte molecole, le costanti di velocità della conversione interna S1→S0 e dell'intersystem crossing T1→S0 hanno lo stesso ordine di grandezza. In un numero sempre maggiore di casi, questa apparente anomalia della disattivazione fisica verso lo stato fondamentale è infatti una conseguenza della formazione di fotoprodotti isomerici instabili, che si riconvertono per via termica nel materiale di partenza a una velocità sufficientemente rapida da sfuggire alla rivelazione.
Il benzene è un sistema che mostra questo fenomeno. Per irraggiamento viene trasformato nel benzovalene:
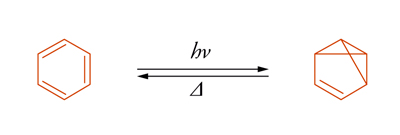
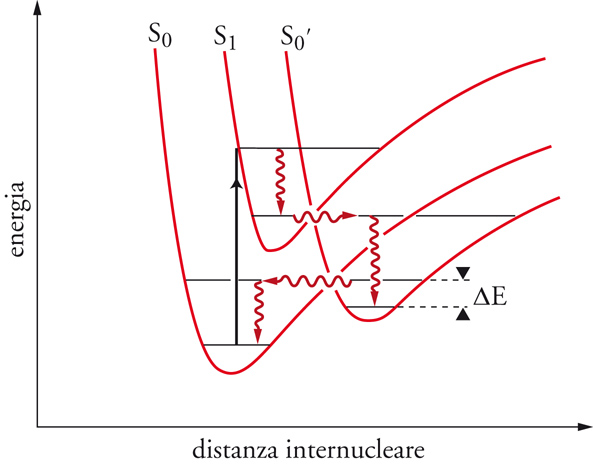
che isomerizza di nuovo a benzene a temperatura ambiente. La situazione è descritta nella fig. 2, dove viene illustrato il passaggio dallo stato eccitato singoletto a quello fondamentale attraverso un intermedio. Se la differenza di energia ΔΕ può essere superata per attivazione termica, l'isomero metastabile si trasforma nel materiale di partenza. Il decadimento di uno stato elettronicamente eccitato, M*, al suo stato fondamentale, M, può essere accelerato mediante interazione con un'altra sostanza Q che viene detta disattivatore (quencher). Tale disattivazione (quenching), espressa nella sua forma più generale dall'equazione
[4] formula
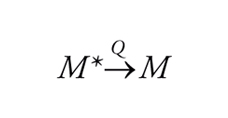
può procedere secondo diversi meccanismi. Eccetto che per certi tipi di trasferimento di energia elettronica, i processi di disattivazione comportano la formazione di un complesso di contatto oppure di un eccimero o di un ecciplesso.
Mentre nel primo caso i componenti hanno orientamenti relativi casuali a distanze di 7 Å o superiori, i secondi sono entità con una geometria definita, che occupano posizioni di minima energia sulla superficie dell'energia potenziale dello stato eccitato. Dal momento che questi processi di disattivazione sono collisionali, essi sono soggetti alla regola di Wigner sulla conservazione dello spin, la quale stabilisce che il momento angolare di spin complessivo del sistema non deve variare. Gli ecciplessi sono complessi eccitati formati dall'associazione di uno stato eccitato con uno stato fondamentale di specie diverse; gli eccimeri sono gli analoghi dimeri eccitati che coinvolgono componenti della stessa specie. Una seconda via per la formazione di eccimeri singoletti consiste nel processo di annichilimento tripletto-tripletto, che può essere provocato dalla collisione di due molecole nello stato di tripletto. Il decadimento di eccimeri e di ecciplessi con ripristino di M può avvenire in due maniere principali, come riassunto nelle equazioni seguenti (X=M: eccimero; X=Q: ecciplesso):
[5] (MX)*→M+X(+hν)
[6] (MX)*→M+X*.
La fluorescenza, o la conversione interna (da un complesso singoletto), e la fosforescenza, o il passaggio da un sistema all'altro (da un complesso tripletto), danno direttamente i due componenti dissociati nello stato fondamentale, come appare dalla prima delle equazioni precedenti. Alternativamente, la dissociazione può anche avere luogo con ritenzione dell'eccitazione elettronica. Questo fatto corrisponde al trasferimento cinetico dell'energia, se viene coinvolto un ecciplesso e la molecola disattivante rimane eccitata (EQ*〈EM*). Nel caso di eccimeri, ciò corrisponde alla migrazione dell'eccitazione elettronica fra molecole uguali (migrazione eccitonica). Il trasferimento elettronico è un processo importante per la disattivazione degli stati eccitati di singoletto, in particolare di idrocarburi aromatici e azocomposti da parte di dieni e di olefine. Tale processo può già avere luogo in un complesso di contatto per dare origine direttamente a coppie ioniche solvatate, sia competendo con un processo che coinvolge un ecciplesso come intermedio, sia prendendone il posto. L'equazione [7] riassume tali possibili alternative.

La disattivazione di una molecola di donatore eccitata D* al suo stato fondamentale e il simultaneo trasferimento della sua energia di eccitazione a una molecola di accettore A avvengono attraverso un processo di trasferimento dell'energia elettronica:
[8] D*+A→D+A*.
Come risultato, si osserva la disattivazione di D* e l'attivazione, con processi simili, di A, benché i fotoni siano stati assorbiti da D. La generazione di processi derivati da A* ha dato origine a una tecnica chiamata fotosensibilizzazione. Fra i principali processi di trasferimento dell'energia elettronica, si possono distinguere quelli che avvengono con irraggiamento e quelli che avvengono senza irraggiamento; questi ultimi comportano interazioni di tipo coulombiano e interazioni basate sullo scambio di elettroni. La probabilità del trasferimento di energia con irraggiamento, secondo lo schema:
[9] D*→D+hν, hν+A→A*
dipende dall'efficienza quantica dell'emissione da parte del donatore, dalla concentrazione delle molecole dell'accettore, dal loro potere di assorbire la luce e dalla sovrapposizione degli spettri di emissione del donatore e di assorbimento dell'accettore. Tale trasferimento di energia, compreso il caso speciale della migrazione dell'energia radiante fra molecole uguali, può avere luogo a distanze piuttosto elevate. Il trasferimento senza irraggiamento può avvenire a distanze molto più grandi delle dimensioni molecolari, qualora venga indotto da interazioni di tipo coulombiano, mentre il meccanismo di scambio elettronico richiede che donatore e accettore si avvicinino a distanze comparabili con i diametri di collisione.
Tecniche sperimentali
Negli strumenti moderni per l'irraggiamento, la luce del Sole, che era stata usata in un primo tempo come la sorgente principale di luce ultravioletta, è stata ovviamente sostituita da lampade artificiali, più efficienti. Tra queste, le più usate sono quelle ad arco di mercurio, poi i tubi fluorescenti a scarica in gas rari (per es., lo xeno) e le lampade combinate a mercurio-xeno. Nel loro insieme, esse forniscono un'illuminazione continua con un'intensità di luce relativamente elevata, distribuita complessivamente su tutto l'intervallo spettrale di maggiore interesse in fotochimica (cioè da lunghezze d'onda inferiori a 200 nm fino al visibile). Circa il 90% dell'emissione delle lampade a mercurio a bassa pressione è concentrata a 253,7 nm. Queste lampade sono usate per reazioni in fase gassosa fotosensibilizzate dal mercurio e forniscono anche una sorgente monocromatica molto conveniente della radiazione di 254 nm da usare in fotolisi dirette.
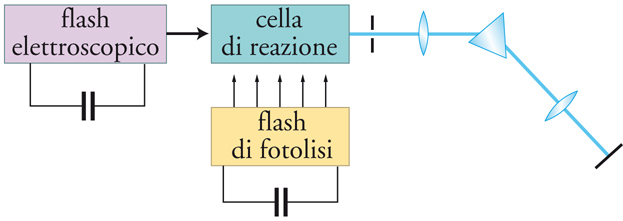
Le lampade a mercurio a media pressione hanno una distribuzione spettrale molto più ampia e perciò un più vasto campo di applicazioni. La presenza di bande intense e larghe è notevole nella regione sopra i 300 nm, con intensità particolarmente elevata intorno all'intervallo di 313÷366 nm. Le sorgenti più intense di radiazione ultravioletta sono le lampade ad alta pressione che, nella versione a sorgente (quasi) puntiforme, vengono particolarmente usate in applicazioni qualitative e semipreparative, nelle quali il campione da irradiare è piccolo. Nelle flash photolysis, che operano secondo lo schema illustrato nella fig. 3, si richiedono impulsi di radiazione di alta energia e di durata brevissima. Le lampade convenzionali di questo tipo sono per lo più basate su tubi a scarica nei gas, caratterizzati dall'erogazione di energia elettrica fino a 5000 J per lampo e da un tempo di decadimento del lampo dell'ordine di 10−3 s. Tuttavia, questi lampi sono policromatici e l'energia degli impulsi più brevi è enormemente ridotta, con una dissipazione dell'ordine di 10−9 J soltanto, per tempi di lampo di circa 10−9 s. Un considerevole progresso è stato raggiunto con l'avvento della tecnica delle sorgenti laser, che sfruttano il principio dell'amplificazione ottenuta mediante l'emissione stimolata di radiazioni, diversamente dalle sorgenti convenzionali, che operano mediante l'emissione spontanea. Si produce un raggio altamente collimato, monocromatico e coerente di enorme luminosità. Mediante appropriati otturatori, gli impulsi relativamente lunghi di laser semplici (ca. 10−6 s) possono essere ridotti a lampi singoli (con durata dell'impulso sempre più piccola), che con particolari accorgimenti raggiungono 10−15 s.
Nella progettazione di un qualsiasi reattore per fotochimica, si devono considerare alcune caratteristiche che sono essenzialmente indipendenti dallo scopo e dal tipo dell'esperimento. Esse riguardano in primo luogo la scelta del materiale da costruzione per il recipiente, o per quella parte di esso che deve essere trasparente alle lunghezze d'onda incidenti desiderate. Tali materiali variano da vetri teneri ordinari (trasmissione di luce con lunghezza d'onda all'incirca >330 nm) a vetri pirex (ca. >280 nm) e quarzo (ca. >170÷190 nm), al fluoruro di calcio (ca. >140 nm) e al fluoruro di litio (ca. >110 nm). Altri fattori determinanti sono: l'opportuno orientamento della sorgente di luce per l'utilizzazione ottimale della radiazione, l'omogeneizzazione del volume di reazione (nell'irraggiamento di materiali non rigidi) e la prevenzione della formazione di depositi sulle pareti trasparenti del reattore. La migliore disposizione geometrica per il lavoro sintetico consiste nel porre la lampada, circondata da una camicia refrigerante, immersa nel reagente.
La determinazione dei rendimenti quantici delle fotoreazioni richiede la valutazione quantitativa dei prodotti formati (o dei materiali di partenza consumati) in un dato periodo di tempo e la corrispondente quantità di radiazione assorbita nello stesso tempo. Misure fisiche dirette dell'intensità della radiazione si basano su metodi radiometrici (termopile, termistori) o fotoelettrici (fotocellule e fotomoltiplicatori). I procedimenti sviluppati più di recente misurano la differenza del flusso di luce prima e dopo la cella contenente il campione, mediante la capacità di assorbimento e fluorescenza di uno scintillatore accoppiato con un appropriato fotoelemento. Una tale misura continua differenziale è indipendente dalla lunghezza d'onda incidente entro un'ampia regione ed è direttamente proporzionale ai quanti di luce assorbiti. Infine, è necessario osservare che i tempi di vita degli stati eccitati sono parametri importanti per la cinetica dei processi fotochimici e di quelli fotofisici. Il metodo più diretto, tra quelli disponibili per determinarli, consiste nella misura del tempo di decadimento dell'emissione mediante fluorimetria e fosforimetria a impulsi, in cui vengono usati, per eccitare il campione, rispettivamente impulsi di luce con tempi di nanosecondi e microsecondi. Il decadimento dell'emissione viene controllato dopo ciascun impulso con un fotomoltiplicatore veloce e con un oscilloscopio.
La flash photolysis è stata introdotta da Ronald G.W. Norrish e George Porter, ai quali fu assegnato nel 1967 il premio Nobel per la chimica, proprio per lo sviluppo di questa importante tecnica, finalizzata allo studio qualitativo e quantitativo degli stati eccitati e degli intermedi a vita breve nei processi fotochimici. Il principio di questo metodo si basa sulla produzione di un'alta concentrazione di molecole eccitate con un impulso luminoso di intensità molto alta e di breve durata. Varie tecniche di rivelazione, basate sulla spettroscopia di assorbimento e di emissione, vengono usate per analizzare il sistema poco tempo dopo il lampo di luce. Lo spettro di emissione di una specie transitoria può essere registrato usando uno spettrografo; similmente, lo spettro di assorbimento può essere misurato se un raggio analitico è predisposto a dare il lampo a un intervallo di tempo predeterminato dopo l'impulso iniziale. Alternativamente, il processo di decadimento della o delle specie transienti può essere seguito cineticamente controllando l'emissione o l'assorbimento (di un raggio di luce continuo invece che di un lampo di luce analitico) a una lunghezza d'onda particolare.
Studio dei meccanismi di reazione
Nell'istante della loro formazione, la maggior parte degli stati eccitati è dotata di un eccesso di energia vibrazionale e, occasionalmente, anche di energia elettronica, se vengono inizialmente prodotti stati eccitati a energia più alta di quella del primo stato eccitato. In fase gassosa, dove la frequenza collisionale è bassa, tali stati eccitati transitori possono reagire prima che abbia luogo il rilassamento energetico. Al contrario, in soluzione l'eccesso di energia viene in genere rapidamente dissipato per collisione con le molecole di solvente per cui sia le reazioni sia la luminescenza hanno luogo di norma a partire dagli stati più bassi S1 e T1. I processi a 2 fotoni mostrano una dipendenza caratteristica della formazione dei prodotti dal quadrato dell'intensità della luce. È noto che parecchi casi del genere si verificano nei vetri rigidi; basti ricordare la fotoionizzazione di composti aromatici a partire da uno stato di tripletto più alto, che viene popolato quando un tripletto più basso metastabile assorbe un altro fotone di sufficiente energia, come è indicato dall'equazione:
[10] formula
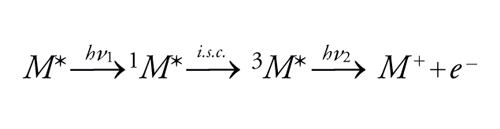
Una situazione simile è osservata nella reazione elettrociclica:
[11] formula
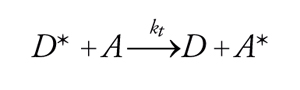
[12] I=[D*](kd+kr+kt[A])
dove I è l'intensità della luce assorbita o velocità di formazione di D* (ammettendo ki.s.c. =1,0 per uno stato eccitato di tripletto D*) e kd rappresenta la somma di tutti i processi di decadimento, sia irradianti sia non irradianti, di D* diversi da kr e kt. Queste ultime sono, rispettivamente, la costante di velocità del processo sotto misura (formazione di prodotto o emissione) e la costante di velocità bimolecolare per la disattivazione. L'irraggiamento simultaneo del reagente con lunghezze d'onda di 254 nm (eccitazione del singoletto) e maggiore di 360 nm (assorbita dal tripletto del naftalene) permette di ottenere il risultato. Esso può essere prodotto anche attraverso una via alternativa, diversa dal percorso basato sui 2 fotoni, che consiste nell'eccitare il reagente solamente con una luce dotata di una lunghezza d'onda ≤229 nm, che fornisce energia sufficiente per passare al tripletto superiore notevolmente reattivo, in modo da competere con il ritorno allo stato S termicamente equilibrato.
Gli studi che impiegano il trasferimento bimolecolare (collisionale) dell'energia in esperimenti scelti di sensibilizzazione e di disattivazione, in generale costituiscono il metodo più valido per differenziare tra di loro e analizzare le specie reattive S1 e T1. Un'oculata scelta del sensibilizzatore o del disattivatore richiede che si conoscano le velocità di trasferimento e le energie di eccitazione. In particolare, l'abilità relativa di un dato composto a disattivare singoletti e tripletti deve essere stabilita, per esempio, mediante disattivazione per emissione. La sensibilizzazione del singoletto è raramente utile, salvo il caso in cui gli stati di singoletto e di tripletto siano ambedue reattivi e sia disponibile un composto capace di trasferire selettivamente l'energia del singoletto, ma che disattiva i tripletti. La sensibilizzazione del tripletto e la sua disattivazione permettono di determinare se una fotoreazione ha origine solo da singoletti o da tripletti, oppure da entrambi. La composizione quantitativa dei fotoprodotti nella reazione diretta e in quella sensibilizzata permette di discriminare fra le ulteriori possibilità, ovvero, che un tempo di vita molto breve (inferiore a 0,1 ns) prevenga la disattivazione di un reagente obbligatoriamente nello stato di tripletto e che il singoletto reagisca prima dell'intersystem crossing nel caso di irraggiamento diretto, dal momento che difficilmente singoletto e tripletto reagiscono in maniera identica.
La resa quantica Φi deve essere nota qualora si vogliano derivare le costanti di velocità dello stato eccitato. In realtà, si tratta di un parametro cinetico associato a una fotoreazione direttamente misurabile in condizioni stazionarie. Le equazioni cinetiche dalle quali si può risalire alle rese quantiche di reazioni mono- e bimolecolari sono rispettivamente la [14] e la [15]:
[13] formula [14]

Φi=ΦSEkrτSEPi
[15] Φi=ΦSEksCSτSEPi
dove CS è la concentrazione del substrato S. Quattro di questi parametri, Φi, ΦSE, τSE e Pi, sono in linea di principio misurabili mediante esperimenti separati. ΦSE è la probabilità che l'assorbimento di luce dia origine allo stato eccitato richiesto; kr e ks sono le costanti di velocità con cui lo stato eccitato dà luogo alla fotoreazione primaria (con il substrato S in processi bimolecolari) necessaria per il processo i, e τSE è il tempo di vita di tale stato. Pi è la probabilità che un qualsiasi stato metastabile intermedio porti a un prodotto stabile, ossia completi il processo i piuttosto che formare altri prodotti o riportare il reagente allo stato fondamentale. L'inclusione di questo fattore non è perciò necessaria se si tratta di un processo irreversibile, come un'emissione o una riorganizzazione di un legame chimico che ha luogo in un solo passaggio. Tuttavia, gli intermedi reattivi contribuiscono molto spesso a determinare le rese quantiche totali. Mentre, nel caso di reazioni bimolecolari, Pi può essere spesso misurato mediante tecniche cinetiche dirette, questo non è possibile in reazioni monomolecolari e l'intermedio deve essere quantitativamente intrappolato in maniera misurabile. Un agente adatto per l'intrappolamento deve essere selettivamente reattivo nei confronti dell'ipotetico intermedio, ma non reattivo nei confronti del materiale di partenza, dello stato eccitato, di ogni altro intermedio e del prodotto finale.
Probabilità di formazione di stati reattivi eccitati
Nell'irraggiamento diretto, la probabilità che l'assorbimento della luce produca lo stato eccitato richiesto ΦSE è uguale a 1, se si sta studiando una reazione di singoletto e se la resa quantica della reazione non dipende dalla lunghezza d'onda. Nel caso che sia coinvolto uno stato di tripletto, ΦSE risulta uguale alla resa quantica dell'intersystem crossing, Φi.s.c, che viene comunemente misurata per sensibilizzazione di un appropriato processo di tripletto, facendo riferimento a un sensibilizzatore avente un Φi.s.c noto (tecnica di conteggio del tripletto). I tempi medi dello stato eccitato possono essere misurati direttamente mediante flash spectroscopy, o indirettamente mediante tecniche di quenching. In entrambi i metodi, τSE può essere valutata dalla somma delle velocità di tutti i processi di disattivazione che competono tra loro, ovvero τSE−1=∑k. Una disattivazione quantitativa comporta determinazioni di rese quantiche Φr che sono anche richieste per scomporre τSE−1 nei suoi vari componenti. L'espressione di Stern-Volmer:
[16] formula
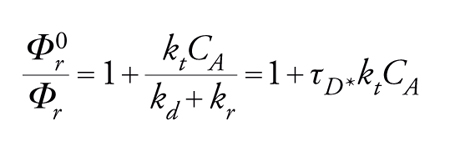
predice una relazione lineare tra Φ0r/Φr e la concentrazione del disattivatore CA, con una pendenza uguale a τD*kt, dove τD* è il tempo di vita dello stato eccitato da disattivare (in assenza del disattivatore A). Se in un dato sistema un solo stato eccitato è reattivo, i rapporti tra i prodotti non variano con l'entità della disattivazione. L'inverso della resa quantica dipende linearmente dalla concentrazione del disattivatore
[17] formula.
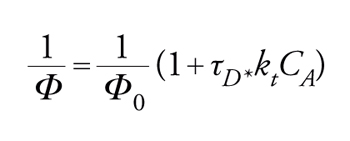
La pendenza di tale retta divisa per l'intercetta 1/Φ0 è uguale a τD*kt. Le velocità specifiche per il trasferimento di energia, kt, vengono determinate nel modo migliore mediante esperimenti con luce lampeggiante. Tuttavia, specialmente quando si devono studiare tripletti, lo stato eccitato reattivo ha vita troppo breve. Per fortuna, il trasferimento di energia esotermico in solventi mobili è in generale molto vicino a quello controllato dalla diffusione, cioè kt≈kdiff. Un'espressione approssimata per la costante di velocità del processo controllato dalla diffusione, kdiff, è data dall'equazione di Debye:
[18] formula
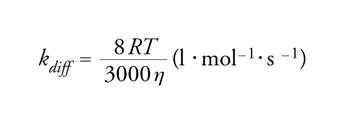
dove R è la costante dei gas, T la temperatura assoluta e η la viscosità in poise. Non è raro che più di uno stato eccitato sia implicato nella cinetica complessiva. L'analisi dei tempi di vita, allora, diventa più complicata poiché viene a dipendere da dettagliati eventi di disattivazione e la relazione di Stern-Volmer può deviare dalla linearità verso l'alto o verso il basso.
Bibliografia
Ashmore 1967: Ashmore, Philip G., Photochemistry and reaction kinetics, Cambridge, Cambridge University Press, 1967.
Bunsen, Roscoe 1857-1863: Bunsen, Robert W. - Roscoe, Henry E., Photochemical researches, London, Taylor & Francis, 1857-1863.
Ciamician 1913: Ciamician, Giacomo, La fotochimica dell'avvenire, Bologna, Zanichelli, 1913.
Cundall, Gilbert 1970: Cundall, Robert B. - Gilbert, Andrew, Photochemistry, London, Nelson, 1970.
Draper 1878: Draper, John W., Scientific memoirs, being experimental contributions to a knowledge of radiant energy, New York, Harper, 1878.
Einstein 1912: Einstein, Albert, Thermodynamische Begründung des photochemischen Äquivalentgesetzes, ‟Annalen der Physik", 37, 1912, pp. 832-838.
Herschel 1842: Herschel, John F.W., On the action of the rays of the solar spectrum on vegetable colours and some new photographic processes, "Philosophical transactions of the Royal Society A", 132, 1842, pp. 181-214.
Jablonski 1935: Jablonski, Alexander, Über den Mechanismus der Photolumineszenz von Farbstoffphosphoren, ‟Zeitschrift für Physik", 94, 1935, pp. 38-46.
Lamola, Turro 1969: Lamola, Angelo A. - Turro, Nicholas J., Energy transfer and organic photochemistry, New York-London, Interscience, 1969.
Mulliken 1964: Mulliken, Robert S., Molecular orbitals in chemistry, physics and biology, New York-London, Academic Press, 1964.
Noyes, Leighton 1966: Noyes, William A. jr - Leighton, Philip A., The photochemistry of gases, New York, Dower, 1966.
Orchin, Jaffé 1971: Orchin, Milton - Jaffé, Hans H., Symmetry, orbitals and spectra, New York-London, Wiley Interscience, 1971.
Porter 1974: Porter, Gerald B. - Balzani, Vincenzo - Moggi, Luca, Primary process and energy transfer: consistent terms and definitions, "Advances in photochemistry", 9, 1974, pp. 147-196.
Turro 1965: Turro, Nicholas J., Molecular photochemistry, New York, Benjamin, 1965.
Von Grotthuss 1819: von Grotthuss, Theodor, Über die chemische Wirksamkeit des Lichtes und der Elektrizität, ‟Jahresverhandlungen der Curlandischen Gesellschaft für Literatur und Kunst", 1, 1819, pp. 119-124.
Wolf 1967; Wolf, H.P. e altri, Apparatus for the measurement of quantum yields and rates of photochemical reactions, "Photochemistry and photobiology", 6, 1967, pp. 321-329.