
Neuroscienze
NEUROSCIENZE
Autocoscienza di Antonio R. Damasio
Sommario: 1. Introduzione. 2. La creazione delle immagini nella mente. a) Definizione di immagine. b) Zone cerebrali di formazione delle immagini. c) Sistemazione ed evocazione delle immagini. d) Meccanismo di formazione delle immagini. 3. La generazione di soggettività. a) Il senso cognitivo di sé. b) La teoria omuncolare. 4. Uno stato del sé, ma non un homunculus. a) Natura del sé cognitivo-neurale. b) Componenti centrali del sé. 5. Gli stati soggettivi: a) reazione del cervello alle immagini. b) Strutture cerebrali implicate nella reazione. c) Origine della soggettività. □ Bibliografia.
1. Introduzione
Sebbene sia difficile formulare una definizione universalmente accettata di ‛coscienza', la maggior parte di noi usa questo termine per riferirsi alla consapevolezza di sé e del proprio ambiente o, anche più specificamente, alla consapevolezza dell'ambiente dal punto di vista del sé. Quando nella mia mente ho un'immagine di un dato oggetto, io sento automaticamente che l'immagine appartiene a me e a nessun altro, che si è formata in me e non altrove e che usa la mia prospettiva e non quella di un altro. In altre parole, io compio un'autoreferenza, cioè genero soggettività. A nostro avviso la generazione di soggettività è la componente critica dei meccanismi biologici che permettono alla coscienza di emergere. La componente che presiede alla formazione di immagini è importante anche perché, dopo tutto, siamo sempre coscienti di qualcosa. Noi abbiamo la sensazione di soggettività nei confronti di ciò che può essere presente nella nostra mente solo sotto forma di immagine. Ma è anche possibile possedere delle immagini e non essere coscienti di esse, come accade in certi stati neurologici anormali. La formazione della soggettività segue la costruzione di immagini e crediamo che tale formazione sia l'ultimo stadio del processo che porta alla consapevolezza.
La comprensione della neurobiologia della coscienza, vale a dire della neurobiologia della soggettività, richiede, quindi, la comprensione delle basi neurali del sé. Il sé neurale si fonda su una continua segnalazione di stati del corpo e sull'attivazione, ripetuta continuamente, di ricordi autobiografici, compresi quei ricordi che si riferiscono a progetti per il futuro. Gli eventi autobiografici e la struttura corporea costituiscono, combinandosi, quel nucleo invariante su cui è possibile costruire un concetto di sé.
L'ipotesi qui prospettata è che la coscienza si determini quando, nel cervello di un organismo che sta formando un'immagine relativa a un dato oggetto, la rappresentazione di sé di quell'organismo viene modificata dal processo di formazione dell'immagine, e che anche il processo di modificazione sia rappresentato in forma di immagini.
È del tutto legittimo, per i biologi, studiare la coscienza, e non c'è ragione per dubitare che molti, o addirittura tutti, i meccanismi che concorrono alla generazione di questo complesso stato biologico possano, alla fine, essere spiegati. Il formare immagini, il costruire un concetto di sé e l'essere in grado di avere una prospettiva soggettiva sulle immagini sono processi emersi e affermatisi nel corso dell'evoluzione, perché hanno favorito la sopravvivenza; non c'è una ragione evidente perché essi debbano essere meno accessibili all'indagine di quelli, per esempio, della memoria o del linguaggio. È, comunque, possibile che, poiché tutti i nostri sforzi investigativi dipendono dalla coscienza stessa, alcuni aspetti del processo rimangano nascosti e inaccessibili, sfuggendoci così almeno per qualche tempo.
Gli strumenti moderni delle neuroscienze cognitive hanno esteso la gamma dei mezzi a nostra disposizione per indagare la coscienza negli esseri umani; d'altronde, entro certi limiti, anche le ricerche sugli animali possono dare risultati interessanti. L'uomo ha certamente una mente più ricca di qualunque altra specie, ma credo che anche negli individui di molte altre specie operino gli stessi meccanismi di base che permettono la formazione della coscienza negli esseri umani.
Nell'affrontare la neurobiologia della coscienza, dobbiamo chiarire che il termine ‛coscienza' è usato con molti significati, che vanno da ‛consapevolezza di sé e dell'ambiente circostante' a ‛coscienza morale'. Prenderemo in considerazione solo un aspetto del primo di questi significati, e cioè il processo di formazione della soggettività, considerato a livello di sistemi neurali macroscopici; pertanto non ci occuperemo di tutti gli altri aspetti della coscienza e non considereremo i dati neurobiologici a livello cellulare o molecolare. Questo articolo, inoltre, si basa sull'assunzione che la coscienza, nel senso sopra precisato, possa essere studiata dal punto di vista neurobiologico e sulla convinzione che, per progredire in questo campo, la neurobiologia debba innanzitutto capire come il cervello crei le immagini nella mente e come le immagini acquisiscano il carattere della soggettività, cioè che cosa permetta al soggetto di riconoscere queste immagini come proprie. È chiaro che la risposta della neurobiologia a entrambe le questioni non equivale a fornire una spiegazione esauriente delle basi neurali della coscienza, ma può rappresentare un proficuo inizio.
2. La creazione delle immagini nella mente
a) Definizione di immagine
Prima di trattare del modo in cui il cervello crea le immagini nella mente e del luogo in cui, dal punto di vista neuroanatomico, tali immagini si formano, dobbiamo definire il termine ‛immagine'. Con questo termine ci riferiamo a configurazioni di attività mentale basate su varie modalità sensoriali: non esistono soltanto immagini visive, ma anche immagini di suoni, immagini di movimento nello spazio, ecc. Le immagini descrivono sia il mondo esterno sia quello interno all'organismo, cioè vi sono immagini degli stati viscerali, immagini della struttura muscolo-scheletrica, del movimento del corpo, ecc. (v. somatoestesia). Le immagini rappresentano entità sia verbali che non: sono immagini anche le corrispondenze acustiche e visive delle parole e dei segni in generale. Noi usiamo il termine ‛immagine' per riferirci a configurazioni generate sia nella percezione sia nella rievocazione dalla memoria.
b) Zone cerebrali di formazione delle immagini
In questo breve articolo non possiamo esaminare tutte le ragioni per cui siamo convinti che una parte essenziale della costruzione delle immagini dipenda dalle aree corticali primarie, ma qui di seguito ne spiegheremo i motivi principali. Sappiamo che la parziale distruzione della corteccia visiva primaria preclude sia la percezione che il ricordo di alcuni aspetti della visione. Per esempio, in seguito a lesione nelle aree corticali V2 e V4 i colori non possono essere né percepiti né immaginati: non si ha più la consapevolezza del colore, anche se altri aspetti della visione rimangono invariati e anche se si può essere consapevoli dell'assenza dell'esperienza del colore. Il fatto che la percezione e il ricordo siano compromessi da una lesione che interessa una stessa zona e che non si conosca alcun'altra zona la cui lesione produca lo stesso deficit suggerisce che le aree corticali sensoriali primarie siano sedi cruciali per i processi di costruzione delle immagini. A sostegno di questa ipotesi sta anche la constatazione che un danno ad aree corticali associative di ordine superiore, localizzate al di fuori di quelle sensoriali primarie, non impedisce la formazione delle immagini (v. Damasio e Damasio, 1993 e 1994; v. Kosslyn, 1994).
Noi abbiamo ipotizzato che le aree corticali sensoriali primarie relative a ciascuna modalità sensoriale formino le configurazioni neurali che costituiscono la base delle immagini, con la collaborazione di strutture del talamo e dei collicoli. L'ipotesi si fonda sui risultati forniti da studi condotti su individui che presentavano lesioni e da studi neurofisiologici in Primati non umani, nonché su modelli conosciuti di connessione neuroanatomica. Per quanto riguarda la corteccia cerebrale, il processo richiede la cooperazione di varie regioni corticali primarie interconnesse (v. emisferi cerebrali: Interazioni interemisferiche cerebrali). Sebbene gli esatti meccanismi che realizzano tale processo siano sconosciuti, sembra che l'attività coordinata nel tempo di queste aree corticali primarie e delle stazioni sottocorticali a esse interconnesse generi l'elemento rappresentativo di attività corrispondente a ciò che noi chiamiamo ‛immagine' (v. Tononi e altri, 1992; v. Zeki, 1993; v. Churchland e altri, 1994; v. Crick, 1994).
c) Sistemazione ed evocazione delle immagini
Le immagini hanno elementi rappresentativi organizzati spazialmente e temporalmente. Nel caso delle immagini visive, somatosensitive e acustiche, tali elementi sono organizzati topograficamente. La corrispondenza fra la struttura dell'elemento rappresentativo di attività neurale nelle aree corticali sensoriali primarie e la struttura degli stimoli che l'hanno evocato è impressionante, come mostra il lavoro di Roger Tootell e altri (v., 1988). Abbiamo ipotizzato che le rappresentazioni topografiche possano essere affidate alla memoria in forma di sistemi non organizzati topograficamente ed essere immagazzinate in forma inattiva nelle regioni corticali o nei nuclei sottocorticali. Le rappresentazioni organizzate topograficamente possono essere rigenerate dalla successiva riattivazione di questi sistemi inattivi che, quindi, dalle loro sedi, inviano segnali retroattivi verso le aree corticali sensoriali primarie (v. Damasio, The brain binds..., e Time-locked..., 1989; v. Damasio e Damasio, 1994). Le reti neurali artificiali forniscono un quadro concettuale per capire come ciò sia possibile (v., per es., Churchland, 1995). Il processo di ‛retroattivazione' usa gli elementi rappresentativi di connessione a feedforward e a feedback che caratterizzano l'architettura delle regioni corticali e dei nuclei sottocorticali. In breve, le rappresentazioni topografiche possono essere suscitate o da segnali esterni al cervello - nel processo percettivo - o - nel processo di rievocazione - da segnali interni al cervello, provenienti da registrazioni mnemoniche conservate in forma di sistemi.
d) Meccanismo di formazione delle immagini
Quasi tutte le nostre esperienze sono basate su immagini delle diverse modalità sensoriali, che si formano approssimativamente nell'arco della stessa finestra temporale. Poiché le aree corticali sensoriali di ciascuna modalità sensoriale non sono contigue né interconnesse direttamente, le nostre esperienze polimodali devono derivare dall'attività convergente di varie regioni separate del cervello, e non dall'attività di una singola regione. Questo significa che la costruzione delle immagini è un processo le cui componenti sono spazialmente separate; eppure alla nostra mente e alle nostre esperienze appare ‛integrato' anziché parcellizzato. Come avviene questa ‛integrazione'? La nostra idea è che la sincronizzazione di attività separate svolga un ruolo essenziale nell'integrazione. Noi sospettiamo che il meccanismo neurale preposto alla sincronizzazione comporti l'invio di segnali da parte di insiemi di neuroni sia corticali che sottocorticali, capaci di scaricare simultaneamente verso molte popolazioni separate di neuroni. Abbiamo chiamato questi insiemi ‛zone di convergenza' (v. Damasio, The brain binds..., e Time-locked..., 1989): esse ricevono segnali convergenti e danno origine a segnali divergenti verso i siti di provenienza dei segnali convergenti. Le zone di convergenza contengono un deposito di conoscenze in forma di sistemi pronti per essere attivati. Tali zone, interamente localizzate nelle aree corticali associative e nei nuclei sottocorticali, durante la rievocazione generano immagini nelle aree corticali sensoriali primarie, mentre durante la percezione rinforzano le immagini percepite accrescendone la coerenza.
L'idea che la sincronizzazione svolga un ruolo nella costruzione delle immagini nell'ambito di una modalità sensoriale è stata prospettata da alcuni autori (v. Malsburg e Bienenstock, 1986; v. Edelman, 1987; v. Singer e altri, 1990), mentre altri, tra cui noi, sostengono che la sincronizzazione può essere essenziale per l'integrazione polimodale (v. Damasio, Time-locked..., 1989; v. Llinas, 1993).
Ma gli elementi rappresentativi sensoriali organizzati temporalmente e topograficamente qui discussi non bastano a formare l'esperienza di un'immagine, cioè la coscienza di un'immagine. Senza dubbio, rievocazione o percezione non possono avvenire senza l'attività concertata di aree corticali sensoriali primarie, del corpo genicolato laterale e del collicolo superiore, ma non crediamo che l'attività di queste aree sia sufficiente a rendere il soggetto cosciente di un'immagine. L'emergere della consapevolezza necessita di altre strutture, corticali e non, e di altri processi. Le belle immagini di attivazione sensoriale che le moderne tecniche di neuroimmagine permettono di ottenere non corrispondono alle esperienze visive coscienti, sebbene indichino un sottoinsieme delle attività neurali necessarie per rendere le immagini consce.
3. La generazione di soggettività
a) Il senso cognitivo di sé
L'idea che esista un senso cognitivo di sé, che si poggia su strutture e meccanismi neurobiologici, non significa che tutte le immagini, e le manipolazioni delle immagini, che hanno luogo nel cervello siano controllate da un elemento unico e centrale deputato al riconoscimento e al controllo, né che esso sia addirittura situato in una sola parte del cervello. Il nostro punto di vista è in linea con le critiche rivolte all'ipotesi che la coscienza risieda in una sola regione del cervello (v. Churchland, 1984 e 1995; v. Churchland e Sejnowski, 1992; v. Damasio, Time-locked..., e The brain binds..., 1989; v. Dennett, 1991). Semplicemente, intendiamo dire che le nostre esperienze vengono elaborate in una prospettiva coerente - quella del singolo soggetto - che è diminuita o sospesa negli stati patologici del cervello, quali le forme estreme di anosognosia, alcuni tipi di crisi epilettiche, i casi di personalità multipla e la schizofrenia.
L'idea che ciascuno ha di sé non è legata ai pronomi ‛io' o ‛me': è infatti difficile immaginare come i meccanismi di costruzione del linguaggio avrebbero potuto evolversi, se gli animali non avessero posseduto ‛sé' precedenti il linguaggio. Gli animali sprovvisti di linguaggio hanno probabilmente un sé, nel senso descritto sopra, sebbene la complessità del sé sia maggiore nei Primati superiori e ancora più grande negli esseri umani. Il linguaggio arricchisce il sé dell'uomo anche se non è indispensabile alla sua formazione.
b) La teoria omuncolare
La soluzione usuale del problema del sé è rappresentata dall'ipotesi dell'homunculus (v. sistema piramidale: Fisiologia del sistema piramidale), che consiste nel postulare l'esistenza, nel cervello, di una ‛creatura' con una ben definita collocazione spaziale, davanti ai cui occhi scorrerebbero le immagini, e nell'assumere che questa ‛creatura' disponga delle conoscenze necessarie per interpretarle. Ma questa soluzione sarebbe valida solo se l'homunculus pensante avesse un suo cervello e sue conoscenze, sicché si dovrebbero, a loro volta, spiegare le sue immagini: chiaramente la soluzione dell'‛homunculus spaziale' non è accettabile, perché perpetua il problema all'infinito. La fallacia di questa ipotesi è ora ampiamente riconosciuta, ma, poiché il sé è stato tradizionalmente concettualizzato in termini omuncolari, i tentativi di rimuovere il concetto di homunculus hanno spesso comportato l'abolizione della nozione di ‛sé' e, per estensione, della nozione di soggettività. Ma rifiutare l'idea che esista un homunculus nel nostro cervello non modifica il fatto che, nella maggior parte dei casi, le immagini della nostra mente sono elaborate in una prospettiva coerente. Sostenere che il nostro cervello non fa altro che formare immagini e che noi siamo coscienti di queste immagini non è una buona soluzione: la natura dell'entità neurale che è conscia di queste immagini rimane non specificata. Ciò di cui abbiamo bisogno è un'ipotesi plausibile e verificabile circa la struttura neurale che è alla base del sé, così da poter evitare i problemi dell'homunculus.
4. Uno stato del sé, ma non un homunculus
a) Natura del sé cognitivo-neurale
Le linee essenziali della nostra ipotesi sulla natura del sé, che abbiamo descritto dettagliatamente altrove (v. Damasio e Damasio, 1994; v. Damasio, 1994), sono le seguenti.
Il sé è formato da una collezione di immagini riguardanti la maggior parte degli aspetti costanti del nostro organismo e delle sue interazioni, compresi alcuni aspetti della struttura corporea e del funzionamento del corpo (tra cui un repertorio dei movimenti eseguibili con l'intero corpo e con le sue parti), i tratti che definiscono l'identità (legami di parentela, attività, luoghi) e i modelli di risposta - motoria e sensoriale - specifici. Questa collezione di immagini ha un'alta probabilità di essere evocata ripetutamente e continuamente dai vari stati del corpo oppure per attivazione da parte di sistemi rappresentativi, come accade con le registrazioni riguardanti l'identità e i modelli di risposta specifici.
Il sé cognitivo-neurale è quindi l'equivalente cognitivo-neurale di un concetto, non differente, nella sua essenza, dal concetto di un particolare oggetto la cui rappresentazione si basa sulla registrazione separata delle sue proprietà (quali forma, dimensione, colore, tessitura, movimento caratteristico, ecc.) in diversi sistemi neurali, dai quali possono essere rievocate congiuntamente non appena il concetto viene attivato.
b) Componenti centrali del sé
Abbiamo ipotizzato che le componenti centrali del concetto di sé riguardino sia la struttura corporea, cioè i visceri e l'armatura muscolo-scheletrica, sia i fondamenti dell'identità individuale, cioè le attività usuali, le preferenze, le relazioni con gli oggetti e con le altre persone. Queste componenti centrali cambiano in modo sostanziale durante l'infanzia e l'adolescenza, mentre mutano meno e più gradualmente durante la vita adulta. Il concetto di sé è legato agli stati viscerali e al meccanismo neurale che rappresenta e regola i processi biologici fondamentali, la cui modificabilità è minima. La situazione degli stati viscerali viene continuamente segnalata al midollo spinale, alla formazione reticolare del tronco-encefalo e quindi al complesso delle aree corticali somatosensoriali dell'insula, all'operculum parietale e alle zone corticali parietali postrolandiche, nonché a quelle limbiche (la segnalazione è bilaterale, ma, a livello corticale, si ha, nell'uomo, l'effetto dominante dell'emisfero destro; v. emisferi cerebrali: Dominanza cerebrale; sistema reticolare ascendente, somatoestesia).
Gli scettici possono obiettare che di solito noi non siamo consapevoli degli stati del nostro corpo e che, quindi, è strano che la soggettività sia dipendente dai segnali corporei. Ma questa obiezione, come abbiamo sostenuto altrove (v. Damasio, 1994), è debole: è vero che la nostra attenzione generalmente è concentrata su segnali non corporei, ma essa può spostarsi rapidamente, soprattutto in situazioni quali il dolore o l'agitazione emotiva. Inoltre il discorso che stiamo portando avanti riguarda in particolar modo lo sviluppo storico - evolutivo e individuale - del senso di sé, più che la situazione di un adulto. Infine, poiché siamo tutti d'accordo sul fatto che i meccanismi deputati a far emergere la soggettività sono occulti, non vi sono ragioni perché gli stati corporei che abbiamo ipotizzato come loro impalcatura debbano essere facilmente percepiti dalla coscienza. La questione importante è decidere se tali meccanismi siano plausibili, non se ne siamo, o ne potremmo essere, consapevoli.
5. Gli stati soggettivi
a) Reazione del cervello alle immagini
È importante indicare come la soggettività possa emergere dal meccanismo neurale sopra descritto. Il cervello reagisce alle immagini corrispondenti a un'entità percepita di recente - per esempio, un viso - non appena queste si formano nelle aree corticali sensoriali primarie. Secondo la nostra teoria, ciò accade perché segnali indotti da quelle immagini sono trasmessi a diversi nuclei sottocorticali (per esempio, l'amigdala e il talamo) e a molte regioni corticali (nei settori temporale, parietale e frontale), e anche perché questi nuclei e queste regioni corticali contengono sistemi atti a rispondere a determinate classi di segnali. Il risultato è che tali sistemi rappresentativi presenti nei nuclei e nelle regioni corticali vengono attivati e, di conseguenza, inducono un insieme di cambiamenti nello stato del soggetto. Questi cambiamenti, a loro volta, alterano momentaneamente lo stato corporeo e perturbano la configurazione corrente del sé. In altre parole, il molteplice processo di riconoscimento di un oggetto genera un insieme di risposte - nervose autonome, ormonali, motorie, ‛eidetiche', ecc. - che cambiano lo stato dell'organismo per un certo intervallo di tempo. Noi suggeriamo che l'essenza dei meccanismi di coscienza neurali risieda nella perturbazione degli stati del sé da parte di immagini formatesi di recente.
Sebbene il processo di risposta che abbiamo delineato sopra implichi conoscenza (conoscenza registrata in tutto il cervello sotto forma di rappresentazioni sistematizzate innate o acquisite), esso certamente non implica che qualsiasi componente del cervello ‛sappia' che le risposte vengono stimolate dalla presenza di un'entità. Quando il cervello di un organismo genera un insieme di risposte all'immagine di un'entità, l'esistenza di una rappresentazione di sé non può far sì che il sé sappia che il suo organismo sta rispondendo. Il sé, come descritto sopra, non può sapere: non è un homunculus. Arriviamo così all'interrogativo cruciale di questa teoria: come possono generare soggettività l'immagine corrente di un'entità, da una parte, e un insieme di immagini dello stato dell'organismo, dall'altra, l'una e l'altro esistenti come momentanee attivazioni di rappresentazioni? Una possibile risposta è rappresentata dal seguente processo: 1) il cervello crea una descrizione della perturbazione dello stato dell'organismo, conseguente alle risposte del cervello stesso alla presenza di un'immagine; 2) la descrizione del processo di perturbazione assume una forma ‛eidetica' e diventa l'immagine del sé perturbato; 3) questa immagine è mostrata accanto, o in rapido avvicendamento, all'immagine che ha scatenato la perturbazione.
b) Strutture cerebrali implicate nella reazione
Per realizzare questo processo il cervello deve avere: 1) strutture neurali che sostengono l'immagine di un oggetto; 2) strutture neurali che sostengono le immagini del sé; 3) strutture neurali interconnesse con le prime e con le seconde, costituite da un terzo insieme di neuroni, che abbiamo chiamato ‛zona di convergenza' e che abbiamo indicato come il substrato neurale necessario per costruire i sistemi rappresentativi nelle regioni corticali e nei nuclei sottocorticali.
Questo terzo insieme di neuroni riceve segnali sia dalla rappresentazione di un oggetto sia da quelle del sé, quando queste ultime sono perturbate dalla reazione all'oggetto. In altre parole, il terzo insieme di neuroni può costruire una rappresentazione sistematizzata del sé nel processo di cambiamento, mentre l'organismo risponde a un oggetto. Questa rappresentazione sistematizzata sarebbe proprio dello stesso tipo di quelle che il cervello ininterrottamente costruisce, alimenta e rimodella nelle condizioni generali di apprendimento. L'informazione necessaria a costruire una tale rappresentazione sistematizzata è prontamente disponibile: poco dopo aver visto un oggetto e ottenuto una sua rappresentazione nelle aree corticali visive primarie, noi possediamo anche molte rappresentazioni dell'organismo che reagisce all'oggetto, in varie regioni somatosensoriali.
Come tutti gli altri sistemi rappresentativi, la rappresentazione sistematizzata in esame possiede la potenzialità, una volta formata, di riattivare un'immagine in qualsiasi area corticale sensoriale primaria con cui sia connessa. L'immagine fondamentale sarebbe quella del corpo dell'organismo nel processo di risposta a un particolare oggetto, cioè un'immagine somatosensoriale.
c) Origine della soggettività
Noi abbiamo avanzato l'ipotesi che tutti gli elementi sopra descritti - un oggetto che viene rappresentato, un organismo che risponde all'oggetto della rappresentazione e una descrizione dell'organismo nel processo di cambiamento in risposta all'oggetto - siano presenti simultaneamente nella memoria di lavoro e siano situati, l'uno accanto all'altro o in rapido avvicendamento, nelle aree corticali sensoriali primarie. La soggettività emergerebbe durante l'ultimo stadio, quando il cervello produce contemporaneamente non solo l'immagine di un'entità, del sé e delle risposte dell'organismo, ma anche un altro tipo di immagine: quella dell'organismo nell'atto di percepire un'entità e di rispondere a essa. Quest'ultimo tipo di immagine è all'origine della soggettività (v. Damasio, 1994).
La rappresentazione sistematizzata elaborata dal terzo insieme di neuroni di cui sopra fornisce una visione schematica dei principali protagonisti, l'oggetto e il sé, da una prospettiva esterna a entrambi, una descrizione non verbale di cosa stia avvenendo a quei protagonisti, realizzata con gli strumenti base dei sistemi sensoriale e motorio. Ciò che abbiamo in mente non è creato né percepito da un homunculus e non necessita di linguaggio.
Il cervello degli Uccelli e dei Mammiferi può essere in grado di costruire descrizioni dello stesso tipo. In effetti, la soggettività emergerebbe in ogni organismo fornito di una certa rappresentazione di sé, della capacità di formare immagini e di rispondere a esse, e della capacità di generare un qualche tipo di descrizione-sistema rappresentativo in un terzo insieme di neuroni. Le capacità narrative del second'ordine fornite dal linguaggio permetterebbero agli esseri umani di creare descrizioni verbali, oltre a quelle non verbali, e porterebbero a quella forma sofisticata di soggettività che emerge dall'ultimo processo. La macchina seriale virtuale proposta da Dennett (v., 1991) opererebbe a tale alto livello piuttosto che al livello elementare al quale funzionerebbe il meccanismo di base della soggettività da noi ipotizzato. La nostra ipotesi è abbastanza diversa da quella, avanzata da Crick (v., 1994), relativa alla base neurale della consapevolezza visiva, mentre ha alcuni importanti aspetti in comune con l'ipotesi di Edelman (v., 1992) sulla coscienza primaria, ipotesi che sviluppa il concetto di valore biologico e postula un sé radicato in sistemi omeostatici.
Una macchina fornita di dispositivi di costruzione di immagini, della capacità di rappresentare con immagini la propria struttura fisica e i propri stati fisici, e di una conoscenza dei sistemi rappresentativi del proprio passato probabilmente non sarebbe in grado di generare soggettività, anche se dovesse costruire immagini del proprio ‛sé perturbato', come descritto sopra. A meno che, naturalmente, il corpo della macchina non fosse vivente, con proprietà derivate dal proprio stato interno instabile, da proprie inerenti necessità di sopravvivenza e dalla propria intrinseca cognizione che ciò che favorisce la sopravvivenza è prezioso. Il dispositivo neurale che secondo la nostra ipotesi presiede alla formazione della soggettività ha la funzione di connettere le immagini con lo svolgersi della vita.
BIBLIOGRAFIA
Churchland, P. M., Matter and consciousness: a contemporary introduction to the philosophy of mind, Cambridge, Mass., 1984.
Churchland, P. M., The engine of reason, the seat of the soul: a philosophical journey into the brain, Cambridge, Mass., 1995.
Churchland, P. S., Neurophilosophy: toward a unified science of the mind-brain, Cambridge, Mass., 1986.
Churchland, P. S., Ramachandran, V. S., Sejnowski, T. J., The critique of pure vision, in Large-scale neuronal theories of the brain (a cura di C. Koch e J. L. Davis), Cambridge, Mass., 1994, pp. 23-60.
Churchland, P. S., Sejnowski, T. J., The computational brain: models and methods on the frontiers of computational neuroscience, Cambridge, Mass., 1992 (tr. it.: Il cervello computazionale, Bologna 1995).
Crick, F., The astonishing hypothesis: the scientific search for the soul, New York 1994 (tr. it.: La scienza e l'anima, Milano 1994).
Damasio, A. R., The brain binds entities and events by multiregional activation from convergence zones, in ‟Neural computation", 1989, I, pp. 123-132.
Damasio, A. R., Time-locked multiregional retroactivation: a systems level proposal for the neural substrates of recall and recognition, in ‟Cognition", 1989, XXXIII, pp. 25-62.
Damasio, A. R., Descartes' error: emotion, reason and the human brain, New York 1994 (tr. it.: L'errore di Cartesio: emozione, ragione e cervello umano, Milano 1995).
Damasio, A. R., Damasio, H., Cortical systems underlying knowledge retrieval: evidence from human lesion studies, in Exploring brain functions: models in neuroscience. Dahlem workshop (a cura di T. A. Poggio e D. A. Glaser), New York 1993, pp. 233-248.
Damasio, A. R., Damasio, H., Cortical systems for retrieval of concrete knowledge: the convergence zone framework, in Large-scale neuronal theories of the brain (a cura di C. Koch e J. L. Davis), Cambridge, Mass., 1994, pp. 61-74.
Dennett, D. C., Consciousness explained, London 1991 (tr. it.: Coscienza, Milano 1993).
Edelman, G. M., Neural darwinism: the theory of neuronal group selection, New York 1987 (tr. it.: Darwinismo neurale: la teoria della selezione dei gruppi neuronali, Torino 1995).
Edelman, G. M., Bright air, brilliant fire: on the matter of the mind, New York 1992 (tr. it.: Sulla materia della mente, Milano 1993).
Kosslyn, S., Image and brain: the resolution of the imagery debate, Boston 1994.
Llinas, R., Coherent 40-Hz oscillation characteristics dream state in humans, in ‟Proceedings of the National Academy of Sciences", 1993, XC, pp. 2078-2081.
Malsburg, C. von der, Bienenstock, E., Statistical coding and short-term synaptic plasticity: a scheme for knowledge representation in the brain, in Disordered systems and biological organization (a cura di E. Bienenstock, F. Fogelman Soulié e G. Weisbuch), Berlin-New York 1986, pp. 247-272.
Singer, W., Gray, C., Engel, A., Konig, P., Artola, A., Brocher, S., Formation of cortical cell assemblies, in ‟Cold Spring Harbor symposia on quantitative biology", 1990, LV, pp. 939-952.
Tononi, G., Sporns, O., Edelman, G., Reentry and the problem of integrating multiple cortical areas: simulation of dynamic integration in the visual system, in ‟Cerebral cortex", 1992, II, pp. 310-335.
Tootell, R. B. H., Switkes, E., Silverman, M. S., Hamilton, S. L., Functional anatomy of macaque striate cortex. II. Retinotopic organization, in ‟The journal of neuroscience", 1988, VIII, pp. 1531-1568.
Zeki, S., A vision of the brain, Cambridge, Mass., 1993.
Basi molecolari della comunicazione neuronale di Francesco Clementi
Sommario: 1. Organizzazione del sistema nervoso. a) Composizione cellulare del tessuto nervoso. b) Organizzazione neuronale. 2. La sinapsi. 3. I neurotrasmettitori. a) Neurotrasmettitori classici. b) Amminoacidi. c) Neuropeptidi. 4. Canali ionici voltaggio-dipendenti. a) Trasmissione dell'informazione attraverso il potenziale d'azione. b) Struttura dei canali voltaggio-dipendenti. c) Composizione dei canali ionici. d) Regolazione dei canali ionici. 5. Recettori. a) La scoperta dei recettori. b) Superfamiglie recettoriali. c) Recettori canale. d) Recettori accoppiati a proteine G. e) Recettori con attività tirosinchinasica. 6. Modulazione delle risposte ai neurotrasmettitori. 7. Modulazione patologica e farmacologica della comunicazione neuronale. a) Integrazione di più informazioni nella risposta neuronale. b) Canali ionici. c) Recettori nicotinici. d) Trasduzione del segnale. e) I farmaci come strumenti specifici per analizzare e modificare la comunicazione nervosa. 8. Conclusioni. □ Bibliografia.
1. Organizzazione del sistema nervoso
La costruzione e il funzionamento di un tessuto o di un organo presuppongono che le cellule siano tra loro connesse non solo fisicamente e meccanicamente, ma anche funzionalmente. Questo significa che ciascuna cellula può non solo rendersi conto dello stato morfofunzionale di cellule vicine e di conseguenza modificare il suo atteggiamento funzionale, ma anche modificare la risposta di altre cellule a stimoli esterni.
Nel tessuto nervoso la comunicazione intercellulare è di estrema importanza proprio perché attraverso di essa questo tessuto può svolgere la sua funzione essenziale di integrazione e di coordinamento. Inoltre, a differenza di altri tessuti, il tessuto nervoso non solo coordina le informazioni al suo interno, ma svolge un coordinamento generale tra tutti gli organi dell'organismo attraverso una fitta rete di connessioni.
a) Composizione cellulare del tessuto nervoso
L'unità fondamentale di base del sistema nervoso è il neurone, una cellula specializzata per ricevere ed elaborare informazioni e trasmetterle ad altri neuroni, a cellule muscolari o ghiandolari (v. fig. 1). Il neurone è in genere una cellula molto allungata, costituita da un corpo cellulare, che contiene il nucleo e la maggior parte dei ribosomi, del reticolo endoplasmatico rugoso e dei lisosomi. Al suo interno si attua la sintesi di quasi tutte le proteine e degli organuli neuronali; le macromolecole sintetizzate vengono assemblate negli organuli citoplasmatici e in complessi proteici e quindi trasportate nei dendriti e nell'assone. Dal corpo cellulare si dipartono una serie di propaggini, i dendriti, che possono essere considerati una sorta di antenne deputate a ricevere i segnali dagli assoni di altre cellule nervose. La superficie ricevente del neurone, tra dendriti e corpo cellulare, diviene così molto estesa, fino a poter accogliere talvolta più di 100.000 sinapsi. Da una regione precisa del neurone, chiamata asson hillock, si diparte un lungo e stretto prolungamento, l'assone, la cui funzione è quella di condurre il segnale informativo del neurone alle altre cellule; esso alla fine può sfioccarsi in numerosi assoni più piccoli, che possono quindi prendere contatto e trasmettere un segnale contemporaneamente a più cellule (v. fig. 2). La lunghezza di questo prolungamento neuronale, il cui diametro è spesso inferiore al micron (µm), può andare da qualche micron (interneuroni) a molti centimetri (motoneuroni), e arrivare anche a più di un metro: si pensi a un motoneurone posto nel midollo spinale che debba innervare un muscolo delle dita dei piedi. Esso contiene alla sua estremità una delle strutture più delicate e complesse di questa cellula, la sinapsi, il punto nel quale avviene la trasmissione dell'impulso nervoso tra una cellula e l'altra. Per consentire i continui contatti, non solo elettrici ma anche metabolici, tra il corpo cellulare e le parti più distali dei dendriti e dell'assone, nel neurone sono particolarmente sviluppati i microtubuli, i ‛motori' cellulari, e il citoscheletro, cioè l'apparato necessario per attuare il trasporto del materiale cellulare in tutte le direzioni (v. neurobiologia: Trasporto assonale).
Naturalmente, nel tessuto nervoso sono presenti anche altre cellule, le cellule gliali, le quali, fornendo soprattutto supporto meccanico e nutritivo ai neuroni ed esercitando un controllo metabolico del milieu intercellulare, permettono il regolare funzionamento dei neuroni. Le cellule gliali possono essere differenziate in diversi sottotipi: tra questi ricordiamo le cellule di Schwann, responsabili della costruzione della guaina mielinica attorno agli assoni, e le cellule microgliali, che hanno una funzione simile a quella dei macrofagi e attraverso la secrezione di interleuchine possono influenzare non solo la risposta immune a livello del sistema nervoso, ma anche l'attività e la reattività neuronale.
Le strutture attraverso le quali si attua l'interazione cellulare sono le giunzioni cellulari, classificabili in tre gruppi funzionali: giunzioni occludenti (tight junctions), comunicanti e ancoranti (v. fig. 3A). Le giunzioni occludenti sono deputate a sigillare le cellule tra di loro in modo che la permeabilità tra di esse sia praticamente nulla e neppure molecole di piccole dimensioni possano attraversarle: questo è reso possibile dall'esistenza di una struttura complessa di particelle intramembrana poste in file ordinate nelle cellule adiacenti e combacianti perfettamente tra loro (v. fig. 3B); queste giunzioni sono tipiche dei tessuti epiteliali e vengono utilizzate per isolare completamente il mondo esterno dal milieu intérieur. Le giunzioni comunicanti permettono alle cellule di comunicare tra loro da un punto di vista metabolico, controllando il passaggio di ioni e piccole molecole: sono infatti costituite da raggruppamenti di piccoli canali che mettono in comunicazione il citoplasma di due cellule vicine e permettono il passaggio bidirezionale di molecole fino a un peso molecolare di 1.500 circa (v. fig. 3C); attraverso di esse passano, per esempio, gli ioni (e tra essi il più importante è il calcio), AMP e cAMP, ATP e altre molecole utili per il metabolismo cellulare; l'apertura e la chiusura di queste giunzioni è controllata dalla concentrazione di ioni Ca2+ intracellulari. L'adesione meccanica tra le varie cellule si ottiene, invece, attraverso le giunzioni ancoranti, che legano le cellule alla membrana basale, e i desmosomi, che le tengono legate tra loro.
Nel tessuto nervoso sono presenti tutte e tre le strutture di interazione intercellulare sopra descritte, che sono poste sia tra i neuroni, sia tra i neuroni e le cellule gliali, sia tra le cellule gliali. Per quanto riguarda la comunicazione neuronale, le giunzioni più interessanti sono quella comunicante e soprattutto un altro tipo di giunzione, la sinapsi, elaborata appositamente dalla cellula nervosa per comunicare in modo controllato e specifico con altre cellule, della quale discuteremo più avanti e alla quale è dedicato uno specifico articolo (v. sinapsi). Gli altri tipi di giunzione sono invece importanti non solo durante lo sviluppo, in quanto permettono a ogni cellula sia di trovare la giusta via durante la migrazione, sia il riconoscimento reciproco (v. Shatz, 1992), ma anche dopo che il tessuto nervoso si è sviluppato poiché consentono di mantenere e solidificare i rapporti tra le cellule. Nel cervello svolgono un ruolo importante le giunzioni occludenti poste tra le cellule endoteliali dei capillari, in quanto garantiscono la tenuta della barriera ematoencefalica che costituisce una sorta di isolamento biochimico tra il liquido extracellulare del cervello e il sangue. (Per una descrizione più esauriente del neurone, delle cellule gliali e dell'organizzazione del sistema nervoso, v. neurone e impulso nervoso; neurobiologia).
b) Organizzazione neuronale
Due delle fondamentali funzioni del sistema nervoso sono la coordinazione della risposta a uno stimolo e la modulazione della risposta dell'organismo agli stimoli esterni. Schematicamente abbiamo, quindi, un sistema per riconoscere lo stimolo (i neuroni sensitivi), un sistema per ordinare la risposta, in genere motoria (i neuroni motori), e vari sistemi per coordinare, modulare e integrare le informazioni tra questi due sistemi (gli interneuroni).
Negli organismi più semplici, per esempio in molti Invertebrati, i neuroni sensitivi si connettono direttamente con i neuroni motori, e non vi sono quindi grandi possibilità di modulare la risposta (v. fig. 4A). Negli organismi che hanno raggiunto un certo grado di sviluppo, i neuroni sono collegati tra loro a formare dei circuiti o delle vie neuronali. Un esempio di circuito neuronale tra i più semplici è quello dell'arco riflesso, scoperto fin dagli inizi delle ricerche in neuroscienze. Lo scopo dell'arco riflesso è quello di collegare i neuroni sensitivi con i neuroni motori attraverso una serie di interneuroni: in tal modo un neurone sensitivo può mandare la sua informazione a più neuroni motori e un motoneurone può modificare la sua attività ricevendo le informazioni provenienti da più neuroni sensitivi; gli interneuroni integrano le informazioni che vengono dai motoneuroni e dai neuroni sensitivi, favorendo quindi la coordinazione e la modulazione della risposta dell'organismo allo stimolo ambientale (v. fig. 4B). Nel sistema nervoso centrale (SNC), che è il punto più alto d'integrazione dell'informazione e la sede delle elaborazioni intellettive ed emotive, i circuiti nervosi sono assai più complessi ed è molto difficile dissecarli nelle diverse parti, anche se le metodiche moderne di immunocitochimica e di biologia cellulare e molecolare hanno permesso di identificare le vie nervose più importanti.
La caratteristica dei circuiti nervosi consiste soprattutto nella specificità delle connessioni neuronali che si realizzano attraverso le sinapsi; tale specificità è ottenuta sia attraverso collegamenti precisi sul piano spaziale, sia attraverso la compatibilità tra il neurotrasmettitore secreto dalla parte presinaptica e i recettori presenti in quella postsinaptica.
2. La sinapsi
La sinapsi è la struttura che permette il passaggio unidirezionale dell'informazione da una cellula a un'altra. Questa struttura si trova alla fine dell'assone e rappresenta il punto di contatto tra l'assone stesso e la cellula innervata ed è qui che avviene la trasmissione dell'informazione (v. fig. 1). La sinapsi ha le seguenti proprietà, che sono essenziali per formare un corretto circuito neuronale: 1) la trasmissione del segnale è unidirezionale, dall'assone alla cellula innervata e non viceversa; 2) la trasmissione è focalizzata, avviene cioè non solo con una cellula ben precisa, ma anche con una porzione precisa della cellula, in modo che il segnale sia localizzato e puntiforme; 3) la trasmissione è specifica, in quanto si basa sull'interazione tra il neurotrasmettitore liberato dalla parte presinaptica e un recettore specifico presente sulla membrana postsinaptica: solo quando i due elementi, trasmettitore e recettore, sono compatibili avviene la trasmissione nervosa; inoltre, la risposta postsinaptica dipende dal tipo di recettore attivato: per esempio l'acetilcolina può indurre sia una trasmissione assai rapida dell'impulso nervoso, se eccita recettori di tipo nicotinico, sia una risposta lenta e di tipo metabolico, se eccita recettori di tipo muscarinico; 4) la trasmissione del segnale può essere modulata nell'intensità attraverso sofisticati meccanismi che controllano sia la secrezione di neurotrasmettitore, sia la presenza e la reattività dei recettori, sia l'interazione tra neurotrasmettitore e recettore. Per ottemperare a queste funzioni la sinapsi ha una struttura particolare che è già stata descritta precedentemente (v. sinapsi).
Le sinapsi hanno dimensioni diverse (da qualche micron2 nelle giunzioni tra neuroni fino a molti micron2 nella giunzione tra motoneurone e cellula muscolare), ma la loro struttura di base è simile e consiste in: a) una parte presinaptica, che contiene gli enzimi per sintetizzare i neurotrasmettitori e il complesso apparato per immagazzinarli e secernerli; b) uno stretto spazio intrasinaptico di 200 Å nel quale viene rilasciato il neurotrasmettitore, ricco di proteine della matrice extracellulare e, talvolta, di enzimi implicati nel metabolismo del neurotrasmettitore: le sue funzioni sono quelle di fissare meccanicamente il contatto sinaptico tra le due cellule attraverso le numerose molecole di adesione, di rendere disponibile uno spazio limitato nel quale il mediatore può temporaneamente rimanere in alta concentrazione senza essere diluito dal liquido extracellulare, di cooperare al mantenimento della struttura della membrana postsinaptica, soprattutto favorendo l'addensamento dei recettori attraverso molecole che collegano la matrice extracellulare ai recettori; c) una membrana postsinaptica, nella quale sono presenti i recettori specifici per il neurotrasmettitore liberato dalla presinapsi, la cui struttura può essere semplice, come nella giunzione tra due cellule nervose, oppure più elaborata, come nella giunzione neuromuscolare (v. fig. 5); la sua funzione più importante è quella di contenere recettori per i neurotrasmettitori e altre strutture necessarie alla trasduzione del segnale, come canali ionici e pompe; essa inoltre garantisce che queste strutture molecolari si trovino nella giusta posizione per svolgere il loro lavoro: per esempio, i recettori debbono essere localizzati proprio in corrispondenza del punto in cui il neurotrasmettitore è secreto dalla parte presinaptica; ciò è possibile in quanto nella parte citoplasmatica, sotto la membrana plasmatica, è presente un citoscheletro, con un'organizzazione spaziale ben precisa, al quale i recettori o le altre molecole di membrana si ancorano attraverso proteine specifiche che fanno da ponte tra queste strutture. Tale complesso citoscheletro-recettori conferisce alla membrana postsinaptica, vista al microscopio elettronico, il particolare aspetto mostrato nella fig. 5.
La funzione della sinapsi è relativamente semplice (v. fig. 6). Nel momento in cui la membrana presinaptica è invasa dal potenziale d'azione, essa si depolarizza e questo cambiamento di voltaggio viene percepito dai canali per il calcio voltaggio-dipendenti, che si aprono lasciando entrare il Ca2+: attraverso un meccanismo complesso recentemente delucidato, questo ione permette che le vescicole sinaptiche ricche di neurotrasmettitore si fondano con la membrana plasmatica e liberino il neurotrasmettitore nello spazio sinaptico. Il neurotrasmettitore liberato attiva i recettori postsinaptici e in questo modo il segnale si trasferisce dalla fibra presinaptica alla cellula innervata. In seguito, le vescicole sinaptiche svuotate del neurotrasmettitore ritornano all'interno della presinapsi per ricaricarsi ed essere pronte per un nuovo ciclo (v. McPherson e De Camilli, 1994). Il neurotrasmettitore liberato nello spazio sinaptico viene rimosso attraverso diversi meccanismi che possono anche funzionare contemporaneamente: può essere ripreso dalla presinapsi, attraverso trasportatori specifici recentemente clonati (v. Attwell e altri, 1993; v. Attwell e Mobbs, 1994), e immagazzinato nelle vescicole; oppure distrutto da enzimi, come per esempio l'acetilcolina dall'acetilcolinesterasi; o catturato dalle cellule gliali che circondano la giunzione; o ancora, diluito dal liquido extracellulare.
La risposta che la cellula innervata produce quantitativamente è funzione del numero di molecole di neurotrasmettitore uscito dalla parte presinaptica e del numero di recettori postsinaptici attivati, mentre qualitativamente dipende dal tipo di recettore attivato.
Nei capitoli seguenti analizzeremo in modo più dettagliato i meccanismi molecolari che permettono alla sinapsi di funzionare, soffermandoci soprattutto sui neurotrasmettitori, sui canali ionici e sui recettori.
3. I neurotrasmettitori
Come abbiamo detto, le molecole che trasportano l'informazione neuronale attraverso la sinapsi sono i neurotrasmettitori, che possono quindi essere chiamati messaggeri sinaptici. Essi vengono sintetizzati nel citoplasma della presinapsi, immagazzinati in vescicole e, allorché l'impulso depolarizzante invade la membrana presinaptica, vengono secreti nello spazio sinaptico, ove raggiungono i recettori e li stimolano. Contemporaneamente alla secrezione, si pongono in atto i meccanismi deputati alla rimozione dei neurotrasmettitori dallo spazio sinaptico, mettendo fine alla trasmissione sinaptica.
I neurotrasmettitori possono essere divisi in due gruppi fondamentali: i neurotrasmettitori classici, o convenzionali, e i neuropeptidi (v. McQueen, 1987).
a) Neurotrasmettitori classici
Appartengono a questo gruppo l'acetilcolina (le sinapsi del SN sono per il 5-10% colinergiche), le monoammine (catecolammine, serotonina e istamina, secrete dall'1-2% delle sinapsi cerebrali) e gli amminoacidi (GABA, glicina, glutammato, aspartato, taurina, secreti da circa il 60% delle sinapsi centrali). Essi sono sintetizzati nel citoplasma, soprattutto a livello della sinapsi, per azione di una serie di enzimi specifici, partendo da precursori molto comuni e che facilmente possono essere disponibili nel neurone (v. tab. I). Ogni neurone può sintetizzare solamente un neurotrasmettitore di tipo convenzionale; si hanno così neuroni colinergici, dopamminergici, GABA-ergici, ecc., che sono raggruppati in nuclei specifici del SNC dai quali proiettano assoni a zone anche distanti dove stabiliscono le connessioni sinaptiche. Si hanno pertanto delle vie neuronali ben precise destinate a convogliare i neurotrasmettitori ai siti particolari nei quali devono agire. Alcune di queste vie sono schematizzate nella fig. 7.

L'acetilcolina è sintetizzata a partire dalla colina - che entra nei neuroni attraverso uno specifico trasportatore per azione di un enzima, la colinoacetiltransferasi - ed è inattivata per idrolisi dalle acetilcolinesterasi, presenti nello spazio sinaptico. I neuroni colinergici sono raggruppati soprattutto nel nucleo magnocellulare di Maynert, nel nucleo del setto mediale e in alcuni nuclei pontini. Da lì essi proiettano alla corteccia in modo diffuso, al talamo e all'ippocampo. I recettori per l'acetilcolina si dividono in due tipi: i nicotinici, canali ionici per il sodio e il calcio, e i muscarinici, accoppiati alle proteine G (v. cap. 5). In genere l'acetilcolina è un mediatore eccitatorio.
Le monoammine costituiscono una grande famiglia della quale fanno parte la noradrenalina, l'adrenalina, la dopammina, la serotonina e l'istamina. La loro sintesi coinvolge più enzimi, come esplicitato nella tab. I. Le fibre monoamminergiche, nonostante siano minoritarie, sono molto diffuse e possono avere un'influenza assai importante su molte funzioni cerebrali. Molti farmaci psicotropi, dagli antidepressivi agli antiparkinsoniani, ai farmaci usati nella schizofrenia, agiscono modulando la secrezione o la sintesi o la liberazione di questi neurotrasmettitori.
La noradrenalina è sintetizzata da neuroni confinati nel midollo allungato, nel ponte e nel nucleo del tratto solitario. Questi neuroni proiettano in modo diffuso a quasi tutte le regioni del SNC, soprattutto alla corteccia, all'ipotalamo e al midollo spinale. L'adrenalina è meno diffusa della noradrenalina; i neuroni adrenergici sono localizzati soprattutto nel midollo allungato e proiettano a vari nuclei ipotalamici e al midollo spinale. I recettori adrenergici sono diversi, raggruppati in due grandi famiglie - α e β - e sempre accoppiati a proteine G. L'effetto dell'adrenalina o della noradrenalina dipende dal tipo di recettore eccitato e in genere è di tipo inibitorio.
La dopammina è sintetizzata soprattutto in neuroni concentrati nella substantia nigra, nell'ipotalamo e nel bulbo olfattivo: nella via più conosciuta, quella tra la substantia nigra e lo striato, essa controlla la regolazione del movimento. La degenerazione delle cellule dopamminergiche porta a una patologia imponente, come quella del morbo di Parkinson, che può essere parzialmente alleviata somministrando un precursore della dopammina, la L-DOPA. Si conoscono almeno quattro recettori per la dopammina, i quali agiscono tutti attraverso proteine G.
La serotonina è sintetizzata a partire dal triptofano attraverso diverse tappe metaboliche, ed è inattivata per ossidazione dalle monoamminossidasi (v. tab. I). I neuroni che la producono sono localizzati soprattutto nel rafe e nel midollo allungato e proiettano in modo diffuso alla corteccia, al midollo spinale e al cervelletto. La serotonina è certamente connessa con importanti processi affettivi e conoscitivi nonché con il controllo di alcune funzioni importanti, come la regolazione della temperatura e l'appetito.
Si pensa che molti farmaci antidepressivi agiscano attraverso un'attivazione delle vie serotoninergiche. I recettori serotoninergici sono di almeno cinque tipi: i recettori 5-HT1,2,4,5 sono legati a proteine G; il 5-HT3 è invece un canale ionico.
b) Amminoacidi
Questi neurotrasmettitori, tra i più abbondanti nel SNC, sono costituiti da amminoacidi inibitori, come il GABA (acido γ-amminobutirrico) e la glicina, o eccitatori, come il glutammato e l'aspartato.
Il GABA è il più importante neurotrasmettitore inibitorio del SNC, essendo contenuto in circa il 40% delle sinapsi. È sintetizzato soprattutto da piccoli interneuroni distribuiti in quasi tutto il SNC; la sua sintesi avviene anche in alcuni neuroni che proiettano dal corpo striato alla substantia nigra. I recettori per il GABA sono di due tipi, i GABAA e i GABAB: i recettori GABAA sono dei canali ionici selettivi per il cloro; i recettori GABAB sono accoppiati a proteine G e il loro ruolo funzionale non è ancora chiaro. Farmaci assai attivi, come ansiolitici e ipnotici (le benzodiazepine e i barbiturici) o come antiepilettici (l'acido valproico), agiscono nel sistema nervoso potenziando il sistema GABA-ergico. La glicina è il neurotrasmettitore inibitorio presente nel midollo spinale; anch'essa attiva recettori-canale per il cloro.
L'acido glutammico, che è il mediatore eccitatorio più diffuso nel SNC, è sintetizzato da neuroni sparsi in molti nuclei del cervello; particolarmente studiati sono i nuclei ippocampali, che forniscono un modello prezioso per analizzare i meccanismi con i quali si instaura e si mantiene la memoria.
c) Neuropeptidi
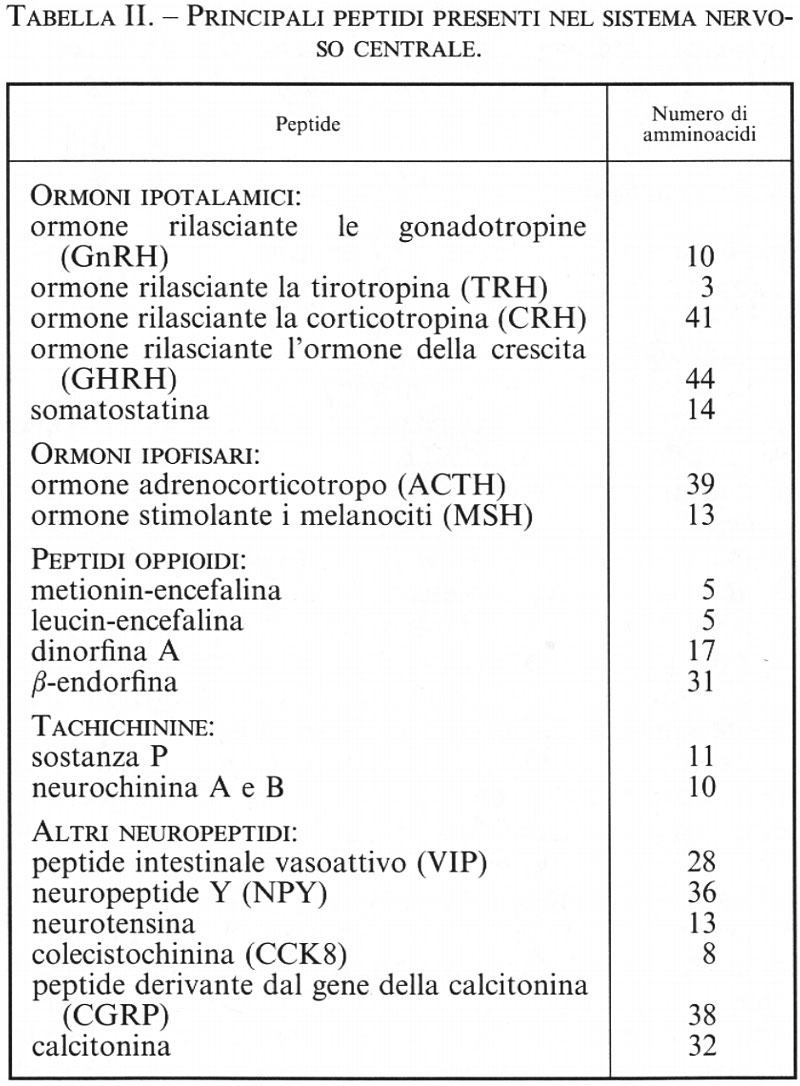
Dagli anni settanta si è cominciato a isolare dal SNC alcuni piccoli peptidi con attività di neurotrasmettitori (v. Harmor, 1987). Questi peptidi possono essere sintetizzati dagli stessi neuroni che sintetizzano i neurotrasmettitori classici; si realizza così per alcuni neuroni un duplice codice d'informazione attraverso mediatori differenti. Sembra infatti che i peptidi possano avere una funzione più modulatrice che non proprio neurotrasmettitrice: tra le associazioni più comuni ricordiamo quelle tra acetilcolina e galanina, noradrenalina e neuropeptide Y (NPY), acetilcolina e calcitonin gene-related peptide. Tra i peptidi più studiati vi sono l'ossitocina e la vasopressina, gli ormoni ipotalamici - quali la somatostatina, il fattore per la liberazione della tirotropina (TRH), il fattore per la liberazione delle gonadotropine (GnRH) -, la colecistochinina, il peptide vasoattivo interstiziale (VIP), ecc. Ma la scoperta più rilevante in questo settore ha riguardato i peptidi oppioidi, come le endorfine e le encefaline, che stimolano i recettori per la morfina: la loro individuazione ha permesso di capire il meccanismo d'azione della morfina, in che modo, cioè, essa produca gli effetti analgesici, stimolanti o depressivi nel SNC. Si sperava anche di poter arrivare a sintetizzare farmaci analgesici che non avessero contemporaneamente gli effetti indesiderati della morfina, quali ad esempio la depressione dei centri respiratori o la dipendenza; purtroppo, nonostante molte ricerche, questo risultato non è stato ancora raggiunto.
La scoperta di peptidi neurotrasmettitori così numerosi (un elenco dei più importanti è riportato nella tab. II) ha permesso non solo di ampliare notevolmente le conoscenze sulla trasmissione a livello del SNC, ma soprattutto di fornire delle basi funzionali per attività nervose non spiegabili solo attraverso l'azione dei neuromediatori classici. I neurotrasmettitori peptidici agiscono, in genere, su recettori accoppiati a proteine G.
Dal punto di vista della biologia cellulare vi è una grande differenza tra i neurotrasmettitori classici e i neuropeptidi: i primi sono sintetizzati a livello del citoplasma presinaptico e veicolati poi nelle vescicole sinaptiche tramite trasportatori, mentre i neuropeptidi sono sintetizzati nel corpo cellulare e nel reticolo endoplasmatico, e da lì trasferiti nei granuli di secrezione che vengono poi trasportati fino alla sinapsi; inoltre, le vescicole sinaptiche possono essere ricaricate di neurotrasmettitore subito dopo averlo liberato, mentre i granuli che contengono i peptidi non possono più essere ricaricati e la sinapsi deve essere rifornita di nuovi granuli dal corpo cellulare; infine, sembra che per liberare i neuropeptidi siano necessarie stimolazioni nervose più intense e prolungate (v. fig. 6).
Queste differenze di organizzazione cellulare e di secrezione distinguono quindi ulteriormente i due tipi di neurotrasmettitori. In genere, si può ipotizzare che i veri neurotrasmettitori siano quelli di tipo classico, mentre i neuropeptidi agirebbero soprattutto da modulatori dell'attività neuronale. Solo le endorfine possono essere considerate dei veri neurotrasmettitori.
4. Canali ionici voltaggio-dipendenti
I neurotrasmettitori rinchiusi nelle vescicole sinaptiche vengono liberati nello spazio extrasinaptico attraverso una depolarizzazione della membrana plasmatica (potenziale d'azione) che permette l'entrata di ioni calcio e la fusione delle vescicole con la membrana presinaptica. Il flusso degli ioni attraverso la membrana sinaptica dipende dai canali ionici voltaggio-dipendenti.
a) Trasmissione dell'informazione attraverso il potenziale d'azione
Nonostante che i vari tipi neuronali trasmettano segnali con diverso significato e che alla fine si possano ottenere informazioni tanto numerose e tuttavia ben definite, il segnale è sempre rappresentato da cambiamenti del potenziale elettrico della membrana cellulare del neurone. La propagazione del segnale lungo l'assone può avvenire in quanto una piccola perturbazione elettrica prodotta in una parte della cellula si propaga a tutta la membrana plasmatica del neurone. Questa propagazione diviene naturalmente sempre più debole man mano che si allontana dal punto d'insorgenza, a meno che la cellula non impieghi energia per amplificarla durante il tragitto. L'attenuazione del segnale non ha molta importanza per comunicazioni a breve distanza, ma diviene assai significativa per distanze lunghe. Il neurone e le altre cellule eccitabili hanno pertanto elaborato un sofisticato sistema di amplificazione del segnale: una depolarizzazione che superi una certa intensità (e vedremo più avanti come essa possa prodursi in seguito all'azione dei neurotrasmettitori) attiva una serie di canali ionici, detti appunto voltaggio-dipendenti, permeabili soprattutto agli ioni Na+; l'ingresso di questi ultimi depolarizza ulteriormente la membrana, permettendo l'apertura di altri canali che fanno entrare una quantità ancora maggiore di Na+ che a sua volta fa aumentare la depolarizzazione della membrana. Si innesca così una ‛esplosione' di attività elettrica che si propaga rapidamente e autonomamente su tutta la membrana plasmatica del neurone: questa eccitazione elettrica è chiamata potenziale d'azione (v. elettrofisiologia). Il potenziale d'azione è un sistema assai efficiente e veloce per trasmettere l'informazione attraverso il neurone; esso può viaggiare, infatti, con una velocità di più di 100 metri al secondo. La genesi e il mantenimento del potenziale d'azione sono più complessi di quanto qui delineato e coinvolgono una fine interazione tra diversi canali ionici voltaggio-dipendenti e con altre molecole necessarie per la ripolarizzazione della membrana.
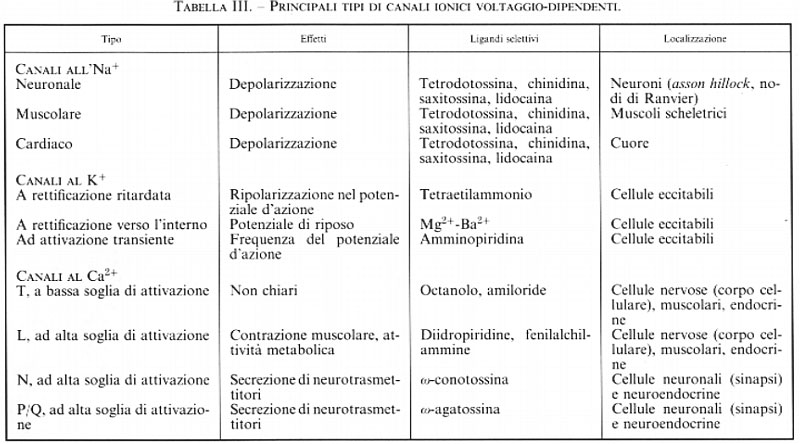
I canali voltaggio-dipendenti non solo sono importanti per la conduzione dell'impulso nervoso lungo l'assone, ma sono anche essenziali per la funzionalità della sinapsi. A questo livello i canali coinvolti sono soprattutto quelli permeabili al calcio, in particolare quelli sensibili alla ω-conotossina e alla ω-agatossina, denominati N e P/Q, rispettivamente, responsabili dell'accoppiamento tra l'arrivo dell'impulso nervoso e la secrezione di neurotrasmettitore. Nel corpo cellulare del neurone sono presenti anche altri tipi di canali voltaggio-dipendenti per il Ca2+, soprattutto quelli di tipo sensibile alle diidropiridine, probabilmente responsabili di altri fenomeni calcio-dipendenti, quali il trofismo neuronale, l'attivazione genica e, in condizioni non fisiologiche, la neurotossicità (v. Varadi e altri, 1995).
b) Struttura dei canali voltaggio-dipendenti
La chiave della trasmissione dell'impulso nervoso, sia a livello assonale e dendritico sia a livello sinaptico, risiede, quindi, nel corretto funzionamento dei canali ionici. Le molecole costitutive di questi canali, che probabilmente derivano da uno stesso canale ancestrale, si sono evolute poi in diverse classi, schematizzate nella tab. III. Poiché la biofisica classica dei canali è stata già trattata in precedenza (v. neurone e impulso nervoso; sinapsi; trasporto attraverso membrane biologiche), ci limiteremo a dare una breve descrizione della struttura molecolare e delle implicazioni funzionali che questa comporta, in quanto si tratta di acquisizioni assai recenti (v. Hille, 19922; v. Jan e Jan, 1992; v. Catterall, 1993; v. Volterra e Sher, 1996).
La prima intuizione della presenza dei canali ionici si è avuta nella metà degli anni cinquanta, ma solo in questi ultimi anni si è riusciti a ottenere molte informazioni di carattere molecolare e strutturale, e anche biofisico, che hanno permesso di comprenderne la funzione nonché la regolazione in situazioni fisiologiche e in determinate patologie. Si è così aperta la possibilità di un intervento farmacologico che in molti casi ha portato a grandi progressi terapeutici: basti pensare agli inibitori dei canali al Ca2+ (calcioantagonisti) e all'Na+ (antiaritmici) nelle patologie cardiovascolari. Uno schema della struttura di un canale ionico è presentato nella fig. 8. Il canale appare come una macromolecola proteica che attraversa il doppio strato lipidico della membrana cellulare formando un poro acquoso che si estende per tutto lo spessore della membrana. Il poro ha dimensioni assai più ampie di quelle di uno ione per la maggior parte della sua lunghezza, ma si restringe a dimensioni atomiche in un piccolo tratto, detto ‛filtro di selettività', dove viene stabilita la ‛selettività ionica', ovvero il tipo di ione con carica e raggio atomico adatti a passare attraverso quel punto. Nel canale vi è un sensore del voltaggio che è in grado di ‛sentire' la differenza di potenziale attraverso la membrana. Dal sensore dipende la possibilità che un canale voltaggio-dipendente si apra o si chiuda in funzione del potenziale di membrana. Nella porzione intracellulare vi sono inoltre siti importanti per l'inattivazione e per la modulazione dei canali stessi e per il legame con il citoscheletro che garantisce la loro localizzazione precisa a livello della membrana plasmatica.
c) Composizione dei canali ionici
I canali ionici voltaggio-dipendenti sono costituiti da proteine integrali di membrana altamente omologhe tra loro. La componente principale del canale è rappresentata da una struttura proteica il cui peso molecolare si aggira sui 200-250 kDa. Nel caso dei canali all'Na+ e al Ca2+ questa struttura è generata da una singola grossa catena polipeptidica (subunità α) organizzata in forma simile a un tetramero, mentre nel caso del canale al K+ quattro distinti polipeptidi più piccoli si associano per ottenere un vero tetramero con lo stesso ruolo funzionale (v. fig. 8). Oltre a queste subunità principali, che formano il canale vero e proprio, esiste una serie di subunità accessorie (chiamate α2, β, γ e δ a seconda dei canali), variabili sia come numero che come tipo nei diversi canali, che contribuiscono al corretto assemblaggio, trasporto e localizzazione nella membrana plasmatica e al corretto funzionamento del canale stesso. Esistono numerose isoforme delle subunità costitutive del canale come di quelle accessorie: esse vengono espresse in modo variabile nei diversi tessuti e sono all'origine delle singole proprietà di ciascun canale.
La subunità α è un'unica proteina di grandi dimensioni formata da quattro territori molto simili tra loro, organizzati in modo da formare il canale all'interno della subunità. Si tratta di strutture che si sono conservate nell'arco evolutivo e che contengono le caratteristiche fondamentali per la funzionalità del canale, quali il poro ionico e la sensibilità al voltaggio. I quattro territori transmembrana (I, II, III e IV), di 300-400 amminoacidi, hanno una omologia di sequenza molto alta non solo nel singolo canale, ma anche nei diversi tipi di canali. Ciascuno dei quattro territori contiene sei segmenti presumibilmente transmembrana (da S1 a S6), formati prevalentemente da amminoacidi idrofobici. Ognuno di questi segmenti, nonché ciascun tratto di congiunzione tra i segmenti, svolge un ruolo specifico nelle varie attività del canale, come attivazione, inattivazione, sensibilità al voltaggio e selettività ionica.
Recentemente è stato dimostrato che il tratto che congiunge i segmenti transmembrana S5 e S6 (chiamato in genere P) contiene una sequenza di una ventina di amminoacidi, che si insinua nello strato lipidico (pur senza attraversarlo) e che presumibilmente forma il contorno del poro acquoso attraverso il quale passano gli ioni (v. fig. 8, C e D).
Queste informazioni di correlazione tra struttura e attività sono state ottenute attraverso esperimenti di mutagenesi, che consentono di modificare singoli amminoacidi della proteina e di valutare poi le conseguenze delle diverse mutazioni sulle caratteristiche biofisiche del canale o sulla alterazione di tali caratteristiche determinata da diverse sostanze farmacologiche.
d) Regolazione dei canali ionici
L'inattivazione, cioè la capacità di una canale voltaggio-dipendente di chiudersi spontaneamente anche se permane la variazione di potenziale di membrana che aveva portato alla sua apertura, è una proprietà assai importante del canale grazie alla quale esso rimane aperto solo per un tempo prestabilito, a prescindere dallo stimolo. Questa proprietà può essere modificata da farmaci o da neurotrasmettitori, che cambiano così le caratteristiche della risposta neuronale: basti pensare ai farmaci antiaritmici, ai β-stimolanti, alle catecolammine, la cui attività si esplica proprio interferendo con le caratteristiche di apertura e chiusura dei canali ionici. La perfusione intracellulare dell'assone gigante di calamaro con enzimi proteolitici (come la proteasi alcalina di tipo B o la tripsina) o con anticorpi diretti contro piccole parti del tratto intracellulare di congiunzione tra i territori III e IV del canale all'Na+ è risultata in grado di bloccare completamente l'inattivazione del canale. Inoltre, diversi esperimenti di mutagenesi puntiforme in questi settori del canale hanno confermato l'ipotesi che questa zona idrofobica intracellulare rappresenti la zona che controlla l'inattivazione. Altri modelli di inattivazione, in parte simili, sono stati proposti per i canali al K+ e al Ca2+ (v. fig. 9; v. Catterall, 1993).
5. Recettori
Altre molecole chiave nella trasmissione neuronale sono i recettori, che trasducono il messaggio portato dal neurotrasmettitore all'interno della cellula postsinaptica. Per recettore si intende una molecola che lega in modo specifico, definito e con affinità precisa uno o più mediatori endogeni e che in seguito a questo legame subisce una trasformazione conformazionale capace di far scaturire un effetto biologico. Non è quindi sufficiente che una proteina leghi un ormone o un neurotrasmettitore per definirla un recettore: per esempio, l'albumina, che lega gli acidi grassi, o la ceruloplasmina, che lega il rame, non sono recettori.
I recettori per i neurotrasmettitori sono attivati da mediatori che, essendo sostanze idrofile, difficilmente attraversano la membrana cellulare; per tale ragione è necessario che questi recettori trasducano il segnale dall'esterno all'interno della cellula. I meccanismi impiegati a tal fine variano grandemente da recettore a recettore: è attraverso questa diversità che si attua la specificità della risposta cellulare ai vari neurotrasmettitori.
a) La scoperta dei recettori
La prima ipotesi di recettore (receptive substance) fu avanzata da J. N. Langley, attorno al 1880, in base a esperimenti eseguiti sul sistema autonomo del gatto, nel corso dei quali aveva scoperto l'antagonismo tra nicotina e curaro. Contemporaneamente, P. Ehrlich arrivava alle stesse conclusioni studiando un sistema diverso e più semplice, quale le interazioni tra tossine e antitossine e tra coloranti e Batteri. A lui si deve la famosa frase ‟Corpora non agunt nisi fixata". Negli anni venti vi furono poi le ricerche, effettuate da H. H. Dale in Inghilterra e da O. Loewi in Germania, sui mediatori chimici e sui loro effetti biologici, che vennero spiegati mediante l'interazione con diversi e specifici recettori. In quel periodo, inoltre, A. J. Clark introdusse la legge di massa che diede una prima spiegazione modellistica dell'interazione tra farmaco e recettore. Negli anni quaranta e cinquanta la chimica farmaceutica, attraverso fini modifiche della struttura chimica dei neurotrasmettitori o dei loro agonisti e antagonisti, giunse a definire alcune caratteristiche del sito di legame di molti recettori, fornendo la base per un disegno logico e non casuale dell'azione dei farmaci; è in questo periodo che nei laboratori di E.-F.-A. Fourneau prima e di D. Bovet poi, furono ottenuti gli anestetici locali, i curari, gli antistaminici. Ma il grande progresso nello studio dei recettori è stato possibile solo con l'aiuto della biologia molecolare. Nel 1982 S. Numa, in Giappone, clonava dall'organo elettrico della torpedine il recettore colinergico nicotinico, e da quel momento si apriva una nuova prospettiva nella comprensione di queste molecole. Successivamente, sono stati clonati molti altri recettori, dei quali si comincia a capire la struttura molecolare, i meccanismi della trasduzione del segnale e quelli attraverso cui le cellule regolano il numero di recettori e la loro funzione.
Un dato assai importante, che è emerso sin dall'inizio, è che ogni neurotrasmettitore è capace di attivare molti sottotipi di recettori, ciascuno con diverse caratteristiche biofisiche e farmacologiche, e la biologia molecolare ha permesso di scoprire che essi sono più numerosi di quanto si fosse supposto in base all'osservazione dei soli effetti dell'attivazione recettoriale e delle caratteristiche di legame dei farmaci. Questo permette al sistema nervoso di attivare un numero molto elevato di risposte assai specifiche con un numero limitato di neurotrasmettitori. Inoltre, come riportato schematicamente nella fig. 7, ogni neurotrasmettitore attiva delle precise vie nervose specifiche. L'esatta localizzazione del neurotrasmettitore e il fatto che la trasmissione dell'informazione avvenga solo quando nella sinapsi si trovano i recettori per quel trasmettitore garantiscono la specificità della trasmissione nervosa. La molteplicità delle risposte è garantita invece dal numero di neurotrasmettitori e dalla diversità di recettori per ogni tipo di neurotrasmettitore.
b) Superfamiglie recettoriali
I recettori per i neurotrasmettitori si possono raggruppare in quattro grandi famiglie, composte ciascuna da numerosi membri: recettori canale, recettori accoppiati alle proteine G, recettori che attivano una tirosinchinasi intrinseca, recettori che attivano una guanilatociclasi intrinseca (v. fig. 10). La maggioranza dei neurotrasmettitori agisce attivando recettori associati a canali o a proteine G, mentre i fattori di crescita attivano i recettori con proteinchinasi (v. Heinemann e Stühmer, 1993).
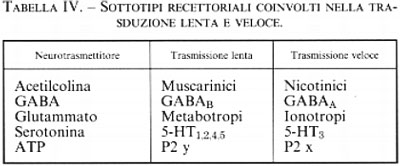
La stimolazione da parte di un neurotrasmettitore di un tipo di recettore e non di un altro non è evento indifferente. Come vedremo più avanti, l'apertura di un canale comporta una trasduzione del segnale molto rapida, mentre l'attivazione di una proteina G o di una proteinchinasi porta a una risposta più lunga e più lenta. Quasi tutti i neurotrasmettitori possono attivare sia canali sia proteine G (v. tab. IV) e indurre quindi nella cellula bersaglio risposte rapide o lente. È pertanto importante conoscere quali sottotipi recettoriali sono presenti in una cellula per capire quale sarà la sua risposta a un determinato neurotrasmettitore (v. anche farmacologia molecolare).
c) Recettori canale
I recettori di questo tipo sono costituiti da un canale ionico che viene aperto in seguito al legame con il neurotrasmettitore o con farmaci agonisti, e perciò la loro attivazione porta a rapidi cambiamenti delle concentrazioni ioniche intracellulari e quindi del potenziale elettrico transmembrana (v. De Lorey e Olsen, 1992; v. Sargent, 1993; v. Unwin, 1993; v. Bertrand e Changeux, 1995). Appartengono a questa famiglia i recettori nicotinici, il recettore A per il GABA, il recettore per la glicina, i recettori ionotropi per il glutammato, il recettore HT3 per la serotonina. Essi hanno tutti una struttura simile e una particolare storia evolutiva (v. Ortells e Lunt, 1995): sono composti da 4 o 5 subunità che delimitano un canale idrofilo attraverso il quale passano gli ioni (v. fig. 11); ogni subunità è formata da una catena polipeptidica che attraversa quattro volte la membrana plasmatica in corrispondenza di altrettante regioni ricche di amminoacidi idrofobici (chiamate regioni M); il canale è delimitato dalle regioni M2 di ciascuna subunità; la selettività della carica ionica che attraversa il canale è data dalla presenza di amminoacidi elettricamente carichi posti nella regione M2 di ciascuna subunità, in posizione tale da costituire degli anelli di carica positiva o negativa all'interno del canale.
Il sito di legame per il neurotrasmettitore o per i farmaci è posto all'esterno della membrana cellulare, in un territorio vicino al terminale amminico nella subunità che, per convenzione, viene chiamata α. Sulla superficie extracellulare del recettore sono spesso presenti siti di legame per altre sostanze regolatrici, detti ‛siti allosterici', in quanto la loro occupazione modifica le caratteristiche di attivazione recettoriale da parte dell'agonista principale. Nel citoplasma i recettori possono contenere dei siti di fosforilazione, importanti per la regolazione delle cinetiche di apertura e chiusura del canale ionico, e dei siti di legame con le proteine del citoscheletro, che ne garantiscono la stabilità e la giusta localizzazione nella membrana cellulare.
I farmaci attivi su questa classe di recettori possono avere come bersaglio il sito di legame per l'agonista naturale (per esempio, i curari sul recettore nicotinico), oppure possono legarsi al sito allosterico (per esempio, le benzodiazepine sul recettore A del GABA). Infine, alcuni farmaci possono interferire con le proprietà biofisiche e funzionali del canale legandosi a siti posti nel lume del canale stesso (per esempio, l'esametonio nel recettore nicotinico gangliare).
d) Recettori accoppiati a proteine G
Questa è la famiglia più numerosa di recettori e il bersaglio della maggior parte dei farmaci utilizzati a scopo terapeutico. La caratteristica peculiare di questi recettori è quella di trasdurre il segnale generato dal legame con il mediatore attivando una proteina G (v. Hille, 1992; v. Linder e Gilman, 1992; v. Coughlin, 1994; v. Vallar e Vicentini, 1996). Le proteine G rappresentano una famiglia di molecole proteiche eterotrimeriche e derivano il loro nome dalla capacità di legare il GTP (guanosintrifosfato) e di possedere un'attività GTPasica intrinseca. Delle tre subunità che costituiscono ciascuna proteina G, chiamate α, β e γ, solo la subunità α è capace di legare il GTP e idrolizzarlo in GDP (guanosindifosfato) (v. fig. 12).
In seguito all'attivazione di uno di questi recettori da parte del suo agonista, esso subisce una modificazione conformazionale grazie alla quale riesce ad attivare una specifica proteina G che, a sua volta, lega una molecola di GTP presente nel citoplasma; il legame con il GTP provoca la dissociazione delle tre subunità e l'attivazione della subunità α, che rimane attiva finché non riesce a idrolizzare il GTP. Durante la fase di attivazione, la subunità α modula l'attività di effettori quali le adenilatociclasi, le fosfolipasi C e A2 e alcuni canali ionici (v. Hille, 1992; v. Tang e Gilman, 1992; v. Berridge, 1993; v. Vallar e Vicentini, 1996). Ogni proteina G attiva in modo specifico solo determinati effettori (per esempio, fosfolipasi e ciclasi; v. tab. V). L'attivazione dell'adenilatociclasi e delle fosfolipasi produce la sintesi di secondi messaggeri, quali cAMP, IP3, DG, acido arachidonico e altri, che a loro volta attivano numerose proteinchinasi. La fosforilazione di substrati specifici provoca la reazione cellulare che sfocia nella risposta biologica indotta dall'attivazione recettoriale (v. fig. 13). Quindi il legame dell'agonista con un recettore accoppiato a proteine G può portare alla produzione di molte molecole di secondi messaggeri, ciascuna delle quali può attivare numerose altre molecole enzimatiche in una cascata che si amplifica sempre più. Lo sviluppo di questa cascata può essere controllato a ogni livello, sia positivamente sia negativamente, da fattori endogeni e da farmaci.

Una conseguenza di questa cascata amplificatrice di eventi biochimici è che la durata degli effetti indotti dall'attivazione di questa classe di recettori può essere anche dell'ordine di minuti e dipende non solo dalla durata dell'interazione farmaco-recettore, ma soprattutto dall'efficienza di meccanismi cellulari specifici preposti alla riduzione della concentrazione del secondo messaggero e all'abolizione delle modificazioni post-traduzionali da questo indotte.
I recettori accoppiati a proteine G sono inclusi in un'unica superfamiglia genica, in quanto hanno un'organizzazione molecolare comune: sono formati da una singola catena polipeptidica che attraversa sette volte la membrana plasmatica in corrispondenza di altrettante regioni idrofobiche e che si organizza spazialmente nella membrana in modo da costituire una particella globulare (v. fig. 12). Il sito di legame per il neurotrasmettitore si trova nelle porzioni transmembrana o extracellulari della sequenza amminoacidica. Il tratto di sequenza compreso tra le regioni transmembrana 5 e 6 è rivolto verso il citoplasma e presenta siti di fosforilazione, importanti per la funzionalità del recettore, e un sito per il riconoscimento delle proteine G; quest'ultimo permette a ciascun recettore di riconoscere e di legarsi solo ad alcuni tipi di proteina G ed è quindi responsabile della specificità d'interazione tra recettore e proteina G.
e) Recettori con attività tirosinchinasica
A questa famiglia appartengono i recettori non di neurotrasmettitori ma di fattori responsabili del trofismo, della differenziazione e della sopravvivenza dei neuroni (v. Fantl e altri, 1993; v. Comoglio e Boccaccio, 1996): il primo di questi fattori a essere individuato e studiato è stato quello per la crescita neuronale (Nerve Growth Factor, NGF), scoperto da Rita Levi-Montalcini, al quale se ne sono associati altri come il Brain Derived Growth Factor (BDGF) e i Neurotrophic Factors (NT3 e NT4). I recettori con attività tirosinchinasica sono costituiti da una catena polipeptidica che attraversa una sola volta la membrana cellulare e sono caratterizzati dal fatto di possedere un'attività tirosinchinasica intrinseca, così che sono capaci di fosforilare substrati proteici in corrispondenza di residui tirosinici (v. figg. 10 e 14). L'interazione ligando-recettore avviene a livello della porzione extracellulare della sequenza amminoacidica e porta alla dimerizzazione del recettore, che è la tappa responsabile dell'attivazione della tirosinchinasi intrinseca. L'autofosforilazione di residui tirosinici presenti nella porzione citoplasmatica del recettore porta alla sua associazione con una serie di proteine citoplasmatiche, alcune delle quali sono enzimi che iniziano una complessa serie di eventi a cascata, inducendo la cellula a proliferare o differenziarsi. Molti oncogeni codificano recettori per fattori di crescita che, a causa di mutazioni o delezioni di parte della proteina, hanno perso la loro attività regolatrice fisiologica (v. neoplasie: Oncologia sperimentale).
6. Modulazione delle risposte ai neurotrasmettitori
Perché la risposta cellulare agli stimoli portati dai diversi neurotrasmettitori sia la più adeguata possibile, è necessario che essa sia controllata in termini quantitativi e temporali. Le strategie attuate per esercitare questo controllo sono molteplici e comprendono soprattutto meccanismi di regolazione della capacità del recettore di rispondere al mediatore.
Controllo a livello della produzione e della degradazione del neurotrasmettitore. - Il primo livello di controllo viene attuato a livello del neurotrasmettitore. Circuiti neuronali e altri sistemi di feedback locali e a distanza controllano la sua sintesi e il suo rilascio, mentre sistemi enzimatici degradativi o sistemi di recupero provvedono all'eliminazione dell'ormone o del neurotrasmettitore in modo da ridurne la concentrazione nello spazio sinaptico e quindi la possibilità di incontro con il recettore.
Per dare un'idea della rilevanza di questo meccanismo di controllo si possono ricordare gli inibitori della colinesterasi, l'enzima deputato all'inattivazione dell'acetilcolina nelle sinapsi colinergiche. Queste sostanze provocano una lunga persistenza dell'acetilcolina nella sinapsi: l'acetilcolina non più idrolizzata continua ad attivare i recettori colinergici provocando dapprima una loro risposta molto intensa e, in seguito, una completa desensitizzazione e quindi un blocco della trasmissione. L'effetto finale è la morte dell'organismo per completa paralisi del sistema nervoso autonomo e della trasmissione neuromuscolare. Gli inibitori della colinesterasi sono infatti usati come insetticidi e come armi da guerra.
Controllo dell'interazione neurotrasmettitore-recettore. - Il secondo livello di controllo viene attuato a livello dell'interazione neurotrasmettitore-recettore, che è, tranne rare eccezioni, di tipo reversibile. Poiché il recettore può essere allo stato attivo (in grado cioè di indurre la cascata di eventi che costituiscono il suo sistema di trasmissione del segnale) solo quando è stato legato dal suo mediatore, la sua attività è controllata dalla costante di equilibrio (Kd) del complesso mediatore-recettore, variabile da 10-3 a 10-12 M e tipica per ogni complesso mediatore-recettore. In genere, quei recettori che mediano risposte rapide (per esempio depolarizzazione) sono caratterizzati da basse affinità per i loro ligandi naturali (Kd compresa tra 10-3 e 10-5 M): la loro attività non è quindi continua, ma caratterizzata da cicli di attività-inattività corrispondenti all'occupazione del recettore da parte del ligando e al rilascio di quest'ultimo. Recettori che mediano risposte più lente, come il differenziamento o l'ingresso della cellula nel ciclo mitotico, sono in genere caratterizzati da tempi di occupazione molto più lunghi e quindi da Kd più basse.
Spesso le cellule sono in grado di modificare la Kd di un recettore (in genere attraverso la sua fosforilazione): esempio tipico è quello del recettore β-adrenergico, la cui affinità per l'adrenalina viene aumentata in risposta a una stimolazione prolungata o di eccessiva intensità.
Controllo sulla trasduzione del segnale recettoriale e sulla sua persistenza. - Il terzo livello di controllo si attua a livello di trasduzione del segnale. Le strategie messe in atto per questo controllo sono molteplici e vanno dalla regolazione della capacità della singola molecola di recettore di trasdurre il segnale alla regolazione del sottotipo e del numero di recettori espressi dalla cellula. La riduzione della capacità di un recettore di trasdurre il segnale, anche se legato dall'agonista, è definita ‛desensitizzazione', mentre le variazioni del numero di molecole recettoriali espresse dalla cellula vengono definite con i termini inglesi upregulation (aumento) e downregulation (riduzione). Questi tre tipi di eventi regolatori sono molto comuni e hanno una rilevanza fisiologica e farmacologica-terapeutica notevole. Infatti, l'esposizione abbastanza lunga di un recettore al suo agonista, come abbiamo già detto, provoca desensitizzazione: è questo, per esempio, ciò che accade al recettore nicotinico nella placca neuromuscolare dopo avvelenamento con anticolinesterasici. L'esposizione cronica di una cellula all'agonista di un recettore è spesso associata a downregulation di quello stesso recettore e quindi a riduzione dell'entità dell'effetto del farmaco stesso: la downregulation e la desensitizzazione del recettore β-adrenergico sono alla base della riduzione dell'efficacia broncodilatatoria dei β2-agonisti che compare nel corso di una terapia antiasmatica condotta per lungo tempo con questi farmaci. La upregulation è inducibile dalla riduzione dell'attività del recettore causata da assenza dell'agonista (per esempio, a seguito di denervazione) o da trattamento cronico con un antagonista recettoriale (per esempio, β-bloccanti nell'ipertensione o antagonisti del recettore H2 dell'istamina nell'ulcera). In questi casi, al momento della sospensione della terapia, la cellula, che ha un numero maggiore di recettori alla sua superficie, può rispondere in modo esagerato a normali concentrazioni dell'agonista naturale (supersensitività).
Sono compresi in questo livello di regolazione anche quei fenomeni di adattamento cellulare che si attuano attraverso regolazione dell'attività del sistema di trasduzione accoppiato al recettore. Per esempio, la tolleranza (riduzione dell'intensità delle risposte a una certa concentrazione di farmaco) agli oppiacei sembra attuarsi attraverso un controllo dell'attività del complesso proteina G-ciclasi senza che intervengano variazioni del numero e della Kd dei recettori.
Controllo sullo spegnimento dell'attivazione recettoriale. - Il quarto livello di controllo si attua a livello dei meccanismi responsabili dello spegnimento del segnale generato dal recettore. Lo spegnimento rapido delle risposte indotte dall'attivazione recettoriale è assai importante, in quanto permette che la cellula sia di nuovo eccitata e quindi una più graduale modulazione delle risposte allo stimolo. I meccanismi di spegnimento sono molteplici, ma sono in genere controllati nella loro attività dall'entità del segnale generato dal recettore. Per esempio, l'attività della fosfodiesterasi e delle fosfatasi (gli enzimi responsabili dell'eliminazione del cAMP e della rimozione delle fosforilazioni da questo indotte) è controllata dalla concentrazione dello stesso secondo messaggero, mentre l'attività di canali, pompe e trasportatori preposti al ripristino del potenziale di riposo è modulata dalla concentrazione ionica e dallo stesso potenziale di membrana.
7. Modulazione patologica e farmacologica della comunicazione neuronale
a) Integrazione di più informazioni nella risposta neuronale
Come abbiamo visto all'inizio, un neurone può ricevere sulla sua superficie più di 100.000 sinapsi, ognuna portatrice di una particolare informazione da un particolare circuito nervoso. Se supponiamo che per ogni sinapsi ci possa essere più di un tipo di trasmettitore e/o più di un recettore, si può ben immaginare quante informazioni possano arrivare contemporaneamente a un neurone e influenzarne la risposta. Inoltre, l'attivazione di un recettore non rimane isolata, ma può riflettersi sulla funzionalità di altri recettori o canali. Vediamo, per esempio, che l'attivazione di un recettore β-adrenergico produce un aumento di AMP ciclico e un'attivazione di proteinchinasi A, che a loro volta possono fosforilare non soltanto il recettore β disattivandolo, ma anche altri recettori e altri canali ionici di membrana, modificandone le proprietà. Vi è quindi spesso un controllo reciproco tra i vari recettori che può portare a un aumento o a una diminuzione delle loro attività.
La cellula neuronale quindi è una sorta di computer che riceve informazioni molteplici, le elabora in modi non ancora del tutto chiariti e, infine, produce la sua risposta. La compromissione della funzione di una sola molecola neuronale, sia un canale o un recettore, per opera di farmaci o di agenti patogeni, può quindi portare a grandi modificazioni della risposta di un neurone e determinare un grave squilibrio nell'omeostasi dei circuiti neuronali.
Come è facile ipotizzare, molte patologie umane nascono da modificazioni del numero o della struttura delle molecole coinvolte nella trasmissione dell'impulso nervoso. L'argomento potrebbe essere estremamente ampio, ma spesso le alterazioni sono più supposte - sulla base, ad esempio, dell'azione positiva di farmaci - che non sperimentalmente provate. Ci limiteremo quindi a quelle patologie nelle quali le modificazioni sono conclamate e sperimentalmente provate, in quanto esse possono dare un'idea precisa di come l'omeostasi di un sistema può venire perturbata con la modifica di una di queste molecole; forniremo inoltre alcuni esempi che coinvolgono canali voltaggio-dipendenti, recettori e sistemi di trasduzione del messaggio.
b) Canali ionici
Le patologie più conosciute che hanno come bersaglio i canali ionici sono a carico non solo delle cellule neuronali, ma anche delle cellule muscolari o ghiandolari da queste innervate. Ne riportiamo alcune come esempio.
Nella fibrosi cistica alcune mutazioni geniche provocano modificazioni a carico del canale del cloro, la cui apertura è regolata da ATP, rendendolo non più apribile: ciò determina un grave squilibrio ionico, il quale causa la secrezione di muco denso e colloso che ostruisce i dotti pancreatici e biliari e le vie respiratorie; sono presenti inoltre disturbi a livello della trasmissione nervosa che tuttavia, data la gravità dei sintomi secretori, sono ritenuti di minore gravità.
La paralisi periodica iperpotassiemica è una malattia autosomica dominante caratterizzata da episodi di debolezza muscolare, associati ad aumento del potassio serico. Studi sui muscoli dei pazienti hanno dimostrato che le cellule sono cronicamente depolarizzate in seguito al cattivo funzionamento del canale all'Na+ e mostrano una conduttanza all'Na+ abnormemente ‛non inattivante'. Il locus genico responsabile della malattia è stato localizzato nella zona del cromosoma 17, dove si trova il gene per la subunità α1 del canale all'Na+ muscolare: si ipotizza che il danno al canale sia dovuto ad alterazioni del segmento S5 del territorio II e del segmento S6 del territorio IV conseguenti a due mutazioni puntiformi.
La paramiotonia congenita è una patologia paralitica simile alla precedente, la cui sintomatologia viene scatenata dal freddo o dall'esercizio al freddo. Anche in questo caso il gene responsabile è colocalizzato nel genoma con il gene che codifica la subunità α1 del canale all'Na+; sono state identificate diverse mutazioni che provocano modificazioni specialmente a carico del tratto intracellulare di congiunzione fra il segmento S6 del territorio III e il segmento S1 del territorio IV.
Sembra quindi che in entrambe queste patologie le mutazioni inducano alterazioni a carico delle zone del canale all'Na+ muscolare specificamente deputate al controllo della sua inattivazione.
Recentemente si è osservato che una mutazione che modifica il canale all'Na+ cardiaco è responsabile di una grave patologia della trasmissione cardiaca, la sindrome del QT lungo, che porta a morte improvvisa.
La disgenesia muscolare è una patologia murina caratterizzata da un difetto genetico a carico della subunità α1 del canale al Ca2+ muscolare, la cui espressione viene inibita. Il muscolo malato, quindi, non possiede né ‛sensore' del voltaggio per l'accoppiamento eccitazione-contrazione, né correnti al Ca2+.
Un'altra patologia del canale al Ca2+, non dovuta però a errori genetici, è la sindrome miastenica di Lambert-Eaton (v. Sher e altri, 1991). La patogenesi di questa malattia è di tipo autoimmunitario, in quanto i pazienti producono autoanticorpi in grado di riconoscere e indurre la degradazione del canale al Ca2+ voltaggio-dipendente. In questo caso il canale selettivamente colpito è quello neuronale, senza alcuna alterazione dei vari canali al Ca2+ di tipo muscolare. L'aumentata degradazione del canale al Ca2+ fa sì che, all'arrivo del potenziale d'azione, entri meno Ca2+ nei terminali nervosi e di conseguenza venga rilasciato meno neurotrasmettitore: è questa la base della sintomatologia paralitica presente sia a livello neuromuscolare che neurovegetativo.
c) Recettori nicotinici
La miastenia grave è una patologia che colpisce la giunzione neuromuscolare provocando difficoltà della trasmissione tra nervo e muscolo; ciò porta ad affaticabilità dei muscoli e a una loro progressiva paralisi. In questi ultimi anni si è scoperto che la paralisi è dovuta alla diminuzione del numero di recettori colinergici nicotinici presenti nella giunzione neuromuscolare; l'acetilcolina, quindi, secreta in quantità e con modalità normali dalla sinapsi, trova a livello postsinaptico un numero minore di recettori con i quali interagire e non può garantire un'efficiente trasmissione dell'impulso. La diminuzione dei recettori è provocata da anticorpi antirecettore nicotinico che ne operano la distruzione. La terapia che oggi si attua consiste, fondamentalmente, nel bloccare la produzione di autoanticorpi attraverso farmaci immunosoppressori (v. Drachman, 1994) e, in parte, nell'aumentare la vita dell'acetilcolina nello spazio sinaptico attraverso l'inibizione dell'acetilcolinesterasi. Sono state inoltre descritte molte altre forme di miastenia, meno diffuse, nelle quali la affaticabilità muscolare è prodotta da alterazioni dei geni che codificano per il recettore nicotinico muscolare (v. Engel e altri, 1997).
d) Trasduzione del segnale
In questi anni sono state descritte molte patologie legate a mutazioni che provocano modificazioni dei recettori o delle proteine G a questi associate: tra le prime ricordiamo la retinite pigmentosa causata da rodopsina alterata (v. visione, voll. VII e XI), il diabete insipido nefrogenico causato da un recettore mutato per la vasopressina, alcune forme di nanismo dovute all'alterazione del recettore per l'ormone della crescita; tra le seconde, vi sono gli adenomi dell'ipofisi dovuti ad attivazione congenita della proteina Gsα e i tumori della tiroide dovuti ad attivazione della proteina G1α. Recentemente sono state descritte anche mutazioni che alterano i recettori α1, α2, β2 per le catecolammine, che potrebbero fornire una spiegazione per alcune patologie del SN periferico. Con la diffusione sempre più ampia delle tecniche di biologia molecolare è prevedibile che patologie del SNC con alta familiarità possano essere spiegate attraverso mutazioni che inducono modificazioni strutturali di proteine coinvolte nella trasmissione nervosa.
Oltre a queste patologie conclamate, vi sono poi numerose situazioni patologiche nelle quali si sospetta che vi sia un'alterazione in alcuni punti della trasmissione nervosa, senza averne però delle prove precise. Le maggiori indicazioni vengono dall'uso dei farmaci: per esempio, è ben nota l'azione positiva delle benzodiazepine nell'ansia; sapendo che esse agiscono sul recettore GABA-ergico, si suppone che nell'ansia vi sia un'alterazione di questo sistema di trasduzione dell'informazione nervosa.
e) I farmaci come strumenti specifici per analizzare e modificare la comunicazione nervosa
Questo argomento necessiterebbe di una vastissima trattazione, ma riteniamo importante almeno menzionarlo in questa sede, in quanto i farmaci non solo sono stati strumenti essenziali per capire il funzionamento del SN, ma hanno anche permesso di intervenire terapeuticamente su molte delle sue patologie, recuperando, almeno in parte, alla vita e alla dignità umana persone che sarebbero altrimenti relegate ai confini della società o schiacciate da grandi dolori e angosce. Si pensi, per esempio, all'evoluzione della cura della schizofrenia, della depressione, della paranoia: con l'introduzione degli psicofarmaci è stato possibile rivoluzionare anche l'ospedalizzazione dei malati di queste patologie, abolendo quasi completamente gli ospedali psichiatrici oppure trasformandoli radicalmente.
La complessa struttura responsabile della trasmissione nervosa fa sì che vi siano molti possibili bersagli per un intervento farmacologico. Abbiamo riportato nella tab. VI alcuni esempi di farmaci che vengono comunemente usati in terapia per mostrare come essi agiscano su un bersaglio ben preciso, e come agendo su bersagli comuni si possano avere effetti diversi: per esempio, i farmaci che bloccano i canali all'Na+ voltaggio-dipendenti possono svolgere sia un'azione insetticida (bloccando i canali all'Na+ negli Insetti), sia un'azione anestetica locale, se impiegati per bloccare i canali presenti nei nervi, sia un'azione antiaritmica, se bloccano i canali cardiaci, o antiepilettica, se bloccano i canali nel SNC. Spesso questi effetti possono essere provocati da una stessa molecola, ma più frequentemente si è saputo sfruttare alcune piccole differenze nelle molecole o una loro diversa localizzazione tissutale per rendere il farmaco il più specifico possibile. Nonostante i grandi progressi della biologia molecolare e della farmacologia, siamo però ancora molto lontani dal comprendere come si instauri gran parte delle malattie del SNC e quale ne sia la causa, cosicché appare ancora molto lontana una loro terapia causale e specifica. I progressi fin qui fatti ci permettono però di alleviare molti sintomi delle malattie nervose e quindi di tenerle sotto controllo, consentendo di intervenire assai positivamente anche se l'approccio non può dirsi ottimale.
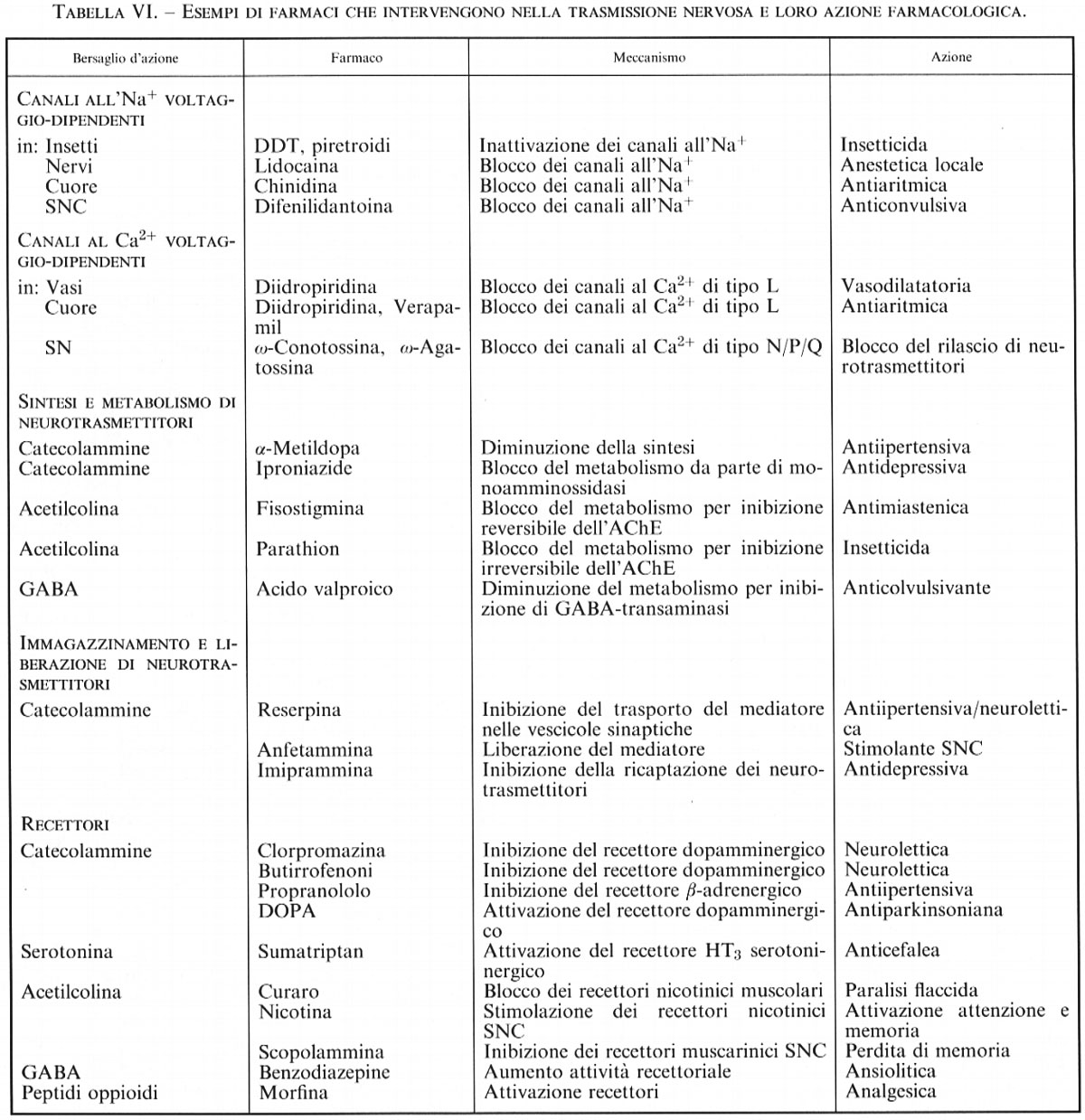
8. Conclusioni
Il SN è organizzato in circuiti ben precisi che esigono un sistema di comunicazione molto puntuale, discreto nello spazio e nel tempo, e ben modulabile. I circuiti neuronali non sono sempre fissi, ma probabilmente possono variare in ampiezza e in moduli, a seconda degli stimoli esterni. Un esempio tipico di questa organizzazione è fornito dalla memoria, che probabilmente si basa sull'attivazione e inattivazione di circuiti neuronali (v. memoria: studi sperimentali). Vi sono quindi due aspetti della comunicazione neuronale: i meccanismi con i quali le cellule nervose comunicano tra loro e i meccanismi e i principî con i quali si stabiliscono i circuiti nervosi durante lo sviluppo e durante l'apprendimento (v. Hinton, 1992; v. Kandel e Hawkins, 1992). In questo articolo ci siamo occupati solo del primo aspetto, in quanto il secondo è già trattato in altre parti dell'opera (v. neurogenesi; neurobiologia: Organizzazione neuronale cerebrale e cerebellare).
Abbiamo illustrato soprattutto il meccanismo di funzionamento della sinapsi, mettendo in luce, relativamente alla parte presinaptica, la struttura e la funzione dei canali ionici voltaggio-dipendenti, responsabili della propagazione del potenziale d'azione, e le modalità di sintesi, immagazzinamento e secrezione dalle sinapsi dei messaggeri sinaptici; quindi, relativamente alla parte postsinaptica, ci siamo soffermati sui recettori, che sono responsabili dell'ultima parte della trasmissione nervosa, cioè la trasduzione nella cellula postsinaptica del messaggio portato dai neurotrasmettitori; abbiamo illustrato la loro struttura e i meccanismi mediante i quali funzionano, soffermandoci sulle ultime conquiste rese possibili dalla biologia molecolare e cellulare e sulle modalità con le quali la cellula può regolare la funzione dei recettori e quindi, in ultima analisi, la sua risposta agli stimoli. Sono state accennate alcune patologie del SN che riconoscono la loro causa in una precisa alterazione delle molecole responsabili della comunicazione neuronale e, infine, abbiamo visto come sia possibile modulare la trasmissione nervosa attraverso i farmaci e indicato qualche meccanismo che può spiegare il loro effetto terapeutico.
L'aspetto molecolare è quello su cui ci siamo maggiormente soffermati, in quanto è da questo approccio che sono scaturite le novità più importanti in questi ultimi anni, ed è da una sempre più feconda interazione tra la biologia molecolare e cellulare da un lato e l'osservazione clinica e comportamentale dall'altro che scaturiranno le basi per una maggior comprensione del funzionamento di quella macchina stupenda che è il cervello.
BIBLIOGRAFIA
Attwell, D., Barbour, B., Szatkowski, M., Nonvesicular release of neurotransmitter, in ‟Neuron", 1993, XI, pp. 401-407.
Attwell, D., Mobbs, P., Neurotransmitter transporters, in ‟Current opinion in neurobiology", 1994, IV, pp. 353-359.
Berridge, M. J., Inositol trisphosphate and calcium signalling, in ‟Nature", 1993, CCCLXI, pp. 315-325.
Bertrand, D., Changeux, J. P., Nicotinic receptor: an allosteric protein specialized for intracellular communication, in ‟Seminars in neurosciences", 1995, VII, pp. 75-90.
Catterall, W. A., Structure and function of voltage-gated ion channels, in ‟Trends in neurosciences", 1993, XVI, pp. 500-506.
Comoglio, P. M., Boccaccio, C., I recettori per i fattori di crescita, in Farmacologia generale e molecolare (a cura di F. Clementi e G. Fumagalli), Torino 1996, pp. 83-92.
Coughlin, S. R., Expanding horizons for receptors coupled to G proteins: diversity and disease, in ‟Current opinion in cell biology", 1994, VI, pp. 191-197.
De Lorey, T. M., Olsen, R. W., Gamma-amino-butyric acid A receptor structure and function, in ‟Journal of biological chemistry", 1992, CCLXVII, pp. 16747-16750.
Drachman, D. B., Myasthenia gravis, in ‟New England journal of medicine", 1994, CCCXXX, pp. 1797-1810.
Engel, A. G., Ohno, K., Milone, M., Sine, S. M., Congenital myasthenia syndromes caused by mutations in acetylcholine receptor genes, in ‟Neurology", 1997, XLVIII, suppl. 5, pp. 528-535.
Fantl, W. J., Johnson, D. E., Williams, L. T., Signalling by receptor tyrosine kinases, in ‟Annual review of biochemistry", 1993, LXII, pp. 453-481.
Harmor, A. J., Neuropeptides, in Transmitter molecules in the brain (a cura di E. Fluckiger, E. Müller e M. O. Thorner), Berlin-New York 1987, pp. 17-26.
Heinemann, S., Stühmer, W., Signalling mechanisms, in ‟Current opinion in neurobiology", 1993, III, pp. 269-400.
Hille, B., G protein-coupled mechanisms and nervous signaling, in ‟Neuron", 1992, IX, pp. 187-195.
Hille, B., Ionic channels of excitable membranes, Sunderland, Mass., 19922.
Hinton, G. E., How neural networks learn from experience, in ‟Scientific American", 1992, CCLXVII, 3, pp. 104-109 (tr. it.: L'apprendimento delle reti artificiali di neuroni, in ‟Le scienze", 1992, IL, 291, pp. 117-123).
Jan, L. Y., Jan, Y. N., Tracing the roots of ion channels, in ‟Cell", 1992, LXIX, pp. 715-718.
Kandel, E., Hawkins, R. D., The biological basis of learning and individuality, in ‟Scientific American", 1992, CCLXVII, 3, pp. 52-61 (tr. it.: Apprendimento e individualità: le basi biologiche, in ‟Le scienze", 1992, IL, 291, pp. 49-59).
Linder, M. E., Gilman, A. G., G proteins, in ‟Scientific American", 1992, CCLXVII, 1, pp. 36-43 (tr. it.: Le proteine G, in ‟Le scienze", 1992, IL, 289, pp. 40-49).
McPherson, P. S., De Camilli, P., Recycling and biogenesis of synaptic vesicles, in ‟Seminars in neurosciences", 1994, VI, pp. 137-147.
McQueen, J. K., Classical transmitters and neuromodulators, in Transmitter molecules in the brain (a cura di E. Fluckiger, E. Müller e M. O. Thorner), Berlin-New York 1987, pp. 7-16.
Ortells, M. O., Lunt, G., Evolutionary history of the ligand-gated ion-channel superfamily of receptors, in ‟Trends in neurosciences", 1995, XVIII, pp. 121-127.
Sargent, P. B., The diversity of neuronal nicotinic acetylcholine receptors, in ‟Annual review of neuroscience", 1993, XVI, pp. 403-443.
Shatz, C. J., The developing brain, in ‟Scientific American", 1992, CCLXVII, 3, pp. 34-41 (tr. it.: Lo sviluppo del cervello, in ‟Le scienze", 1992, IL, 291, pp. 27-34).
Sher, E., Biancardi, E., Passafaro, M., Clementi, F., Physiopathology of neuronal voltage-operated calcium channels, in ‟FASEB journal", 1991, V, pp. 2677-2683.
Söllner, T., Rothman, J. E., Neurotransmission: harnessing fusion machinery at the synapse, in ‟Trends in neurosciences", 1994, XVII, pp. 344-348.
Tang, W. J., Gilman, A. G., Adenylylcyclases, in ‟Cell", 1992, LXX, pp. 869-872.
Unwin, N., Neurotransmitter action: opening of ligand-gated ion channels, in ‟Cell", 1993, LXXII, pp. 31-41.
Vallar, L., Vicentini, L., Recettori accoppiati alle proteine G, in Farmacologia generale e molecolare (a cura di F. Clementi e G. Fumagalli), Torino 1996, pp. 60-71.
Varadi, G., Mori, Y., Mikala, G., Schwartz, A., Molecular determinants of Ca2+ channel function and drug action, in ‟Trends in pharmacological sciences", 1995, XVI, pp. 43-49.
Volterra, A., Sher, E., Canali ionici voltaggio-dipendenti, in Farmacologia generale e molecolare (a cura di F. Clementi e G. Fumagalli), Torino 1996, pp. 115-139.
Neuropatologia di Vincenzo Bonavita, Simone Sampaolo
Sommario: 1. Strutturazione della disciplina e classificazione delle malattie di interesse neuropatologico. 2. Cenni storici. 3. Metodiche d'indagine. 4. Cenni di anatomia funzionale. 5. Anatomia microscopica del SNC. 6. Neuropatologia generale: processi elementari. a) Processi neuronali regressivi. b) Processi assonali regressivi e progressivi. c) Processi mielinici regressivi e progressivi. d) Processi astrocitari regressivi e progressivi. e) Processi oligodendrogliali, microgliali e monocitari. 7. Neuropatologia generale: processi patologici tissutali. 8. Principali sindromi neurologiche lesionali. a) Lobi frontale, parietale, temporale e occipitale. b) Gangli basali. c) Talamo. d) Asse ipotalamo-ipofisario. e) Troncoencefalo e cervelletto. f) Midollo spinale. g) Nervo periferico. h) Sindrome di ipertensione endocranica. 9. Neuropatologia speciale. a) Malattie degenerative. b) Malattie prioniche. c) Malattie da Retrovirus (HIV-1). □ Bibliografia.
1. Strutturazione della disciplina e classificazione delle malattie di interesse neuropatologico
La neuropatologia studia le alterazioni strutturali cui vanno incontro il sistema nervoso centrale (SNC) e periferico in seguito all'azione di noxae patogene esogene e/o endogene, nelle varie fasi del ciclo vitale (sviluppo embrionale, crescita, maturazione e invecchiamento). Sue finalità sono la descrizione della localizzazione, estensione, tipologia, qualità ed evoluzione di dette alterazioni a livello macroscopico (organo), microscopico (tessuti e cellule), ultrastrutturale (organelli subcellulari) e biomolecolare, e l'individuazione delle condizioni e delle connessioni causali degli eventi responsabili di tali modificazioni (eziopatogenesi). Nel corpo della materia rientra, comunque, anche la morfologia patologica del muscolo scheletrico: la frequente associazione causale tra disordini del SN e alterazioni muscolari ne ha storicamente motivato, infatti, lo studio unitario.
Si distinguono la neuropatologia generale e la neuropatologia speciale: la prima tratta dei processi elementari a livello cellulare e tissutale, la seconda dei complessi quadri morfologici lesionali conseguenti a specifiche malattie. Settori superspecialistici vengono individuati nella paido-neuropatologia, in ragione della peculiarità dei processi tissutali che si verificano nel tessuto nervoso immaturo sotto l'azione delle varie noxae patogene, e nella neuropatologia forense, in ragione dell'approccio metodologico differente rispetto a quello della routine neuropatologica, perché finalizzato alle necessità dell'indagine giudiziaria e della particolare prospettiva che orienta l'interpretazione critica dei reperti.
Una classificazione dettagliata delle malattie che hanno una base neuropatologica definita esula dagli obiettivi della presente trattazione e inoltre uno schema internazionalmente accettato non è attualmente disponibile. È in atto, infatti, una revisione in chiave eziopatogenetica della nosografia neuropatologica classica, basata in larga parte su criteri anatomopatologici, e cioè su distribuzione, tipologia e momento di comparsa delle lesioni. Ciò è dovuto alla sempre più rapida acquisizione di nuove conoscenze sulla natura e le cause delle malattie del sistema nervoso, frutto dei progressi delle metodiche biochimiche, immunologiche e, soprattutto, sul versante della biologia molecolare, della messa a punto delle cosiddette tecniche del DNA ricombinante (v. biotecnologie). L'inquadramento nosografico secondo criteri anatomopatologici rimane, ovviamente, l'unico possibile per i disordini la cui patogenesi è ancora sconosciuta. Le classificazioni attuali seguono pertanto un criterio misto, cioè anatomopatologico ed eziopatogenetico: a tale modello si ispira anche quella adottata nel presente capitolo (v. tab. I).
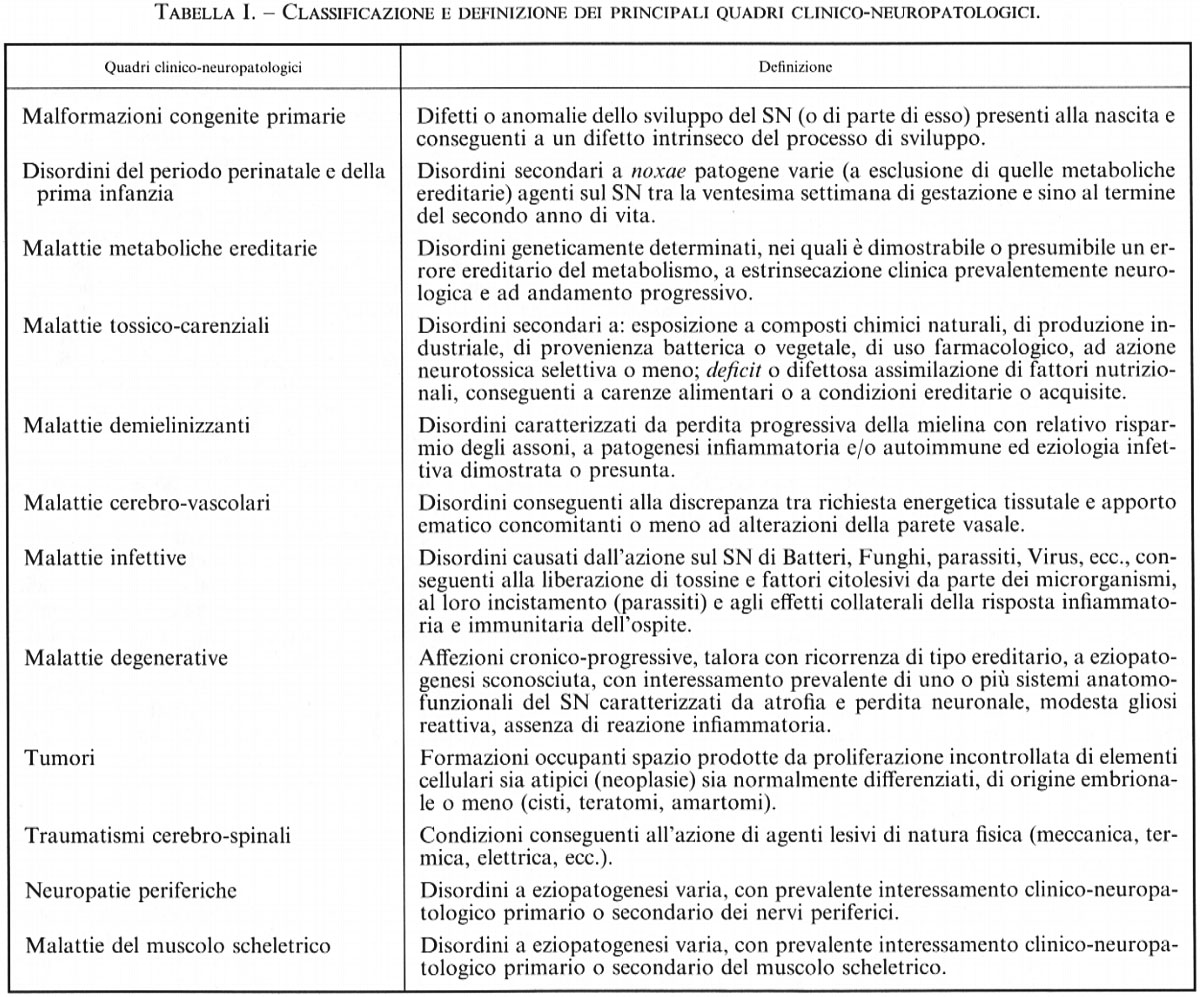
2. Cenni storici
Le prime osservazioni sistematiche sulla patologia del sistema nervoso risalgono al XVII secolo, grazie all'opera di Johann Jakob Wepfer (De Apoplexia, Schaffhausen 1658), Thomas Willis (Cerebri anatome, Londra 1664; Pathologiae cerebri et nervosi generis specimen, Oxford 1667; De anima brutorum, Oxford 1672) e soprattutto, quasi un secolo più tardi, a quella di Giambattista Morgagni (De sedibus et causis morborum per anatomen indagatis, Venezia 1761). In sostanza, però, la natura e l'origine delle malattie del sistema nervoso furono studiate solo in misura assai limitata sino ai primi decenni del 1800, periodo in cui gli anatomopatologi e i frenologi iniziarono a occuparsi dello studio macroscopico dei reperti neuropatologici. A opera di studiosi quali Robert Hooper, Jean Cruveilhier, sir Robert Carswell e Richard Bright fu pubblicata una serie di atlanti illustrati di notevole interesse scientifico e valore artistico, il più importante dei quali, per la precisione delle immagini e la varietà dei quadri descritti, è comunque quello di Bright, intitolato Diseases of the brain and nervous system (1831), che costituiva il secondo volume del trattato Reports of medical cases. In questo lavoro Bright espone la teoria secondo la quale i disordini del sistema nervoso sono riconducibili a cinque fenomeni principali: infiammazione febbrile, pressione da lesione occupante spazio, concussione meccanica, irritazione del cervello o delle sue meningi, inanizione o deficit circolatorio. Nello stesso periodo, John Cheyne, pupillo di Charles Bell e noto anche per aver descritto una modificazione patologica della respirazione (respiro periodico di Cheyne-Stokes), pubblicò una monografia sull'idrocefalo acuto (1808) - la prima in assoluto sull'argomento - e una sull'apoplessia (1812), nella quale vengono descritti e illustrati, per la prima volta, i reperti di un caso di emorragia subaracnoidea, oltre ad alcuni esempi di infarto ischemico cerebrale.
Una pietra miliare nella storia della neuropatologia macroscopica è rappresentata dalla monografia di John Abercrombie, Pathological and practical researches on diseases of the brain and spinal cord (1828): questo lavoro, in cui vengono descritti oltre 150 casi clinico-neuropatologici suddivisi in quadri di malattie infiammatorie, apoplettiche, organiche e del midollo spinale, ebbe una vasta diffusione e fu tradotto in francese e tedesco. Determinante per l'ulteriore sviluppo della neuropatologia fu l'enunciazione nel 1838 - da parte di Matthias Jakob Schleiden, ordinario di botanica a Jena, e Theodor Schwann, ordinario di anatomia e fisiologia a Liegi - della teoria cellulare di strutturazione dei viventi, che diede impulso agli studi di anatomia microscopica e alla messa a punto di apparecchiature, quali il microtomo e il microscopio composto, oltre che di metodi specifici di fissazione e colorazione dei tessuti biologici.
Da questo periodo in poi la storia della neuropatologia descrittiva corre parallela a quella della neuroistologia, dal momento che la corretta definizione della lesione istopatologica richiede l'esatta conoscenza del normale substrato anatomico. Già nel 1836 C. G. Ehrenberg aveva identificato le cellule dei gangli spinali e simpatici, e nel 1838 Johannes Evangelista Purkinje aveva descritto l'esistenza di ‛corpi gangliari a forma di fiasco' nel cervelletto. Nel 1856, Otto Friedrich Karl Deiters, grazie all'impiego di nuovi metodi di colorazione, identificò la struttura generale dei neuroni, con i prolungamenti dendritici e l'assone, ma solo nel 1873 Camillo Golgi, utilizzando il metodo di impregnazione al cromato d'argento da lui inventato, fornì una rappresentazione accurata della morfologia dei neuroni e dell'istoarchitettura del sistema nervoso. Golgi fu protagonista, con lo spagnolo Santiago Ramón y Cajal, della più interessante controversia della storia delle neuroscienze: il primo, infatti, sosteneva l'ipotesi che i neuroni formassero un reticolo cellulare continuo, mentre Cajal, utilizzando una modificazione del metodo di Golgi, dimostrò irrefutabilmente l'indipendenza individuale dei neuroni e raccolse elementi indicativi che l'impulso nervoso viene trasmesso da cellula a cellula attraverso strutture di contatto. Studi sulla retina e sul bulbo olfattorio lo portarono a formulare la teoria della polarizzazione dinamica dei neuroni: i processi neuronali di maggiore spessore, i dendriti, portano l'impulso dalla periferia al corpo cellulare; l'assone, sottile, porta invece l'impulso lontano dal neurone. Nella sua inesauribile attività Cajal non tralasciò di esplorare i fenomeni di degenerazione e rigenerazione del sistema nervoso e il suo Manual of pathological anatomy, pubblicato nel 1909, fu riedito per ben 7 volte. Carl Weigert, professore di anatomia patologica a Francoforte, mise a punto un metodo specifico per la colorazione della mielina e migliorò i metodi di fissazione e di inclusione del tessuto nervoso; in particolare perfezionò il metodo di inclusione in celloidina, aprendo così la strada allo studio microscopico su sezioni del cervello in toto. Il suo metodo - insieme a quello di Vittorio Marchi per la mielina, a quello di Augustus Volney Waller per le fibre nervose in degenerazione, alla colorazione di Franz Nissl per i neuroni e ai metodi di impregnazione di Cajal - fornì la base tecnica per lo sviluppo della moderna neuropatologia microscopica.
La teoria cellulare fu applicata alla patologia da Rudolf Ludwig Carl Virchow - ordinario di anatomia patologica a Würzburg e poi a Berlino - che nel 1855 pubblicò il manifesto di quella che chiamò ‛patologia del futuro', intitolato con un significativo aforisma Omnis cellula a cellula, in cui sostenne che la cellula è la sede dei processi normali e patologici. Virchow, il quale personalmente era un appassionato dello studio microscopico dei reperti patologici, tuttavia attribuì pari valore alle indagini biochimiche sui tessuti e considerò fondamentali la correlazione delle osservazioni anatomopatologiche con la clinica e la ricerca della patogenesi delle lesioni. Tra i suoi numerosi contributi alla neuropatologia, i più importanti sono la scoperta della neuroglia, la descrizione della capacità fagica di cellule di natura gliale in prossimità di una lesione malacica, gli studi su trombosi, embolia ed emorragia cerebrale, e, ancora, sulle meningiti, sui tumori, sulle anomalie congenite.
Sebbene intorno alla metà del XIX secolo l'anatomia patologica del sistema nervoso avesse già solide basi, la nascita della moderna neuropatologia non si considera anteriore al 1872, anno in cui Jean Martin Charcot successe a Félix-Alfred Vulpian sulla cattedra di anatomia patologica della facoltà di medicina di Parigi. Charcot, infatti, fu il più grande neurologo clinico francese del secolo e un eccellente anatomopatologo: attivo in questa duplice veste presso l'ospedale Salpêtrière dedicò, insieme ai suoi allievi, un incredibile impegno allo studio delle relazioni tra le lesioni organiche del sistema nervoso e le manifestazioni cliniche delle malattie. Cardini di questi studi erano, per un verso, il rilievo periodico e la registrazione metodica dei segni e sintomi neurologici durante il decorso della malattia, per l'altro, lo studio sistematico al microscopio di sezioni istologiche seriate del sistema nervoso. Charcot definì le basi istopatologiche di numerose malattie neurologiche, tra le quali la sclerosi laterale amiotrofica, la paralisi bulbare, la sclerosi multipla, la tabe dorsale e le atrofie muscolari; insieme a Henri Bouchard descrisse gli aneurismi miliari e ne rimarcò l'importanza nella genesi delle emorragie cerebrali; con Alex Joffroy avanzò l'idea che nella poliomielite vi fosse un fattore ‛irritante' che colpiva i neuroni delle corna anteriori ledendone la funzione, e provò inoltre che in questa malattia la perdita dei motoneuroni delle corna spinali anteriori è responsabile della progressiva atrofia muscolare.
Sulle orme di Charcot, del quale ricalcò l'approccio clinico-anatomopatologico alla neuropatologia, si mosse Aloisius Alzheimer, professore di psichiatria a Monaco di Baviera. In questa città egli fondò una scuola di neuropatologia che rappresenta la prima istituzione nella quale sia stata riconosciuta l'autonomia disciplinare della materia. I contributi di Alzheimer alla neuropatologia comprendono studi sulle alterazioni istologiche corticali in pazienti affetti dalla forma di demenza presenile denominata, da Emil Kraepelin, demenza di Alzheimer, quelli sulle lesioni organiche cerebrali che accompagnano l'arteriosclerosi e la senilità, studi sulla paralisi agitante e sulla corea di Huntington. Stretto collaboratore di Alzheimer, e successore di Kraepelin sulla cattedra di psichiatria di Heidelberg, fu Franz Nissl, il quale mise a punto nuove tecniche di fissazione dei tessuti nervosi e introdusse un metodo di colorazione dei neuroni con blu di metilene, che gli consentì di identificare nel loro interno le cosiddette ‛zolle di Nissl' e di elaborare una classificazione dei neuroni normali e patologici. Il successore di Alzheimer a Monaco, Walther Spielmeyer, professore e direttore del laboratorio anatomico dell'istituto di psichiatria, migliorò numerose tecniche istopatologiche e diede contributi scientifici rilevanti su argomenti che variano dalla idiozia amaurotica familiare alla malattia di Wilson, dalla sclerosi tuberosa alla tabe dorsale. Fondamentali sono i suoi studi sulle lesioni dei nervi periferici e sulle alterazioni delle funzioni cerebrali conseguenti ai disordini temporanei della circolazione e all'ipossia cerebrale. Il suo manuale di laboratorio e il testo di istopatologia del sistema nervoso sono classici della letteratura neuropatologica. Importanti contributi allo sviluppo della disciplina furono dati, ancora, da Alfons Maria Jakob e da Max Bielschowsky: il primo studiò, in particolare, le alterazioni traumatiche cerebrali, le condizioni degenerative secondarie, le malattie extrapiramidali e, insieme a Hans Gerhard Creutzfeldt, descrisse la malattia detta di Creutzfeldt-Jakob; Bielschowsky mise a punto un metodo di impregnazione argentica per le neurofibrille che soppiantò il metodo equivalente di Cajal ed è tuttora in uso.
Tra gli scienziati che contribuirono all'introduzione della neuropatologia nei vari paesi europei vanno ricordati: Heinrich Obersteiner ed Emil Redlich a Vienna; Károly Schaffer, professore a Budapest, fondatore della neuropatologia ungherese, autore di studi pionieristici sulle malattie ereditarie del sistema nervoso; il rumeno Georges Marinesco, allievo di Charcot e professore di neurologia a Bucarest, i cui studi sono pubblicati nell'atlante di neuropatologia di Victor Babes e P. O. Blocq, che coniò il termine cromatolisi, descrisse per primo le placche senili, gli aspetti clinici e neuropatologici delle lesioni talamiche e, insieme a Blocq, le alterazioni della substantia nigra nella sindrome parkinsoniana, offrendo, così, la base per la teoria di Éduard Brissaud sull'origine nigrale di questo disordine; Arnold Pick, allievo di Theodor Hermann Meynert e professore di psichiatria a Praga, che descrisse l'atrofia corticale lobare o malattia di Pick e compì studi fondamentali sull'aprassia; Giovanni Mingazzini, direttore di un laboratorio di neuropatologia e in seguito professore di neurologia e psichiatria a Roma, cui si devono importanti studi sul cervelletto (1895) e sul nucleo lenticolare e le sue connessioni (1908).
Nel XX secolo la neuropatologia è riconosciuta e praticata in tutto il mondo come disciplina autonoma, in quanto possiede oggetto e metodiche di studio specifici. La storia contemporanea delle conquiste delle neuroscienze applicate, almeno per quanto riguarda la comprensione della natura e degli eventi causali delle malattie del sistema nervoso, si identifica con quella della neuropatologia. Alle tradizionali scuole neuropatologiche europee si sono affiancate, e per alcuni versi hanno assunto una posizione preminente rispetto a esse, quella americana, di radice culturale anglosassone, e quella giapponese, legata, invece, alla scuola tedesca. La storia e i protagonisti della neuropatologia nel XX secolo possono essere conosciuti prendendo in esame i principali testi prodotti nell'ambito delle esperienze di dette scuole.
3. Metodiche d'indagine
La neuropatologia diagnostica classica studia i tessuti del SNC, il nervo periferico, il muscolo scheletrico - ottenuti mediante autopsia o biopsia - e il liquido cefalo-rachidiano prelevato nel corso dell'iter diagnostico neurologico. Essa utilizza, essenzialmente, metodiche morfologiche macro- e microscopiche (istologia, istochimica, immunoistochimica, microscopia elettronica), biochimiche (spettrofotometria, elettroforesi, cromatografia, ecc.) e, da alcuni anni, metodiche di biologia molecolare derivate dalle cosiddette tecniche del DNA ricombinante, in particolare l'ibridazione in situ e la PCR (Polymerase Chain Reaction), utilizzate soprattutto in neurovirologia e neuroncologia (v. biotecnologie; genetica).
Le metodiche di immagine del sistema nervoso, sviluppate negli ultimi 20 anni - cioè la tomografia computerizzata (TC) e l'imaging di risonanza magnetica (MRI) - hanno migliorato le possibilità e la precisione della diagnostica neurologica, grazie alla capacità di dimostrazione diretta delle alterazioni strutturali del tessuto nervoso. La tomografia a emissione di positroni (PET), che ha finalità essenzialmente sperimentali, consente di ottenere informazioni sul metabolismo e la neurochimica del sistema nervoso in condizioni normali e patologiche. Queste possono essere considerate, a tutti gli effetti, metodiche neuropatologiche in vivo. La correlazione tra i reperti di TC, MRI e PET e quelli neuropatologici ex vivo rappresenta uno dei filoni più affascinanti delle neuroscienze contemporanee (v. radiologia medica; tomografia).
L'esame neuropatologico e l'interpretazione critica dei risultati devono essere indirizzati dai dati clinici (anamnesi, esame obiettivo, esami strumentali). Uno degli obiettivi primari della neuropatologia è rappresentato dalla definizione del rapporto tra lesioni tissutali e reperti clinici: esso non può essere raggiunto senza un'adeguata familiarità con la neurologia clinica, oltre che con l'anatomia e la patologia del sistema nervoso. Lo studio delle alterazioni tissutali può essere effettuato su materiale prelevato nel corso di autopsia e su campioni bioptici. Per procedere all'esame autoptico, l'encefalo e il midollo spinale devono essere rimossi in toto dal cadavere e fissati, a meno di esplicita diversa indicazione (esigenze forensi, richiesta d'esame a fresco), rapidamente, per immersione in soluzione neutra di aldeidi (formalina al 10%); il materiale va poi sezionato, dopo almeno 10 giorni di fissazione: la scarsa consistenza del tessuto nervoso fresco ne preclude un preciso esame macroscopico e una manipolazione priva di artefatti. Nei casi in cui può tornare utile eseguire indagini speciali (immunoistochimiche, biochimiche o biomolecolari), ad esempio in disordini ereditari e/o del metabolismo, è indicato il congelamento rapido di parte dell'encefalo e/o del midollo spinale, o di campioni tissutali prelevati a fresco.
Il prelievo dei campioni di tessuto da utilizzare per gli esami microscopici di routine deve contemplare, sistematicamente, aree anatomiche codificate (lobo frontale, temporale, parietale, occipitale, gangli della base, talamo, mesencefalo, tre livelli del ponte, cervelletto, due livelli del bulbo, sezioni rappresentative del midollo cervicale e, se necessario, di quello dorsale, lombare e sacrale), aree sede di alterazioni visibili a occhio nudo e, infine, aree che, seppure macroscopicamente indenni, sulla scorta dei dati clinici potrebbero essere sede di alterazioni. A differenza di altri organi in cui l'omogeneità strutturale consente un prelievo a campione, l'encefalo e il midollo sono costituiti da più strutture, istologicamente e funzionalmente differenziate, che presentano vulnerabilità variabile alle differenti noxae patogene: solo l'esame d'insieme delle diverse strutture consente una corretta diagnosi neuropatologica.
L'esame microscopico viene eseguito al microscopio ottico su sezioni fini (4-10 µm) o semifini (0,5-0,7 µm) dopo inclusione dei tessuti in paraffina o in resine sintetiche, rispettivamente, oppure al microscopio elettronico su sezioni ultrafini (0,05-0,07 µm). Per l'osservazione microscopica le sezioni tissutali devono essere colorate con metodi istologici generali (ematossilina-eosina, colorazione tricromica secondo van Gieson, ecc.) e con metodi specifici per i neuroni e/o i loro prolungamenti (Nissl, Golgi, Bodian, ecc.), per l'astroglia (Holzer), per la microglia (del Rio-Hortega), per la mielina normale (Klüver-Barrera) o in degenerazione (Marchi, Waller). Negli ultimi 15 anni le colorazioni istologiche sono state integrate, e in taluni casi soppiantate, da quelle immunocitochimiche che utilizzano antisieri o anticorpi monoclonali diretti contro componenti strutturali o composti specifici dei vari elementi cellulari del tessuto nervoso (neurofilamenti, enolasi neurono-specifica, sinaptofisina nei neuroni; proteina gliofibrillare acida negli astrociti; anidrasi carbonica negli oligodendrociti e negli epiteli dei plessi corioidei; proteina basica della mielina nelle guaine mieliniche, ecc.). A queste si affiancano le metodiche per la dimostrazione degli elementi coinvolti nelle reazioni infiammatorie e dei microrganismi potenziali patogeni nei processi meningo-encefalitici.
Un vastissimo settore del corpus metodologico della neuropatologia è quello della diagnostica neuroncologica, intraoperatoria (preparazioni estemporanee su materiale sezionato al criostato, strisciato o schiacciato e colorato con ematossilina-eosina o blu di toluidina) e su tessuto fissato, in cui alle tecniche suddette si affiancano quelle per i marcatori immunocitochimici tumorali (marcatori di attività mitotica; fattori di proliferazione, di crescita, di necrosi; proteine di membrana; componenti del citoscheletro; neurosecreti; ecc.).
Metodiche istochimiche su sezioni di tessuto congelato vengono utilizzate routinariamente nella diagnostica miopatologica, per la dimostrazione di composti intracitoplasmatici (glicidi e lipidi) e di enzimi (miosin-ATPasi, miofosforilasi, NADH-tetrazolio-reduttasi, citocromo-ossidasi, ecc.). Anche in questo settore sono state introdotte metodiche immunoistochimiche principalmente per la dimostrazione di componenti strutturali della miocellula (distrofina, spectrina, desmina, ecc.).
L'esame bioptico dei nervi periferici si fonda, invece, soprattutto sullo studio qualitativo e quantitativo (morfometria) su sezioni semifini, e su quello ultrastrutturale in microscopia elettronica a trasmissione.
4. Cenni di anatomia funzionale
Il SNC è costituito dall'encefalo e dal midollo spinale che sono racchiusi entro gli involucri ossei del cranio e della colonna vertebrale, rispettivamente (v. fig. 1), e quelli fibroconnettivali delle meningi (dura madre, aracnoide e pia). Il SNC galleggia, inoltre, nel liquido cefalo-rachidiano o liquor, circolante sia esternamente al SNC, accolto nello spazio compreso tra dura madre e pia (spazio aracnoideo), che internamente a esso, entro quattro grosse cavità encefaliche profonde, chiamate ‛ventricoli' (v. fig. 2).
L'encefalo, osservato dall'alto dopo la rimozione della dura, appare costituito da due masse simmetriche, ovoidali, gli emisferi cerebrali (v. fig. 3; v. emisferi cerebrali). L'intera superficie emisferica è strutturata in pieghe a margini ondulati, dette ‛circonvoluzioni', separate da solchi di diversa profondità, dei quali quelli più lunghi e profondi prendono il nome di ‛scissure' (v. fig. 4). Osservando la superficie inferiore dell'encefalo, procedendo antero-posteriormente, si distinguono (v. fig. 5): la superficie orbitaria dei lobi frontali, la faccia inferiore dei lobi temporali e, tra questi, sulla linea mediana, l'infundibolo e i corpi mammillari, che sono parte dell'ipotalamo. Posteriormente si nota l'insieme di peduncoli cerebrali, ponte e bulbo, che viene definito ‛troncoencefalo'. Le strutture sovrastanti quest'ultimo costituiscono il cervello propriamente detto. Posteriormente e inferiormente a ponte e bulbo si riconosce, infine, il cervelletto. Sulla superficie inferiore dell'encefalo risaltano, con aspetto di filamenti di vario calibro e di colore bianco-giallastro, i nervi cranici (v. fig. 5A), così detti perché emergono dall'encefalo a livello intracranico.
Tagliando il cervello, si nota che l'intera superficie cerebrale è formata da una lamina, detta ‛corteccia', il cui spessore varia, nelle diverse parti dell'encefalo, da 3 a 6 millimetri (v. fig. 6). La corteccia - che, essendo di colore grigio-rosa, si dice formata da ‛sostanza grigia' - è costituita prevalentemente dai corpi dei neuroni disposti in lamine e dai prolungamenti afferenti o ‛dendriti' e ricopre una voluminosa massa di tessuto di aspetto bianco-lucente - definito perciò ‛sostanza bianca' - formata da fasci dei prolungamenti neuronali efferenti o ‛assoni' avvolti o meno in una guaina lipoproteica detta mielina (v. neurobiologia: Organizzazione neuronale cerebrale e cerebellare). Nelle sezioni cerebrali risalta la presenza di numerose aree a margini ben definiti di sostanza grigia, denominate ‛nuclei' (v. fig. 7). I principali gruppi di nuclei grigi profondi cerebrali comprendono il corpo striato, formato dal nucleo caudato e dal putamen, e il globo pallido (che nell'insieme vengono denominati ‛gangli della base'), nonché il gruppo formato da talamo, epitalamo e subtalamo. Il talamo anteriore è collegato anatomicamente e funzionalmente all'importante gruppo di nuclei grigi che formano l'ipotalamo. Talamo e ipotalamo costituiscono il diencefalo.
Lobi cerebrali. - Il lobo frontale, mediante la corteccia motoria e quella premotoria, controlla i movimenti e le sequenze di movimenti volontari di singoli muscoli o di più muscoli coordinati, compresi quelli della laringe e della bocca necessari per l'articolazione della parola, di cui è responsabile l'area di Broca. Quest'ultima è funzionalmente presente solamente nell'emisfero definito dominante, che è quello sinistro in circa il 95% degli individui (tutti i destrimani e la metà dei mancini). All'area prefrontale e alla corteccia orbitofrontale sono legati le capacità di prestare attenzione, di formulare programmi per il futuro, di iniziativa, di approfondimento del pensiero e il controllo di alcuni aspetti della personalità (v. fig. 8).
Il lobo parietale è dedicato quasi totalmente alla percezione sensitiva somestesica (tatto, pressione, temperatura, dolore), che avviene in un'area corticale primaria che riceve e distingue i segnali, e in una secondaria che ne interpreta il significato (v. somatoestesia). La restante corteccia partecipa, con settori corticali adiacenti del lobo temporale, alla formazione dell'area di Wernicke, o area recettiva del linguaggio, la cui funzione è quella di integrare informazioni linguistiche - siano esse udite, lette o originate nel cervello stesso - e interpretarne il significato; l'area di Wernicke è funzionalmente sviluppata solo nell'emisfero dominante. Occorre ricordare, infine, che nella sostanza bianca del lobo parietale decorrono le fibre della radiazione ottica che trasportano segnali visivi.
Il lobo temporale, rispetto agli altri lobi cerebrali, ha funzioni relativamente più numerose, che comprendono: alcuni aspetti della memoria (neocorteccia laterale); la recezione del linguaggio, nell'emisfero dominante (area di Wernicke); l'interpretazione dei segnali sonori (corteccia uditiva); un ruolo nella percezione olfattiva e gustativa e nel controllo dei processi emozionali, affettivi, mnesici e comportamentali, che avviene a livello del sistema limbico, di cui fanno parte diverse strutture temporali. Nella sostanza bianca del lobo temporale si svolge parte del decorso delle fibre della radiazione ottica.
Il lobo occipitale è sede della corteccia visiva, nella quale si distingue l'area primaria, localizzata lungo la scissura calcarina, che distingue punti luminosi e neri e l'orientamento di linee e margini di un'immagine, e le aree secondarie che interpretano l'informazione visiva (v. visione).
Gangli della base. - Nell'uomo i gangli della base controllano l'attività motoria di base, cioè quella preposta all'assunzione e al mantenimento di un'adatta postura corporea di sostegno complementare all'esecuzione dei movimenti più fini, controllati dalla corteccia. L'alto grado di coordinazione tra i muscoli del corpo, necessario per l'esecuzione della maggior parte delle funzioni motorie, è possibile, comunque, solo attraverso l'attivazione di molteplici strutture nervose, interconnesse attraverso circuiti nervosi molto complessi, che oltre i gangli della base e la corteccia cerebrale comprendono il talamo e il subtalamo, il nucleo rosso e la substantia nigra, e il cervelletto.
Talamo. - Il talamo ha numerose connessioni bidirezionali con tutte la parti della corteccia cerebrale, riceve quasi tutti i segnali provenienti dai livelli più caudali del mesencefalo e dal midollo spinale ed è in stretto rapporto con i gangli della base. In sostanza, il talamo è una stazione di smistamento per indirizzare segnali sensoriali e di altra natura sia alla corteccia cerebrale che ad aree sottocorticali del cervello. I segnali più importanti che vengono ritrasmessi attraverso il talamo sono: tutti quelli della sensibilità somestesica (tatto, pressione, dolore, temperatura, ecc.), che dall'intero organismo sono diretti alla corteccia parietale; i segnali visivi, che dalle vie ottiche sono diretti alla corteccia calcarina del lobo occipitale; i segnali uditivi, diretti alla circonvoluzione temporale superiore (v. udito); i segnali per il controllo motorio provenienti dal cervelletto, dal mesencefalo e da altri livelli più caudali del tronco encefalico e diretti alla corteccia e ai gangli della base. Il talamo conserva inoltre funzioni di interpretazione sensoriale, soprattutto per le sensazioni dolorifiche, e svolge una funzione di attivazione della corteccia cerebrale.
Ipotalamo e sistema limbico. - L'ipotalamo rappresenta il centro di un sistema assai complesso nel quale interagiscono in stretta associazione, oltre all'ipotalamo stesso, il talamo anteriore, il sistema limbico, l'infundibolo con l'ipofisi e la formazione reticolare del troncoencefalo (v. fig. 9). Questo sistema esercita il controllo sia sulle condizioni interne dell'organismo, attraverso la regolazione della maggior parte delle funzioni endocrine e vegetative, che su diversi aspetti del comportamento, in particolare quelli connessi con le emozioni. Il controllo delle funzioni endocrine si effettua mediante la regolazione della secrezione degli ormoni dell'ipofisi anteriore, che a loro volta regolano attività dell'organismo quali le funzioni delle ghiandole sessuali, della tiroide, del surrene, ecc. (v. neuroendocrinologia). Il controllo delle funzioni vegetative si esplica attraverso la regolazione di diverse funzioni: la pressione arteriosa e la frequenza cardiaca, mediata dai centri della formazione reticolare bulbari e pontini (v. circolazione: Regolazione nervosa della circolazione); la temperatura corporea; il patrimonio idrico dell'organismo, attraverso la sensazione di sete e la diuresi controllata dall'ormone antidiuretico; l'assunzione di alimenti attraverso le sensazioni di fame e di sazietà; il controllo del parto e della lattazione mediante l'ormone ossitocina.
Sul piano comportamentale, le strutture ipotalamiche controllano lo stato di veglia e di allerta, in cooperazione con la porzione mesencefalica del sistema reticolare attivatore; lo stato di sonno (v. sonno); il livello di attività, dalla quiete sino alla rabbia e alla lotta; le sensazioni e le reazioni di paura e di punizione; l'impulso sessuale.
Oltre l'ipotalamo anche diverse strutture limbiche sono coinvolte nel determinare il tono affettivo delle esperienze sensoriali. Queste tonalità affettive vanno anche sotto il nome di sentimenti di gratificazione, o ricompensa, e di punizione. La stimolazione dei centri della gratificazione evoca comportamenti di calma e docilità, mentre quella dei centri della punizione evoca terrore, dolore, paura, reazioni di difesa e di fuga. È interessante notare che la stimolazione dei centri della punizione spesso inibisce i centri della gratificazione. La gratificazione e la punizione hanno un ruolo fondamentale nel determinare l'importanza di un'informazione sensoriale e nel promuoverne la memorizzazione mediata attraverso l'ippocampo. Stimoli affettivamente indifferenti inducono rapidamente abitudine e vengono ignorati.
Tronco encefalico. - Il troncoencefalo è il fusto che sostiene il cervello e lo mette in comunicazione con il midollo spinale. Attraverso di esso si stabiliscono, inoltre, le connessioni tra cervello e cervelletto e tra questo e midollo spinale (v. fig. 10). Il tronco può, quindi, essere considerato una stazione di passaggio di segnali sensoriali dal midollo principalmente verso il talamo e il cervelletto, e di segnali motori provenienti dalla corteccia cerebrale e dal cervelletto verso il midollo, ma esso è soprattutto sede di nuclei di grande importanza per la regolazione di diverse funzioni dell'organismo (v. sistema piramidale; sistema reticolare ascendente). Tra queste ricordiamo il controllo di: a) attività muscolari subcoscienti e coordinazione dei movimenti (substantia nigra e nucleo rosso, in concorso con i gangli della base e con il cervelletto); b) livello globale di attività cerebrale, stato di veglia e tono muscolare (formazione reticolare); c) pressione arteriosa e attività cardiaca, così come attività ritmica dei muscoli respiratori (centri vasomotore e respiratorio della sostanza reticolare); d) motilità oculare (nuclei del III, IV e VI paio di nervi cranici e collicoli superiori); e) reazioni motorie a segnali uditivi (collicoli inferiori); f) sensibilità e movimenti della faccia (nuclei del V e VII paio di nervi cranici); g) udito ed equilibrio (nuclei dell'VIII paio di nervi cranici); h) motilità di laringe, faringe, lingua e alcuni muscoli del collo (nuclei del IX, X, XI e XII paio di nervi cranici) così come attività parasimpatica di organi interni (riduzione della frequenza cardiaca, stimolazione della secrezione gastrica e della peristalsi intestinale, ecc.; nuclei del X paio di nervi cranici); i) segnali sensoriali provenienti dalla faringe, dalla lingua e dai visceri (nuclei del IX e X paio di nervi cranici, rispettivamente).
Cervelletto. - Il cervelletto è formato da due masse laterali, gli emisferi, congiunti sulla linea mediana dal verme cerebellare. La corteccia cerebellare, formata come quella cerebrale da sostanza grigia, è ripiegata in sottili pliche trasversali dette folia (v. neurobiologia: Organizzazione neuronale cerebrale e cerebellare). Il cervelletto è interconnesso con la corteccia motoria, con i gangli della base e con aree motorie del troncoencefalo e del midollo spinale (v. fig. 11). La sua funzione principale è quella di regolare la sequenza temporale di contrazione di differenti muscoli, durante movimenti complessi.
Midollo spinale. - Il midollo spinale, che anatomicamente costituisce la prosecuzione del bulbo, è una struttura allungata, che si estende dal forame magno dell'osso occipitale sino, all'incirca, al margine inferiore della seconda vertebra lombare (v. figg. 12 e 13). Esso assolve, essenzialmente, una funzione di trasporto di impulsi nervosi dal sistema nervoso periferico a strutture sovrastanti di quello centrale e, viceversa, da queste di nuovo al sistema nervoso periferico (v. figg. 14 e 15). I cordoni dorsali trasportano gli impulsi delle sensibilità tattile fine o discriminativa (quella tipica, ad esempio, della mano) e vibratoria (sensibilità somatiche), e di quelle riguardanti il senso di posizione o di movimento del corpo (sensibilità propriocettive coscienti). Le fibre dalle radici spinali posteriori vanno direttamente ai nuclei bulbari gracile e cuneato e formano i fasci gracile (mediale) e cuneato (laterale). Nei cordoni laterali e anteriori decorrono, invece, fasci affiancati di fibre sensitive e motorie. I principali fasci sensitivi somatici (spinotalamici ventrale e laterale) trasportano segnali delle sensibilità tattile grossolana, dolorifica e termica al troncoencefalo e al talamo. Essi originano dalle corna grigie posteriori e raggiungono i cordoni antero-laterali del lato opposto del midollo, incrociando la linea mediana al davanti del canale centrale. Tale decorso li espone a danno nel corso di lesioni che interessano primariamente la porzione centrale del midollo. I fasci propriocettivi (spinocerebellari ventrale e dorsale) trasportano segnali sensitivi, che non vengono percepiti a livello cosciente, dai muscoli scheletrici e dai tendini al cervelletto. I fasci motori possono essere distinti in quelli che trasmettono segnali motori dalla corteccia cerebrale (corticospinali laterale e ventrale) e da strutture troncoencefaliche (fasci rubro-, reticolo-, olivo-, vestibolo- e tettospinale) al midollo. Vanno infine ricordati i fasci propriospinali, che servono a mettere in comunicazione reciproca segmenti diversi del midollo.
La sostanza grigia del midollo spinale funge da stazione di collegamento tra l'encefalo e il sistema nervoso periferico, e da centro d'integrazione per alcune attività motorie riflesse (riflessi spinali). È, infatti, alle corna dorsali che giunge la maggior parte dei segnali sensitivi per essere smistata verso altre strutture del midollo e poi alle aree sensitive encefaliche. A livello delle corna ventrali e laterali vengono, invece, smistati i segnali efferenti che dall'encefalo sono diretti alle radici spinali anteriori. La funzione d'integrazione consiste nel controllo di risposte motorie a vari stimoli, che si compie del tutto indipendentemente dai centri encefalici superiori. Esempi di tale funzione sono i riflessi di allontanamento, quasi istantaneo, di un arto dalla sorgente di uno stimolo dolorifico; di grattamento, evocato dal solletico; di stiramento, che si estrinseca con la contrazione di un muscolo in risposta alla sua brusca estensione (ad esempio, l'estensione della gamba sulla coscia da tensione del tendine rotuleo).
Nervo periferico. - Anatomicamente vengono definiti ‛nervi periferici' le prosecuzioni oltre i forami intervertebrali dei nervi spinali (v. fig. 16). L'unità elementare strutturale del nervo periferico è la fibra nervosa, costituita da un prolungamento neuronale (assone o dendrite), rivestito per la quasi totalità della sua lunghezza da protrusioni citoplasmatiche di cellule specializzate, le cellule di Schwann: questo rivestimento, presente nella maggioranza ma non in tutte le fibre, è costituito da mielina e ha la struttura di un avvolgimento a spirale (v. fig. 17). Schematicamente, ciascun tronco nervoso originato dai nervi spinali emergenti nel tratto midollare compreso tra il primo segmento toracico e il secondo o terzo lombare possiede una componente afferente e una efferente (dette anche, in senso lato, ‛sensitiva' e ‛motoria', rispettivamente), a loro volta distinte in una componente detta ‛somatica' (v. somatoestesia) e una ‛viscerale' o ‛autonomica' (v. sistema nervoso autonomo). La componente afferente somatica conduce al midollo spinale impulsi sensitivi (tattili, termici, dolorifici, pressori, ecc.) generati dalla stimolazione fisica di recettori cutanei (esterocettori), muscolari, tendinei, osteoarticolari (propriocettori); quella afferente autonomica conduce impulsi sensitivi generati dalla stimolazione fisica o chimica di recettori localizzati a livello degli organi interni (enterocettori). La componente efferente somatica conduce impulsi motori ai muscoli scheletrici; quella autonomica conduce impulsi preposti al controllo della muscolatura liscia dell'occhio, dei vasi sanguigni, di alcuni apparati interni; dell'attività secretoria ed escretoria delle ghiandole dell'organismo; dell'attività cardiaca; della motilità dell'apparato gastrointestinale (v. recettori).
5. Anatomia microscopica del SNC
Si riconoscono due classi di cellule proprie del SNC: le cellule nervose, o neuroni, capaci di generare e condurre a distanza impulsi elettrici e responsabili, pertanto, della specificità funzionale del tessuto nervoso (v. fig. 18); le cellule neurogliali (termine utilizzato originariamente per designare cellule con funzione di ‛legame' tra i neuroni), o cellule interstiziali, che svolgono diverse funzioni metaboliche fondamentali per mantenere l'efficienza e l'integrità strutturale e funzionale del tessuto nervoso (v. fig. 19). Vi sono tre tipi di cellule neurogliali: astrociti, oligodendrociti e cellule ependimali.
Altre cellule presenti nel SNC sono la microglia, le cellule costituenti delle meningi e quelle dei vasi sanguigni, che hanno origine embrionale diversa da quella dei neuroni e della neuroglia. Essendo stati i neuroni ampiamente trattati in precedenza, qui prenderemo in esame soprattutto gli altri tipi di cellule.
Neuroglia. - Gli astrociti sono cellule la cui forma ricorda un astro, con raggi numerosi e brevi (protoplasmatici) o rari e lunghi (fibrillari): i primi prevalgono nella sostanza grigia, i secondi in quella bianca cerebrale e spinale. Il nucleo si distingue da quello dei neuroni soprattutto perché, generalmente, non è riconoscibile il nucleolo. Gli organuli citoplasmatici sono identici a quelli neuronali, a eccezione della neuromelanina che è assente. Il citoplasma del corpo cellulare e dei prolungamenti maggiori negli astrociti fibrillari è ricco di granuli di glicogeno e di tipici filamenti astrocitari, il cui principale costituente è la proteina gliofibrillare acida (GFAP), il ruolo della quale non è noto; la sintesi della GFAP, estremamente attiva e rapida, è un indicatore sensibile delle variazioni funzionali degli astrociti. Queste cellule costituiscono complessivamente circa il 35% del volume encefalico e sono più numerose dei neuroni, in misura regionalmente variabile (da 4:1 nel putamen a 100:1 nel pallido). Con gli elementi cellulari circostanti stabiliscono rapporti anatomo-funzionali mediante le terminazioni dei loro prolungamenti, dette ‛podociti', o direttamente tramite il corpo cellulare. Le loro principali funzioni, oltre quella di sostegno dei neuroni, comprendono: riparazione e cicatrizzazione delle lesioni tissutali; partecipazione alla funzione di barriera ematoencefalica (BEE); protezione della superficie neuronale da afferenze aspecifiche; partecipazione allo sviluppo e al differenziamento neuronale e alla regolazione dei fenomeni di plasticità neuronale; mantenimento degli equilibri ionici cerebrali (accumulo e trasporto attivo di K+, Na+ e Cl-); partecipazione al controllo dell'eccitabilità e del metabolismo energetico e neurotrasmettitoriale del tessuto nervoso; detossicazione dell'ammoniaca; rimozione di sostanze, particelle e resti cellulari; partecipazione alla risposta immune intracerebrale.
Gli oligodendrociti si localizzano prevalentemente in prossimità delle fibre nervose (oligodendrociti interfascicolari) nella sostanza bianca e in quella grigia, ma anche intorno al corpo cellulare dei neuroni (oligodendrociti satelliti) o nelle adiacenze dei vasi sanguigni. Sono meno numerosi e più piccoli degli astrociti e si riconoscono per la sottile rima citoplasmatica, poligonale o circolare, che circonda il nucleo, tondo od ovale. I prolungamenti cellulari, rari e delicati, sono visibili solo con i metodi di colorazione speciali (impregnazione argentica). Funzione principale degli oligodendrociti è la formazione e il mantenimento della mielina; possiedono, inoltre, vari enzimi (2′,3′'-ciclico-nucleotide-3′-fosfodiesterasi, anidrasi carbonica, glicerofosfato- e glucosio-6-fosfatodeidrogenasi) coinvolti in processi finalizzati al mantenimento dell'integrità strutturale dei neuroni.
Gli ependimociti sono cellule simil-epiteliali che formano l'ependima, il rivestimento delle cavità interne del SNC (ventricoli, acquedotto cerebrale, canale centrale spinale). Hanno forma cuboidale o cilindrica e aderiscono l'un l'altro mediante strutture di membrana specializzate (giunzioni serrate o tight junctions). La faccia apicale o libera, a contatto con il liquido cefalorachidiano, è fornita di ciglia, microvilli e semplici protrusioni citoplasmatiche che ne aumentano la superficie; le ciglia sono strutture capaci di movimento. Dagli ependimociti ordinari si distinguono i taniciti, la cui porzione basale è in contatto con i capillari subependimali e quella apicale presenta scarse ciglia. L'ependima, grazie alla presenza delle giunzioni serrate intercellulari e al possesso di specifici meccanismi di trasporto cellulare per diversi metaboliti, costituisce una barriera (barriera liquido cefalorachidiano-encefalo) alla libera diffusione di sostanze dai tessuti nervosi al liquido cefalorachidiano e viceversa. Si ritiene che i taniciti siano capaci di trasporto bidirezionale tra vasi sanguigni e liquido cefalorachidiano.
Microglia. - Le cellule microgliali sono ubiquitarie nel SNC, ma relativamente più numerose nella corteccia cerebrale, con tendenza a concentrarsi intorno ai neuroni e ai vasi sanguigni. Hanno forma allungata, scarso citoplasma e lunghi prolungamenti, meno numerosi di quelli degli astrociti, evidenziabili con i metodi di impregnazione argentica. Il nucleo, generalmente dislocato a un'estremità del citoplasma, è triangolare o allungato. Tra gli organuli citoplasmatici sono preminenti i lisosomi, che appaiono in forma di corpi inclusi densi. La microglia è responsabile del mantenimento del normale bilancio idrosalino nello spazio extracellulare e della rimozione (fagocitosi) di sostanze e particelle estranee o residue alla distruzione cellulare nei disordini del sistema nervoso, e ha, verosimilmente, anche un ruolo nelle difese immunologiche. La sua origine è oggetto di controversia: secondo l'ipotesi attualmente più accreditata, una quota deriverebbe da primitive cellule mesodermiche in rapporto con la pia madre, e una quota da elementi ematici, i monociti, che penetrati nel cervello assumerebbero, sotto l'influsso del nuovo microambiente, caratteristiche microgliali.
Microcircolo cerebrale e barriera ematoencefalica. - I rami vascolari, definiti arteriole, si distaccano dalle arterie visibili a occhio nudo sulla superficie del SN e penetrano nei tessuti, dove si ramificano in una rete di vasi via via più numerosi e di calibro progressivamente decrescente, in cui si distinguono, in base a criteri strutturali e funzionali, le metarteriole, i precapillari e, infine, i capillari (diametro 4-6 µm). Da questi ultimi, quindi, il sangue passa nel distretto venoso, i cui componenti, di calibro progressivamente crescente, vengono distinti in venule postcapillari, venule e vene. La rete artero-venosa intratissutale prende il nome di ‛microcircolo'. Arteriole e metarteriole possiedono una parete con componente muscolare e sono in grado di modificare il loro calibro in risposta a variazioni della pressione arteriosa sistemica, in modo tale che il flusso ematico all'encefalo rimanga costante nell'ambito di uno spettro di valori della pressione arteriosa sistolica esteso da 65 a 180 mmHg (autoregolazione). Possiedono, inoltre, una spiccata reattività alle variazioni di concentrazione ematica di anidride carbonica (CO2), che si riflette in una pronta dilatazione in caso di aumento di quest'ultima, come avviene, ad esempio, in condizioni di insufficienza respiratoria: la vasodilatazione consente l'aumento del flusso ematico cerebrale in misura tale da mantenere una ossigenazione adeguata del SN anche in condizioni di relativa ipossia. I capillari sono la sede unica degli scambi metabolici tra sangue ed encefalo, in particolare la loro parete viene attraversata per diffusione dall'ossigeno e, grazie a meccanismi di trasporto specifici, dal glucosio. I capillari encefalici, in virtù della peculiare strutturazione della loro parete (giunzioni serrate interendoteliali; membrana basale continua; intimo legame con i podociti astrocitari) e della presenza, in quest'ultima, di numerosi enzimi in grado di degradare, di frazionare o di legare le sostanze più varie, costituiscono una barriera selettiva nei confronti della maggioranza delle sostanze e dei composti trasportati dal sangue. Tale caratteristica, unica dei capillari cerebrali, viene definita barriera ematoencefalica (BEE) ed è presente in tutto il sistema nervoso centrale a eccezione di alcune strutture periventricolari (area postrema, eminenza mediana, organo subcommissurale e subfornicale, cresta sopraottica, pineale e neuroipofisi). La BEE possiede la capacità di regolare il volume encefalico mediante limitazione della diffusione dell'acqua attraverso la parete vasale; d'impedire il passaggio di gran parte delle sostanze idrosolubili dal sangue all'encefalo; di selezionare le sostanze all'interfaccia sangue-encefalo in base alle loro caratteristiche di liposolubilità, carica elettrica, diametro molecolare, costante di dissociazione, affinità per i sistemi di trasporto specifici presenti a livello della BEE stessa; di trasportare dal sangue all'encefalo sostanze, tra le quali esosi e amminoacidi, mediante sistemi stereospecifici. Prive di BEE sono, invece, le arteriole e le venule. Queste ultime sono la sede di inizio dei principali fenomeni reattivi cellulo- e immunomediati in corso di processi infiammatori a livello del SN: in particolare, del passaggio degli elementi ematici della serie bianca dal letto vascolare al cervello.
Meningi. - La dura madre è un involucro formato da un denso intreccio di spessi fascicoli di collagene e fibroblasti, tra i quali decorrono vasi sanguigni, la cui superficie interna, in condizioni normali, è separata dall'aracnoide da uno spazio virtuale. L'aracnoide, invece, delimita lo spazio subaracnoideo nel quale circola il liquido cerebrospinale, ed è in rapporto con la pia sottostante tramite trabecole aracnoidee: è formata da una trama di cellule di sostegno rivestite da cellule laminari, denominate meningoteliociti, che sono congiunte tra loro da contatti intercellulari specializzati. Diverticoli, denominati villi, e granulazioni aracnoidee penetrano entro le vene e i seni venosi della dura e costituiscono la maggior via di drenaggio del liquido cerebrospinale che percola, attraverso di essi, nel circolo venoso. La pia, formata da cellule simili ai meningoteliociti, ma più sottili, riveste la superficie encefalo-midollare; essa aderisce a manicotto ai vasi superficiali del sistema nervoso, dividendo lo spazio subaracnoideo dagli spazi perivascolari intracerebrali di Virchow-Robin. Aracnoide e pia vengono dette leptomeningi.
Plessi corioidei. - Nell'encefalo vi sono 4 plessi corioidei, due adesi alla parete mediale di ciascuno dei due ventricoli laterali e due al tetto del III e del IV ventricolo, rispettivamente. Sono composti da pliche della pia madre, ciascuna delle quali dotata di un capillare, ricoperte da una lamina epiteliale di derivazione ependimale. L'epitelio, formato da un singolo strato di cellule cuboidali ciliate adese a una membrana basale continua e congiunte tra loro mediante tight junctions, non solo costituisce la barriera plessoliquorale ma è anche sede dei fenomeni di trasporto dei substrati necessari per la produzione del liquido cerebrospinale: i plessi formano, infatti, l'80-90% dei 500-700 ml di liquido cerebrospinale prodotti quotidianamente nell'uomo sotto l'influenza di diversi fattori, tra i quali la pressione intracranica, l'osmolarità serica, la temperatura corporea, l'innervazione dei plessi, l'età dell'individuo e i livelli di prostaglandine. Il volume degli spazi liquorali è di circa 140 ml, di cui 25-30 ml dei ventricoli e il resto degli spazi subaracnoidei. I plessi hanno anche limitate capacità di riassorbimento del liquido cerebrospinale.
Lamina subependimale. - È uno strato cellulare sottostante l'ependima dei ventricoli laterali, residuo della matrice embrionale da cui nel feto originano i neuroni e la glia, formato da elementi che presentano caratteristiche ultrastrutturali tipiche delle forme primitive: elevato rapporto nucleo-citoplasmatico, predominanza di ribosomi liberi rispetto a quelli legati a membrane e scarsi organuli citoplasmatici. Vi sono prove convincenti che in diverse specie, compresi i Primati, le cellule della lamina subependimale - astrociti e oligodendrociti maturi - mantengano la capacità mitotica, a differenza degli altri elementi cellulari del sistema nervoso. Oltre alla lamina subependimale, zone germinative secondarie, temporaneamente attive nel periodo postnatale, sono rappresentate dal giro dentato dell'ippocampo, dal bulbo olfattorio e dallo strato granulare esterno del cervelletto. Nell'uomo, tuttavia, le conoscenze a tale proposito sono limitate.
6. Neuropatologia generale: processi elementari
Si distinguono due gruppi generali di processi cellulari che si manifestano in risposta all'azione di noxae patogene sul SN: regressivi/degenerativi e progressivi/ipertrofico-iperplastici. I primi sono espressione del deterioramento e dell'eventuale morte cellulare, i secondi dei tentativi di difesa e di riparazione da parte delle cellule sfuggite all'evento nocivo. Alcuni processi, ad esempio quelli che accompagnano l'invecchiamento, eludono un chiaro inquadramento nell'una o nell'altra categoria. Si devono ricordare ancora i processi neoplastici, espressione dell'alterazione dei meccanismi di controllo della crescita e proliferazione cellulare (polimorfismo cellulare e nucleare, alterazione del rapporto nucleo-citoplasmatico e di quello nucleolo-nucleare, mitosi anomale, atipia cellulare e/o nucleare, ecc.) che, tuttavia, non discostandosi da quelli descritti in oncologia generale, non verranno descritti (v. chirurgia: Neurochirurgia; neoplasie: Oncologia clinica).
La trattazione separata dei processi elementari a livello neuronale, gliale, vascolare e meningeo è didatticamente conveniente, anche se quasi mai gli eventi patologici a carico del sistema nervoso interessano un singolo tipo cellulare, e raramente le reazioni individuali sono patognomoniche del processo che le ha generate. Le lesioni tissutali devono piuttosto essere studiate in termini di costellazioni cellulari, la cui valutazione d'insieme consente, spesso, la diagnosi eziologica.
a) Processi neuronali regressivi
I neuroni sono gli elementi del sistema nervoso più sensibili alle noxae patogene e quelli che mostrano la maggiore varietà di reazioni regressive/degenerative; la loro capacità di riparazione e rigenerazione è, invece, scarsa. I processi progressivi si limitano, in pratica, alla presenza di neuroni binucleati o multinucleati, con corpo cellulare talora gigante e profilo irregolare, privi di arborizzazioni dendritiche, che si trovano nelle malformazioni congenite, negli amartomi e, occasionalmente, al margine di lesioni focali inveterate, con possibile significato reattivo.
L'interruzione o la lesione dell'assone comporta, a livello del corpo cellulare, un quadro definito ‛cromatolisi centrale', caratterizzato essenzialmente da un processo di disgregazione del reticolo endoplasmatico rugoso (sostanza di Nissl) il cui corrispettivo morfologico, nella fase conclamata, è rappresentato dalla perdita della normale colorabilità della porzione centrale del citoplasma neuronale (v. fig. 20): tale perdita si osserva tipicamente nei neuroni motori spinali il cui assone abbia subito una lesione distale, mentre è rara nei neuroni motori centrali. Essa rappresenta l'espressione del tentativo della cellula di ripristinare l'integrità dell'assone: se ciò avviene, la cellula riprende il suo aspetto normale, passando attraverso uno stadio definito ‛cromatolisi periferica', perché la porzione citoplasmatica ipocolorabile è localizzata alla periferia cellulare. Se la lesione dell'assone avviene in prossimità del corpo cellulare, questo usualmente degenera. Quadri indistinguibili dalla cromatolisi centrale si osservano nella carenza della vitamina PP (pellagra) e in una condizione ‛dementigena' progressiva su base degnerativa del SNC, detta malattia di Pick, di cui patogenesi e significato non sono stati ancora chiariti.
In conseguenza di una carenza acuta dell'apporto ematico di ossigeno (ipossia/ischemia) e/o di glucosio (ipoglicemia), che sono i principali substrati metabolici dei neuroni, si manifesta una sequenza di alterazioni cellulari, che dall'iniziale, reversibile, rigonfiamento dei mitocondri, che appaiono come vacuoli intracitoplasmatici (microvacuolizzazione), attraverso una fase caratterizzata dalla riduzione del volume e dalla modificazione delle proprietà tintoriali e della morfologia del nucleo, del citoplasma e dei prolungamenti cellulari (coagulazione o ischemia cellulare) può progredire verso la distorsione della superficie neuronale, ove compaiono piccole masse basofile (incrostazione) sino alla completa destrutturazione citoplasmatica con disfacimento degli organuli (omogenizzazione). In seguito alla lisi delle membrane neuronali resta riconoscibile solo il profilo del corpo cellulare (neurone fantasma). In questo stadio sono frequenti fenomeni di fagocitosi dei neuroni (neuronofagia). Neuroni fantasma si trovano anche al margine di lesioni traumatiche e in processi degenerativi e infettivi selettivi della sostanza grigia (poliomielite). La successione di questi processi può essere interrotta (reversibilità) se la carenza di ossigeno è di breve durata. La vulnerabilità a tale carenza varia, comunque, da specie a specie: nell'uomo, la microvacuolizzazione e la coagulazione non si manifestano, verosimilmente, prima di 30 minuti di ipossia e di 90 di ischemia. La sensibilità al danno ipossico varia inoltre a seconda del tipo neuronale: i neuroni piramidali delle lamine III, V e VI della corteccia cerebrale, quelli del settore CA1 dell'ippocampo e le cellule di Purkinje nel cervelletto sono gli elementi più vulnerabili in assoluto. A livello dello striato vengono preferenzialmente danneggiati i neuroni di piccola e media dimensione rispetto a quelli grandi (v. neurobiologia: Organizzazione neuronale cerebrale e cerebellare).
Nell'invecchiamento fisiologico e in quello prematuro e nelle condizioni degenerative sistemiche (malattia di Alzheimer, malattia dei motoneuroni, degenerazione multisistemica, corea di Huntington, ecc.), per cause non ben conosciute alcuni complessi enzimatici neuronali vanno incontro a selettiva e progressiva alterazione con conseguenti modificazioni cellulari a evoluzione cronica. Questi processi, nella forma più semplice (atrofia semplice o sclerosi cellulare cronica), sono caratterizzati essenzialmente dalla progressiva atrofizzazione del corpo cellulare con riduzione, prima, e disgregazione, poi, dei prolungamenti cellulari e della mielina, sino alla scomparsa del neurone, cui si associa una reazione astrocitaria e microgliale. Questo tipo di processo degenerativo si osserva in neuroni nei quali sono venuti a mancare o si sono fortemente ridotti gli impulsi afferenti (degenerazione transneuronale o transinaptica) e può estendersi a cascata a neuroni collegati in successione. La sclerosi cellulare può manifestarsi anche in elementi il cui assone è in contatto con neuroni degenerati (degenerazione transneuronale retrograda).
Diverse condizioni patologiche (malattie degenerative, anomalie dello sviluppo, ecc.) si accompagnano alla comparsa di inclusioni intraneuronali, di genesi e morfologia variabili. Sebbene quasi nessuno di tali processi sia specifico di una malattia, essi hanno importanza ai fini della diagnostica e della ricerca neuropatologica: nella malattia di Alzheimer, ad esempio, si osservano formazioni filamentose con una configurazione triangolare o a fiamma (degenerazione neurofibrillare di Alzheimer; v. fig. 21) che presentano caratteristiche istologiche, ultrastrutturali e di distribuzione distinte nei diversi tipi neuronali, ma tipiche della malattia. Infatti, processi morfologicamente identici, ma differenti per numero e localizzazione, si riscontrano in diverse altre condizioni degenerative e negli individui affetti dalla sindrome di Down che raggiungono l'età adulta; in altre rare malattie degenerative (paralisi sopranucleare progressiva; forme sporadiche delle malattie del motoneurone) ricorrono invece degenerazioni neurofibrillari ultrastrutturalmente differenti. Ancora nella malattia di Alzheimer, in altre condizioni degenerative e nella sclerosi tuberosa, i neuroni piramidali dell'ippocampo possono presentare nel citoplasma uno o più vacuoli, ciascuno contenente un piccolo granulo basofilo incluso in una matrice translucente (degenerazione granulo-vacuolare di Simchowicz).
Specifiche, invece, della malattia di Pick sono le omonime cellule di Pick, neuroni tipicamente rotondeggianti, pallidi e omogenei, con inclusioni citoplasmatiche filamentose (corpi di Pick), che si trovano solo negli strati II, III e V della corteccia cerebrale, nell'ippocampo e, talora, nei gangli basali e nella substantia nigra.
Nella malattia di Parkinson, nei neuroni del tronco, pigmentati e non, si possono trovare inclusioni perinucleari e intradendritiche, rotondeggianti, singole o multiple, costituite da materiale filamentoso e granulare (corpi di Lewy), uno dei cui componenti principali è l'ubiquitina, che ha un ruolo chiave in un sistema enzimatico responsabile della degradazione di proteine anomale o a breve emivita (v. biochimica).
Pazienti affetti da epilessia mioclonica presentano - soprattutto nei neuroni del nucleo dentato, ma anche in quelli della substantia nigra, del talamo e talora della corteccia - tipici aggregati tondeggianti di natura mucopolisaccaridica (corpi di Lafora).
Diverse altre inclusioni con struttura e distribuzione variabili (corpi di Hirano; corpi colloidi; corpi di Marinesco; corpi di Bunina), ma di incerto significato diagnostico, si trovano nel cervello senile, nella malattia di Alzheimer, in quella di Pick e nelle malattie del motoneurone.
Nel citoplasma di neuroni prossimi ad aree emorragiche, contusive, infartuali, possono accumularsi, in forma di granuli, ematina (pigmento emoglobinico), ferro, calcio e altri elementi in tracce (fosforo, zolfo, rame, zinco): la natura dei depositi può essere riconosciuta mediante colorazioni specifiche; il meccanismo di accumulo non è noto. I depositi possono essere legati a membrane o liberi nel citoplasma. Indipendentemente dalle condizioni suddette, in alcune rare malattie si possono riscontrare incrostazioni ferrose (malattia di Hallervorden-Spatz) e di calcio (malattia di Fahr e soggetti con ipoparatiroidismo), con una distribuzione selettiva a livello di alcune aree encefaliche. Macrocalcificazioni della corteccia cerebrale e, in minor misura, della sostanza bianca sottostante possono riscontrarsi in una condizione malformativa (angiomatosi encefalotrigeminale di Sturge-Weber) associata ad anomalie del circolo vascolare meningeo.
Un processo che si manifesta con l'età, in misura individualmente variabile e di cui è difficile definire con certezza la natura patologica, è l'accumulo di prodotti del catabolismo cellulare in forma di lipidi, proteine, carboidrati ed enzimi idrolitici stipati nei corpi residui lisosomiali (lipofuscina): i neuroni di alcuni nuclei encefalici (talamo, corpo genicolato laterale, nucleo dentato) ne sono fisiologicamente ricchi, le cellule di Purkinje del cervelletto, invece, ne sono generalmente prive; certamente patologico ne è l'accumulo, che si manifesta in corso di alcuni deficit enzimatici lisosomiali e nelle malattie degenerative, spesso, ma non sempre, associato ad atrofia cellulare (atrofia pigmentaria).
Nei disordini da difetti degli enzimi lisosomiali (lipidosi, mucopolisaccaridosi, mucolipidosi e disordini del metabolismo dei glicoconiugati) si verifica l'accumulo intraneuronale di materiale con composizione biochimica variabile e specifica in relazione ai processi metabolici alterati. Tale accumulo comporta la distensione e il rigonfiamento neuronale. Le caratteristiche istochimiche del materiale accumulato consentono di riconoscere a quale classe di composti esso appartenga, e quelle ultrastrutturali (avvolgimenti spiraliformi, corpi zebrati, ecc.) sono abbastanza tipiche per ciascun disordine.
L'identificazione microscopica di corpi inclusi virali, nucleari o citoplasmatici ha avuto molta importanza per dimostrare la natura di un'infezione del sistema nervoso sino all'avvento dei metodi di biologia molecolare. Le inclusioni nucleari vengono abitualmente classificate, in relazione alla loro struttura, in tipo A e tipo B secondo Cowdry. Inclusioni eosinofile, che ultrastrutturalmente appaiono contenere materiale virale, vengono riscontrate soprattutto nell'encefalite necrotizzante da Herpes simplex e nella panencefalite sclerosante subacuta. Nell'infezione da Citomegalovirus, grosse inclusioni circondate da un alone otticamente vuoto conferiscono ai nuclei un aspetto caratteristico, definito a ‛occhio di civetta'. Il riscontro di inclusi citoplasmatici è meno frequente: alcuni, costituiti da materiale di origine virale e localizzati in prevalenza nelle cellule piramidali del corno di Ammone e in quelle di Purkinje del cervelletto, sono patognomonici della rabbia (corpi di Negri). Talora, il materiale incluso è formato da involucri con aspetto ‛a proiettile', costituiti dalle membrane cellulari, all'interno dei quali non è più dimostrabile materiale virale (corpi di Lyssa).
b) Processi assonali regressivi e progressivi
In seguito a una lesione della fibra nervosa, a carico della porzione distale dell'assone si verifica quel processo di disfacimento tendente a progredire in senso centrifugo, che fu originariamente descritto nei nervi periferici (degenerazione walleriana), con meccanismo e quadro morfologico simili. L'assone inizialmente appare rigonfio e pieno di materiale filamentoso, quindi va incontro a coartazione con condensazione dell'assoplasma, degenerazione degli organuli assoplasmatici e, infine, frammentazione (v. fig. 22). Va sottolineato che la degenerazione assonale è seguita da quella mielinica. I resti tissutali vengono rimossi da cellule ‛spazzino' (macrofagi). Il processo di degenerazione della porzione dell'assone a monte della lesione procede in senso centripeto, cioè dalla lesione verso il corpo cellulare (degenerazione retrograda), e si manifesta inizialmente con una riduzione del diametro e poi con la progressiva frammentazione dell'assone. Se la lesione è prossima al corpo cellulare, anche quest'ultimo degenera. La degenerazione walleriana a livello centrale avviene in seguito a lesioni focali di diversa eziologia o a sezione meccanica dei tessuti; a livello periferico tale degenerazione consegue, tipicamente, all'interruzione traumatica del nervo, ma anche a processi di natura vascolare, infiammatoria o tossico-metabolica.
Quando il corpo cellulare del neurone, per cause varie, va incontro ad atrofia, anche l'assone presenta un processo di lenta, progressiva atrofizzazione, sino all'eventuale disintegrazione, che inizia nella sua porzione più distale e ascende verso il corpo cellulare (atrofia semplice retrograda, dying back).
Un processo caratterizzato morfologicamente da una serie di grossi rigonfiamenti tondeggianti lungo il decorso assonale (distrofia neuroassonale) è tipico di alcune rare condizioni osservate nell'infanzia, ma può realizzarsi anche nell'adulto in corso di carenza di vitamina E, nell'intossicazione da triortocresilfosfato e nell'invecchiamento. Rigonfiamenti fusiformi, singoli (formazioni a torpedine), della porzione prossimale dell'assone delle cellule di Purkinje compaiono spesso nelle malattie degenerative croniche.
Nella fase di recupero di una lesione del nervo periferico, a livello del moncone prossimale si verifica una vigorosa rigenerazione degli assoni interrotti, da cui gemmano ramificazioni capaci di attivo accrescimento. Se la porzione distale della fibra è sufficientemente vicina, le ramificazioni neoformate prendono contatto con essa e le cellule di Schwann proliferano per formare un aggregato cilindrico (bande di Bügner) entro il quale gli assoni neoformati possono crescere. La successiva rimielinizzazione di questi assoni completa la fase riparativa. La rigenerazione assonale a livello del sistema nervoso centrale è trascurabile.
c) Processi mielinici regressivi e progressivi
In alcuni disordini da difetti enzimatici, geneticamente determinati, si verifica un disturbo della formazione della mielina (dismielinizzazione), così che le guaine mieliniche a livello centrale e periferico appaiono estremamente sottili. Un processo di distruzione delle guaine mieliniche primitivamente normali (demielinizzazione primaria; v. fig. 23) si osserva, invece, in diverse condizioni patologiche sia del SNC (sclerosi multipla, leucoencefalopatia acuta disseminata, edema cronico) che di quello periferico (neuropatia diabetica, sindrome di Guillain-Barré, neuropatia uremica) che interessano primariamente la mielina o le cellule che la formano. Il processo ha espressione segmentaria, può colpire cioè in parte o per intero un internodo, o alcuni internodi, in modo variabile e irregolare. Possono così realizzarsi un quadro di demielinizzazione completa oppure uno di demielinizzazione parziale. Nel primo, le lamelle mieliniche vanno incontro, attraverso una serie complessa di alterazioni, a disintegrazione completa e nello stadio finale, a livello del SNC, i tessuti circostanti la lesione presentano la scomparsa degli oligodendrociti e la formazione di una cicatrice astrogliale, mentre nel nervo periferico, a livello della lesione, si riscontra proliferazione delle cellule di Schwann e del connettivo proprio del nervo (fibrosi). Nella demielinizzazione parziale, una parte o l'intero internodo presenta una guaina mielinica assottigliata, ma questa alterazione non è associata alla morte degli elementi che formano la mielina. Un processo di distruzione della mielina può manifestarsi anche secondariamente alla sofferenza, per cause varie, dell'assone e/o del neurone (demielinizzazione secondaria), così come può realizzarsi in lesioni, per esempio l'infarto, che interessano tutti gli elementi del tessuto nervoso.
A livello del nervo periferico, strutture che ricordano la superficie di taglio di una cipolla (formazioni a bulbo di cipolla), formate da strati multipli, concentrici, di cellule di Schwann disposte intorno a una fibra nervosa, rappresentano lo stadio avanzato di fasi ripetute di demielinizzazione e tentativi di rimielinizzazione tipici della neuropatia interstiziale ipertrofica familiare e di alcune neuropatie metaboliche ereditarie, ma frequenti anche in diverse neuropatie croniche demielinizzanti.
d) Processi astrocitari regressivi e progressivi
Nell'ipossia/ischemia acuta e nelle lesioni distruttive acute si manifesta il processo di necrosi acuta, durante il quale l'astrocita può passare dallo stadio iniziale, reversibile, di rigonfiamento del corpo cellulare e frammentazione dei prolungamenti cellulari (clasmatodendrosi) a una fase terminale di possibile disintegrazione.
L'invecchiamento e i processi degenerativi cronici si accompagnano all'accumulo di lipofuscina nel citoplasma astrocitario (degenerazione cronica). Negli anziani si osserva, inoltre, l'ammassarsi entro i prolungamenti astrocitari, prevalentemente in regione subpiale e subependimale, di formazioni tonde (corpi amilacei) i cui principali costituenti sono polimeri di glucosio (poliglucosani). Morfologia e origine del tutto sovrapponibili a quelle delle analoghe inclusioni neuronali hanno le inclusioni astrocitarie virali. Negli errori congeniti del metabolismo (malattia di Tay-Sachs, mucopolisaccaridosi, ecc.) l'accumulo di specifici metaboliti anomali nel citoplasma degli astrociti presenta analogia strutturale e biochimica con gli stessi processi che si osservano a livello neuronale.
Marcatamente negli infarti e nelle alterazioni traumatiche, ma praticamente in tutte le lesioni del SNC, i processi riparativi tissutali sono caratterizzati da incremento del numero (proliferazione) e del volume (ipertrofia) degli astrociti e delle loro fibrille intracellulari (astrocitosi; v. fig. 24). La proliferazione, stimolata da diversi fattori, avviene per divisione mitotica già nelle prime 24 ore dopo un evento lesivo. L'ipertrofia riguarda sia il corpo che i prolungamenti cellulari; si verifica un forte incremento del numero degli organuli citoplasmatici, dell'attività degli enzimi ossidativi e, in particolare, dei filamenti intermedi della proteina gliale fibrillare acida (GFAP). Gli astrociti reattivi partecipano attivamente alla fagocitosi, all'attivazione di enzimi proteolitici tissutali, al riassorbimento dell'edema cerebrale, alla ricostituzione delle membrane basali e, mediante i loro prolungamenti ripieni di GFAP, alla formazione della cicatrice gliale. La sola ipertrofia astrocitaria, senza proliferazione e con abnorme produzione di fibrille astrocitarie (astrogliosi o gliosi fibrillare), rappresenta il correlato morfologico di processi riparativi di vecchia data. In aree di astrogliosi cronica e nei tumori astrocitari ed ependimali a lenta crescita, entro i prolungamenti astrocitari si riscontrano, frequentemente, formazioni filamentose di dimensioni variabili, parzialmente positive alla reazione immunocitochimica per la GFAP, dette fibre di Rosenthal.
Nella malattia di Wilson (degenerazione epatolenticolare) si osservano astrociti abnormemente grossi, con nucleo irregolare, detti astrociti di Alzheimer (senza relazione con la omonima malattia), che vengono distinti in quelli di tipo I, con abbondante citoplasma e prolungamenti brevi, e quelli di tipo II, con nucleo gigante, irregolare, uno o due nucleoli e citoplasma scarso. Le modificazioni degli astrociti di tipo II rappresentano un adattamento all'esigenza di metabolizzare l'ammoniaca in eccesso: solo dopo la saturazione degli astrociti o il decadimento di questa loro capacità metabolica si manifesta l'encefalopatia epatica. Nella malattia di Wilson si riscontrano, inoltre, altre cellule, verosimilmente astrociti, con corpo abnormemente ampio e nucleo relativamente piccolo e denso in posizione periferica (cellule di Opalski).
e) Processi oligodendrogliali, microgliali e monocitari
Lo spettro di reazioni dell'oligodendroglia a eventi lesivi è limitato. I processi di significato certamente patologico sono rappresentati dalla presenza di inclusioni nucleari virali (specialmente nella panencefalite sclerosante subacuta) e di materiale d'accumulo nei deficit enzimatici lisosomiali. La perdita di oligodendrociti descritta nella sclerosi multipla non è un reperto costante. Altri processi descritti (rigonfiamento acuto, satellitosi) sembrano artefatti o comunque privi di significato patologico. Devono essere ricordati, tuttavia, il danno tossico diretto subito dagli oligodendrociti a opera di vari composti chimici (isoniazide, 6-amminonicotinammide, esaclorofene), che si manifesta inizialmente con la vacuolizzazione della guaina mielinica e poi con la morte dell'oligodendrocita, e la vulnerabilità degli oligodendrociti alle radiazioni ionizzanti, che si manifesta con degenerazione e morte tardive.
Diversi processi lesivi del sistema nervoso centrale causano, entro 24-48 ore dal loro inizio, attivazione delle cellule microgliali: queste migrano verso il sito della lesione, si moltiplicano per mitosi, assumono una forma tondeggiante, fagocitano i corpi estranei e i resti dei tessuti in disfacimento e il loro citoplasma appare ripieno di materiale lipidico (cellule schiumose) e/o di pigmenti ematici. Il modo in cui le cellule ripiene di materiale fagocitato abbandonano il cervello non è chiaro. Una intensa proliferazione della microglia in forma di elementi a bastoncello (microglia a bastoncello) viene considerata tipica delle encefaliti e del danno cerebrale subacuto. Focolai di cellule microgliali (noduli microgliali), macrofagi e spesso cellule giganti multinucleate si riscontrano frequentemente nell'encefalite dovuta a infezione da virus HIV-1 e 2, responsabili della sindrome da immunodeficienza acquisita (AIDS); gruppi di cellule microgliali attivate si trovano spesso in prossimità di neuroni lesi o morti.
7. Neuropatologia generale: processi patologici tissutali
I processi cellulari elementari sopra descritti possono non solo manifestarsi in misura tale da provocare modificazioni macroscopiche delle strutture nervose, ma associarsi, inoltre, a processi a carico delle meningi, dei vasi e degli elementi connettivali. Tali processi hanno importanza, oltre che per la diagnostica neuropatologica, anche per quella clinica, in quanto la maggioranza di essi è rilevabile con le moderne metodiche d'immagine, quali la tomografia assiale computerizzata e la risonanza magnetica nucleare.
Atrofia. - L'atrofia è caratterizzata da diminuzione di volume, regionale o generalizzata, delle strutture cerebrali, del tronco-encefalo e/o del midollo spinale. La forma di più frequente osservazione è quella che colpisce la corteccia cerebrale nei processi dementigeni, in cui le circonvoluzioni appaiono assottigliate e i solchi e le scissure dilatati (v. fig. 25). Il substrato istologico di questo processo è la diffusa scomparsa di neuroni, associata a gliosi di variabile intensità. L'atrofia a livello del tronco e del cervelletto ricorre soprattutto in condizioni degenerative plurisistemiche. A livello del midollo spinale un aspetto atrofico può essere secondario alla degenerazione dei tratti motori, o dei cordoni sensitivi posteriori, associata o meno a degenerazione dei neuroni delle corna anteriori.
Dilatazione ventricolare. - Questo processo è ben evidente nelle forme gravi, altrimenti richiede una certa esperienza per essere individuato. La dilatazione degli spazi ventricolari può essere secondaria all'atrofia della sostanza bianca e/o delle strutture grigie periventricolari, o a un accumulo di liquor, per ostacolo (tumorale, malformativo, infiammatorio, ecc.) alla sua circolazione e/o riassorbimento: questa seconda condizione viene definita idrocefalo (v. fig. 26).
Rigonfiamento ed edema. - Potenziali complicazioni a mediazione vascolare di numerose condizioni patologiche del sistema nervoso, di rilevante importanza clinica, sono rappresentate dal rigonfiamento e dall'edema tissutali. Questi fenomeni possono manifestarsi, isolatamente o in associazione, sia a livello encefalico che spinale, in forma localizzata o diffusa, ma hanno maggiore significato clinico-neuropatologico nella localizzazione encefalica, che si accompagna a incremento del volume encefalico. A quest'ultimo fenomeno consegue la dislocazione di un corrispondente volume di liquido cerebrospinale dagli spazi liquorali intracranici (ventricoli e spazi subaracnoidei) verso quelli spinali; quando questo meccanismo di compenso viene saturato, la pressione intracranica aumenta, e compaiono fenomeni di dislocazione e deformazione delle strutture encefaliche. In questa catena di eventi il danno secondario del tronco-encefalo, sede di importanti nuclei regolatori delle funzioni vitali dell'organismo, può avere conseguenze letali.
Un incremento volumetrico cerebrale acuto, legato a dilatazione generalizzata del microcircolo (rigonfiamento cerebrale congestizio), può manifestarsi soprattutto nei traumi cranici. Questo fenomeno è causato da una marcata vasodilatazione cerebrale in combinazione con l'incremento della pressione arteriosa regionale. Il rigonfiamento cerebrale, d'altra parte, può indurre un innalzamento della pressione intracranica con progressivo ostacolo alla circolazione cerebrale e ischemia. Persistendo quest'ultima si instaura una paralisi vasomotoria, cui conseguono rottura della BEE, edema tissutale e un ulteriore incremento della pressione intracranica sino all'arresto della perfusione e alla morte cerebrale.
Il contenuto normale d'acqua è pari a circa l'80% del peso fresco della sostanza grigia e al 68% del peso fresco di quella bianca; per valori superiori si realizza una condizione di edema cerebrale. Macroscopicamente questa si distingue dal rigonfiamento congestizio perché la superficie di taglio del cervello, a fresco, appare umida e lucente. Nelle preparazioni istologiche le regioni edematose appaiono pallide rispetto ai tessuti normali; l'edema è contenuto prevalentemente negli astrociti, a livello della sostanza grigia e, in maggior quantità, negli spazi extracellulari tra le fibre mieliniche a livello di quella bianca. In relazione a caratteristiche patogenetiche e morfologiche vengono distinti cinque tipi di edema: 1) vasogenico (tipico di tumori, ascessi, contusioni e infarti), in cui acqua, elettroliti e proteine si accumulano negli spazi extracellulari in seguito a danno fisico della BEE o ad abnorme incremento dell'attività di trasporto transcellulare (pinocitosi) delle cellule endoteliali capillari; 2) citotossico (tipico delle fasi iniziali dell'ischemia e dell'intossicazione da esaclorofene e composti del metilmercurio), così detto perché l'acqua si accumula, inizialmente, a livello intracellulare a causa dell'alterazione dei meccanismi di osmoregolazione cellulare, principalmente della pompa di membrana Na+-K+ ATP-dipendente; persistendo le condizioni patogene responsabili dell'edema, a quella citotossica, entro poche ore, si aggiunge una componente vasogenica; 3) idrostatico, che è quello associato al rigonfiamento congestizio in cui, per l'elevata pressione intravascolare, l'acqua fuoriesce dai vasi e diffonde nello spazio extracellulare; 4) ipo-osmotico, osservato quasi esclusivamente in condizioni di riduzione della pressione colloido-osmotica del plasma, conseguente, in genere, a eccessiva somministrazione intravenosa di soluzioni ipotoniche, o a carente secrezione di ormone antidiuretico; 5) interstiziale liquorale, rappresentato dall'accumulo di liquido cefalorachidiano nei tessuti periventricolari, frequente nell'idrocefalo ostruttivo acuto.
Necrosi. - Questo processo, che consiste, essenzialmente, nella distruzione del tessuto nervoso, può conseguire alle cause più diverse, quali l'insufficiente apporto ematico (ischemia), un trauma, un'infezione, ecc. I processi necrotizzanti di vecchia data culminano nella formazione di cavità intracerebrali di dimensioni variabili. Nella fase iniziale la necrosi è in genere riconoscibile per la ridotta consistenza dei tessuti accompagnata da edema. La necrosi, che microscopicamente è caratterizzata dalla scomparsa degli elementi nervosi, nelle sue fasi iniziali è sempre mediata, indipendentemente dalla causa scatenante, da cellule ematiche (granulociti neutrofili, monociti, linfociti, ecc.).
Emorragia. - Questo processo, che consiste nella fuoriuscita di sangue dai vasi arteriosi e/o venosi, può avere cause varie - traumatiche, malformative, neoplastiche, infettive, ecc. - che comportano, comunque, la rottura della parete vasale. L'emorragia può essere massiva e dare segni di sé in breve tempo, come si verifica, generalmente, nella lesione di un vaso arterioso, o essere circoscritta e manifestarsi lentamente, come è più facile osservare nelle emorragie venose. Le emorragie possono avvenire a vari livelli: schematicamente, tra involucro osseo e dura (epidurale); tra dura e aracnoide (subdurale); tra aracnoide e pia (subaracnoidea); entro i tessuti del SN (intraparenchimale). Anche gli spazi liquorali possono essere sede, primaria o secondaria, di emorragie. Le emorragie di più frequente riscontro sono quelle da rottura di dilatazioni malformative (aneurismi) della parete delle arterie cerebrali, per lo più subaracnoidee; secondarie a crisi ipertensive, localizzate tipicamente a livello dei gangli basali; post-traumatiche, che danno origine a ematomi (raccolte di sangue) epidurali e/o subdurali.
Demielinizzazione e dismielinizzazione. - La demielinizzazione - che consiste nella perdita delle guaine mieliniche a livello centrale e/o periferico - si verifica a causa di processi acquisiti, che agiscono, cioè, su una mielina primariamente normale e vengono detti mielinoclastici (mielinodistruttivi); la dismielinizzazione si osserva in particolari condizioni, per lo più familiari, dette leucodistrofie, caratterizzate da un'alterazione, geneticamente determinata, dei meccanismi necessari al normale rinnovo della mielina. La malattia demielinizzante più frequente è la sclerosi multipla o sclerosi a placche, che ha natura mielinoclastica. La demielinizzazione può manifestarsi a livello della sostanza bianca in qualsiasi parte del SN, con perdita, in aree limitate o diffusamente, del suo normale aspetto bianco-lucente e delle sue proprietà tintoriali nei preparati istologici, e con modificazione delle sue caratteristiche nelle immagini di risonanza magnetica nucleare.
Infiammazione. - Questo processo consiste essenzialmente nell'invasione, localizzata a livello perivascolare o disseminata con infiltrazione dei tessuti, da parte di cellule ematiche della serie leucocitaria, di tipo variabile a seconda della fase e della causa del processo infiammatorio: per esempio, prevalentemente granulociti neutrofili nella fase acuta, e linfociti e mononucleati in quella cronica delle infezioni batteriche; linfociti e plasmacellule a livello perivascolare nelle infezioni virali; ecc. La presenza di elementi infiammatori a livello del SN non è, comunque, legata solo a processi infettivi, in quanto quasi tutti gli eventi patogeni acquisiti, dall'ischemia ai tumori, alle malattie demielinizzanti, si accompagnano a una risposta infiammatoria dei tessuti nervosi.
Spongiosi. - Si tratta di un processo caratterizzato da una degenerazione microcistica della sostanza grigia e/o di quella bianca del SNC. Spongiosi della sostanza grigia si osserva in malattie dementigene e consiste nella presenza di bolle e aree, apparentemente vuote, negli astrociti e nei neuroni. Spongiosi della sostanza bianca può essere riscontrata, per esempio, in processi degenerativi subacuti o in corso di AIDS, a livello del midollo spinale. Le modalità di insorgenza di questo processo non sono chiare.
Tumori. - I tumori sono formazioni occupanti spazio prodotte da proliferazione incontrollata di elementi cellulari atipici (neoplasie) oppure da moltiplicazione di elementi cellulari normalmente differenziati, di origine embrionale o meno (cisti, teratomi, amartomi). I tumori vengono distinti in primari e secondari a seconda che siano originati, rispettivamente, da elementi cellulari propri del sistema nervoso o da elementi extranevrassiali. Le neoplasie primarie derivano da cellule che perdono o non acquisiscono la maturità morfofunzionale e crescono in quantità incontrollata e con qualità abnormi; le cellule che danno origine a cisti, teratomi, amartomi, presentano invece caratteristiche morfofunzionali di maturità, ma si moltiplicano in sedi anomale e in quantità eccessiva. I tumori causano danno alle strutture del SN principalmente attraverso un'azione meccanica, compressiva (effetto massa; v. fig. 27). I tumori primari del SN e le cellule da cui originano sono elencati nella tab. II; le forme più frequenti sono quelle di origine gliale.

Processi vascolari. - Le alterazioni regressive dei vasi sanguigni costituiscono un vasto e importante capitolo della neuropatologia speciale, il cui esame esula dagli scopi della presente trattazione. Modificazioni di tipo progressivo si realizzano nei processi riparativi delle lesioni del tessuto nervoso e sono rappresentate dalla neoformazione e proliferazione dei capillari a cui può associarsi una imponente proliferazione connettivale. Condizioni di anossia e alcuni disordini metabolici si accompagnano a proliferazione e rigonfiamento delle cellule dell'endotelio capillare, che sembrano rappresentare l'alterazione più rilevante rispetto a quelle a carico delle cellule neurogliali. In alcuni processi infiammatori encefalici i fenomeni a livello vascolare, prevalentemente venulare, hanno un ruolo primario, in quanto la comparsa di infiltrati perivascolari di linfociti precede quella di alterazioni neuronali e microgliali.
Processi meningei. - Le leptomeningi (aracnoide e pia) partecipano spesso ai processi infettivi (meningite) e a quelli emorragici (emorragia subaracnoidea): in entrambi i casi può formarsi un tessuto cicatriziale fibro-connettivale a livello degli spazi subaracnoidei, con conseguente ostacolo alla circolazione e al riassorbimento liquorale.
Gli spazi subaracnoidei possono essere anche sede preferenziale di insediamento e diffusione di processi tumorali (carcinomatosi meningea; leucemia).
Gli spazi perivascolari rivestiti dalla pia (spazi di Virchow-Robin), intorno ai vasi cerebrali superficiali, svolgono un ruolo determinante nella diffusione delle infezioni leptomeningee verso il tessuto cerebrale.
Raccolte ematiche (ematomi) ed essudazioni infiammatorie subdurali vanno incontro a processi di incapsulamento e organizzazione fibrotica per attivazione dei fibroblasti della superficie durale interna. Nella cronicizzazione di lesioni severe, ma anche in soggetti di età avanzata, aree meningee fibrotiche, soprattutto a livello lombo-sacrale, possono andare incontro a calcificazione e persino a ossificazione.
Processi ependimali e dei plessi corioidei. - Le cellule ependimali possiedono una reattività relativamente limitata. Esse vanno incontro a disintegrazione in quasi tutti i processi patologici che interessano i tessuti periventricolari e/o le cavità ventricolari o in condizioni di sovradilatazione di queste ultime (idrocefalo). Nell'idrocefalo, a livello delle discontinuità dello strato ependimale si formano ammassi di cellule gliali subependimali proliferanti (gemme gliali), che protrudono nello spazio liquorale. In condizioni infiammatorie, tipicamente nella sifilide, oppure nelle emorragie intraventricolari pregresse, l'imponente proliferazione di gemme gliali può conferire un aspetto granuleggiante alla superficie ventricolare (ependimite granulare).
Le modificazioni istopatologiche dei plessi corioidei, che si osservano soprattutto in relazione all'invecchiamento, comprendono l'atrofia degli elementi epiteliali, la sclerosi ialina della parete dei vasi sanguigni e la calcificazione dello stroma connettivale. Sempre nel cervello senile possono talora verificarsi degenerazione cistica dello stroma e microemorragie, che rappresentano la base per il deposito di materiale lipidico degenerato e cristalli di colesterolo calcificato, da cui possono avere origine quelle masse tumorali benigne note come colesteatomi o xantomi: il significato clinico di queste alterazioni è scarso. Tumori papillari dei plessi si associano, invece, a iperproduzione di liquido cefalo-rachidiano con conseguente idrocefalo.
8. Principali sindromi neurologiche lesionali
a) Lobi frontale, parietale, temporale e occipitale
Le sindromi neurologiche da lesione del lobo frontale comprendono, schematicamente, la paralisi a livello dell'emilato corporeo controlaterale alla corteccia motoria lesa; l'incapacità o il disturbo dell'espressione verbale (afasia espressiva) per lesione dell'area di Broca nell'emisfero dominante; le alterazioni del comportamento, della personalità, dell'attenzione e dell'ideazione. Lesioni ampie del lobo frontale possono, ovviamente, accompagnarsi al contemporaneo manifestarsi di più di una sindrome o di diversi aspetti di ciascuna di esse. Deve essere ancora ricordato che nelle lesioni della porzione frontale del corpo calloso si manifesta una disconnessione tra l'impulso a iniziare i movimenti necessari per la marcia e l'esecuzione degli stessi: i pazienti, pur avendo una forza normale, sono incapaci di deambulare (aprassia della marcia). Le cause più frequenti di lesione monolaterale del lobo frontale sono le malattie cerebrovascolari, i tumori, gli ascessi; l'interessamento bilaterale, meno frequente, si può avere, invece, nei traumi cranici, nell'idrocefalo, in alcune malattie degenerative dementigene, nel vasospasmo di entrambe le arterie cerebrali anteriori, possibile in corso di emorragia subaracnoidea, e, raramente, nei tumori del corpo calloso e in quelli del piano fronto-orbitario.
Le sindromi da lesione del lobo parietale consistono nella perdita della sensibilità tattile discriminativa nell'emilato corporeo controlaterale alla lesione; nell'incapacità di comprendere il linguaggio parlato (afasia sensoriale) associata a incapacità nel calcolo aritmetico (acalculia), nella scrittura (agrafia) e nella lettura (alessia), in caso di lesione nell'emisfero dominante; nell'indifferenza agli stimoli provenienti dall'emilato corporeo controlaterale alla lesione e nella perdita della percezione della propria immagine corporea, in caso di interessamento dell'emisfero non dominante; sempre nel caso di lesione nell'emisfero non dominante, nell'incapacità di eseguire a richiesta semplici sequenze organizzate di movimenti (aprassia costruttiva), e nell'incapacità di vestirsi (aprassia dell'abbigliamento); in deficit del campo visivo. L'interessamento del lobo parietale è generalmente monolaterale e le lesioni più frequenti sono le ischemie nel territorio dell'arteria cerebrale media, gli ematomi traumatici, gli ascessi e i tumori. La lesione contemporanea di entrambi i lobi è estremamente rara e si osserva, quasi esclusivamente, in forma di lesione ischemica conseguente a episodi ipotensivi generalizzati.
Le lesioni del lobo temporale possono manifestarsi con afasia sensoriale (eloquio fluente) e deficit della memoria verbale, nel caso di lesione del lobo dominante; con deficit della memoria visiva per coinvolgimento del lobo non dominante; in caso di lesione bilaterale della corteccia uditiva con sordità corticale (la lesione monolaterale è infatti asintomatica, perché l'udito ha una rappresentazione corticale bilaterale); con comportamento aggressivo o antisociale, che può associarsi a incapacità di memorizzare informazioni nuove per danno del sistema limbico; con deficit del campo visivo. Le manifestazioni più frequenti di sofferenza del lobo temporale sono, comunque, crisi epilettiche parziali complesse (crisi temporali) che si manifestano come disturbi della memoria, episodi di déjà vu o jamais vu, allucinazioni transitorie olfattive, gustative e visive, associate o meno a movimenti elementari degli arti e del tronco. Una lesione monolaterale del lobo temporale, nella maggioranza dei casi ha origine cerebrovascolare o neoplastica; gli ascessi, relativamente frequenti, si realizzano per lo più per estensione di processi infettivi dell'orecchio medio e/o della mastoide. Una sofferenza bilaterale del lobo temporale si può osservare tipicamente nell'encefalite virale da Herpes simplex, in corso di vasculopatie con episodi di ipotensione profonda e prolungata e nell'encefalopatia di Wernicke da carenza nutrizionale di vitamina B1 (tiamina); raramente come conseguenza di eventi traumatici.
I principali sintomi e segni di lesione del lobo occipitale comprendono la perdita della vista in un emicampo visivo o la cecità, a seconda che l'interessamento della corteccia visiva sia monolaterale o bilaterale, rispettivamente; allucinazioni visive; incapacità di riconoscere gli stimoli visivi (agnosia visiva). Lesioni del lobo occipitale si verificano nella maggioranza dei casi in corso di malattia cerebrovascolare per compromissione del distretto circolatorio vertebro-basilare.
b) Gangli basali
Le malattie dei gangli basali si accompagnano prevalentemente a segni e sintomi motori distinti in ‛negativi' e ‛positivi'. I primi consistono essenzialmente in perdita o rallentamento dei movimenti e in anomalie posturali; i secondi in movimenti involontari. Alcune forme di questi disordini del movimento hanno un definito correlato neuropatologico. Si riconoscono: 1) movimenti coreici (da lesione dei nuclei caudato e putamen), che consistono in contrazioni muscolari involontarie, irregolari e imprevedibili che interessano l'intera muscolatura del corpo o una sua parte; 2) movimenti distonici (prevalentemente da lesione monolaterale dei gangli basali), che consistono in contrazioni prolungate a carico dei gruppi muscolari di singoli distretti corporei e producono frequentemente torsioni, movimenti ripetitivi e posture anomale; 3) ballismo (da lesione bilaterale del nucleo subtalamico), in cui si manifestano movimenti involontari violenti, che interessano la muscolatura prossimale della radice degli arti, soprattutto superiori; l'interessamento monolaterale del nucleo subtalamico comporta movimenti anomali controlaterali (emiballismo) che ricordano quelli compiuti per un lancio; 4) atetosi (da lesione del globo pallido, substantia nigra e corteccia cerebrale), in cui i movimenti involontari sono lenti, aritmici, irregolari, di modesta ampiezza e coinvolgono prevalentemente gli arti, soprattutto quelli superiori; 5) tremore, che consiste in movimenti involontari oscillatori, regolari, ritmici, conseguenti alla contrazione alterna di muscoli agonisti e antagonisti. Il tremore non è un segno di esclusiva pertinenza delle lesioni dei gangli basali, ma nella varietà che compare a riposo costituisce, insieme alla rigidità e al rallentamento ideo-motorio, una delle principali manifestazioni della malattia di Parkinson. La sindrome parkinsoniana è conseguenza del deficit di innervazione dopamminergica striatale secondario alla degenerazione dei neuroni pigmentati di una porzione (pars compacta) della substantia nigra, che forniscono tale innervazione.
Si conoscono diverse condizioni degenerative che interessano primariamente i gangli basali: tra queste la malattia di Parkinson e quella di Huntington sono le forme più frequenti; tuttavia, numerose altre condizioni di natura tossica, vascolare, metabolica implicano l'interessamento dei gangli basali.
c) Talamo
Ai fini anatomo-funzionali, ciascun talamo può essere diviso in 4 regioni - anteriore, posteriore, mediale e laterale - aventi ognuna specifiche caratteristiche. Si distinguono, pertanto, segni e sintomi da interessamento del talamo anteriore (dell'emisfero dominante), rappresentati da afasia, inattenzione e disturbi mnesici e, se è coinvolta la regione subtalamica, emiballismo controlaterale; del talamo posteriore, rappresentati da anestesia associata a una sensazione di dolore parossistico (anestesia dolorosa) nell'emisoma controlaterale e deficit del campo visivo; del talamo mediale, rappresentati da deficit della memoria recente, apatia, agitazione, deficit dell'attenzione, turbe del sonno, talora coma; del talamo laterale, rappresentati solo da anestesia e spesso da una sensazione di dolore parossistico nell'emisoma controlaterale. Il dolore talamico è relativamente raro, ma rappresenta tuttavia un sintomo altamente specifico: descritto come molto sgradevole e talora urente, viene riferito alla superficie corporea ed è esacerbato dalla minima stimolazione tattile della cute; compare, in genere, nella fase di recupero dei deficit sensitivi. Le lesioni talamiche più frequenti sono quelle ischemiche e quelle emorragiche in corso di malattie cerebrovascolari, seguite da quelle neoplastiche.
d) Asse ipotalamo-ipofisario
Schematicamente, una lesione ipotalamica può manifestarsi con una delle 4 sindromi seguenti: ipotalamica anteriore, che si rivela con una rapida, accentuata perdita di peso corporeo (cachessia), diabete insipido caratterizzato da abnorme assunzione di acqua (polidipsia) e abnorme emissione di urine (poliuria), e ipotermia; ipotalamica posteriore, che può manifestarsi con ipotermia, apatia, o coma; ipotalamica mediale, caratterizzata da poliuria e polidipsia per inappropriata secrezione dell'ormone antidiuretico, obesità, deficit della memoria, aggressività; ipotalamica laterale, caratterizzata da insufficiente assunzione d'acqua, emaciazione, apatia. Le cause più frequenti di lesioni ipotalamiche sono i traumi cranici, le malattie vascolari e i tumori.
I segni e sintomi di lesione dell'ipofisi anteriore sono riconducibili, in generale, a iper- o iposecrezione degli ormoni ipofisari. Le sindromi ipersecretive sono per lo più legate alla presenza di tumori (adenomi) ipofisari che rappresentano le lesioni ipofisarie più comuni. Frequentemente le manifestazioni ormonali sono precedute dai segni riferibili all'effetto massa degli adenomi, tra i quali tipico è il deficit bilaterale dell'emicampo visivo temporale (emianopsia bitemporale) da compressione del chiasma ottico.
e) Troncoencefalo e cervelletto
Esistono sindromi cliniche da lesione del tronco (sindromi alterne) molto diverse in rapporto al livello (mesencefalico, pontino, bulbare) interessato, la cui descrizione dettagliata esula dagli scopi della presente esposizione. In pratica, elementi comuni delle diverse forme sono: deficit di uno o più nervi cranici, paralisi e deficit delle sensibilità all'emilato controlaterale alla lesione e talora segni cerebellari omolaterali alla lesione. I disordini che più frequentemente interessano il troncoencefalo sono quelli vascolari, i tumori e i traumi.
I principali segni e sintomi di lesione cerebellare comprendono la perdita del tono muscolare; i disturbi della coordinazione che possono manifestarsi in condizioni statiche (atassia statica), come incapacità di mantenere una determinata posizione, associata a continue oscillazioni intorno al punto di equilibrio, oppure nell'imprecisa esecuzione di movimenti volontari (atassia dinamica); la perdita della forza di grado lieve e l'incapacità di compiere movimenti associati; i disturbi dell'equilibrio e della deambulazione. Disturbi del movimento si osservano anche a carico della motilità oculare (nistagmo) e dei movimenti necessari all'articolazione della parola (disartria). Tra le diverse possibili sindromi cerebellari le principali sono quella emisferica, caratterizzata da disturbi della coordinazione dei movimenti omolaterali all'emisfero leso, instabilità nella marcia, disartria e nistagmo; e quella vermiana, contraddisitinta prevalentemente da disturbi della stazione eretta (astasia) e della marcia (abasia). I disordini che più frequentemente interessano il cervelletto sono quelli vascolari, i tumori, la sclerosi multipla e diverse malattie degenerative.
f) Midollo spinale
Le lesioni del midollo spinale si manifestano con una combinazione di segni e sintomi motori, sensitivi e autonomici. In rapporto alla sede, extra- o intramidollare, e al livello cranio-sacrale della lesione, individuabili grazie all'anamnesi e all'esame neurologico, si realizzano le seguenti sindromi cliniche: 1) transezione midollare completa, che si manifesta con paralisi, perdita delle sensibilità, disturbi delle funzioni autonomiche nei distretti corporei la cui innervazione origina nei segmenti midollari sottostanti il livello di lesione, e perdita del controllo sfinterico; 2) emisezione midollare o sindrome di Brown-Séquard, caratterizzata da paralisi e perdita delle sensibilità dei cordoni dorsali (tattile fine, vibratoria, del senso di posizione) omolateralmente alla lesione e perdita delle sensibilità termica e dolorifica controlateralmente; 3) sindrome centromidollare, il cui elemento tipico è l'iniziale perdita selettiva delle sensibilità termica e dolorifica per lesione delle fibre decussanti dei fasci spinotalamici, ma con conservazione delle sensibilità dei cordoni dorsali; deficit di queste ultime possono manifestarsi per estensione della lesione ai cordoni dorsali; atrofie muscolari e paralisi flaccida possono comparire nei distretti sottostanti la lesione in seguito a interessamento dei neuroni motori delle corna anteriori; 4) sindrome dei cordoni e delle radici dorsali (tipica della tabe dorsale), che si manifesta con deficit sia delle sensibilità cordonali posteriori che di quella termo-dolorifica, in assenza di paralisi; 5) degenerazione posterolaterale (tipica della carenza alimentare di vitamina B12), che comporta deficit delle sensibilità dei cordoni posteriori e paralisi spastica da lesione dei tratti cortico-spinali, frequentemente perdita dei riflessi osteotendinei per interessamento dei nervi periferici; 6) sindrome delle corna anteriori, che può manifestarsi con atrofia muscolare e ipostenia associate a perdita dei riflessi, come accade nella poliomielite o in alcune malattie del motoneurone (atrofia muscolare progressiva; atrofia muscolare spinale), o a iperreflessia, come si osserva nella sclerosi laterale amiotrofica, malattia nella quale all'interessamento delle corna anteriori si associa quello dei tratti cortico-spinali; 7) sindrome dell'arteria spinale anteriore, nella quale i due terzi anteriori del midollo vanno incontro a infarto ischemico, con conseguente paralisi e perdita delle sensibilità spinotalamiche ma conservazione di quelle dei cordoni dorsali.
Le cause più frequenti di lesione midollare sono i traumi vertebro-midollari, le patologie del disco intervertebrale (ernia), alcuni processi infiammatori (sclerosi multipla, mielite trasversa), le neoplasie (meningiomi, neurinomi, carcinomi metastatici, ecc.). Relativamente frequente è, inoltre, l'interessamento del midollo in corso di condizioni degenerative (sclerosi laterale amiotrofica, paralisi spinale progressiva, ecc.), infettive (tubercolosi, AIDS, sifilide), carenziali (deficit di vitamina B12, ecc.).
g) Nervo periferico
Numerose malattie del nervo periferico interessano con una certa selettività una delle sue componenti (motoria, sensitiva o autonomica), altre ne colpiscono più di una contemporaneamente, altre ancora, invece, riguardano preferenzialmente le fibre mieliniche o quelle amieliniche. Le sindromi cliniche sono caratterizzate, in genere, da deficit motori e/o delle sensibilità a prevalente distribuzione distale, associati a perdita dei riflessi osteotendinei. I disturbi sensitivi comprendono anche sensazioni anomale (parestesie) come formicolio, punture, senso di addormentamento. Alcune neuropatie ereditarie si associano a palpabilità dei nervi attraverso la cute e a deformazione del piede (piede cavo). Le cause di neuropatia periferica più frequenti nei paesi industrializzati occidentali sono il diabete mellito, l'abuso di alcool, le malattie del connettivo.
h) Sindrome di ipertensione endocranica
Una grande varietà dei processi patologici a carico del SNC (tumori, ematomi, ascessi, infarti, emorragie, aree di contusione, ecc.) è rappresentata da lesioni occupanti spazio, che tendono a espandersi sia per aumento intrinseco del proprio volume, sia per edema dei tessuti circostanti. Poiché il SNC è racchiuso in un involucro osseo inespansibile, i processi occupanti spazio finiscono, inevitabilmente, per esercitare un'azione meccanica dannosa sui tessuti che si estrinseca dapprima in compressione e distorsione locale, quindi in dislocazione delle strutture nervose dalla loro sede anatomica naturale (ernie; v. fig. 28). Questi fenomeni avvengono sia a livello endocranico che intravertebrale, ma nella prima localizzazione la loro importanza clinica è maggiore. Quando una lesione endocranica occupante spazio si espande, entra in funzione un meccanismo di compensazione rappresentato dalla dislocazione dagli spazi endocranici di una quantità di sangue e/o di liquor pari al volume della lesione, che impedisce l'incremento del volume totale del contenuto endocranico e quindi della pressione intracranica. La quantità di liquidi dislocabili dallo spazio endocranico è comunque limitata e l'espansione di una lesione oltre tali limiti di compensazione comporta una condizione di ipertensione endocranica, che rappresenta una situazione di grave rischio per la sopravvivenza dell'individuo. I sintomi e segni d'ipertensione endocranica sono rappresentati da cefalea, vomito, papilla da stasi (stasi artero-venosa e rigonfiamento della papilla oculare) e quindi depressione del livello di coscienza.
9. Neuropatologia speciale
A partire dagli anni ottanta, in seguito alla messa a punto di nuove metodiche biochimiche, immunoistochimiche e di biologia molecolare, sono stati fatti notevoli progressi nelle conoscenze delle malattie del sistema nervoso, in particolare nel campo della eziopatogenesi delle malattie degenerative, tra le quali la malattia di Alzheimer, che rappresenta uno dei più rilevanti problemi socio-sanitari dell'immediato futuro nei paesi industrializzati. Perlomeno sorprendente, e con conseguenze imprevedibili per l'attuale concezione dei meccanismi di determinazione e trasmissione delle malattie, è stata l'identificazione della natura proteica degli agenti responsabili (prioni) di alcune rare condizioni, inquadrate classicamente tra le malattie degenerative, in un recente passato incluse tra quelle di possibile natura virale, e ora considerate autonomamente come ‛malattie prioniche'. Nello stesso periodo, infine, si è verificato un drastico mutamento dello spettro epidemiologico delle malattie infettivo-infiammatorie del sistema nervoso, sia per la comparsa dell'infezione da HIV (Human Immunodeficiency Virus), sia per il notevole incremento di altre condizioni di immunodeficienza (terapia immunosoppressiva nei trapianti d'organo, prolungata sopravvivenza dei pazienti con neoplasie della serie ematica, terapia steroidea, tossicomanie, ecc.) cui è legata l'aumentata incidenza di quadri infettivo-infiammatori di riscontro solo occasionale in passato.
a) Malattie degenerative
Classicamente vengono definite malattie degenerative del sistema nervoso condizioni a eziologia ignota, decorso cronico progressivo, esordio insidioso, talora con ricorrenza di tipo ereditario, contrassegnate neuropatologicamente dall'interessamento preferenziale, più o meno simmetrico, di neuroni e tratti nervosi funzionalmente correlati (sistemi) che vanno incontro a lenta, progressiva degenerazione ed eventualmente a morte, associata ad alterazioni istopatologiche relativamente aspecifiche, modesta astrogliosi e scarsa o assente reazione infiammatoria. All'origine di alcune malattie incluse in passato tra quelle degenerative sono stati individuati difetti enzimatici specifici geneticamente determinati, e queste condizioni vengono attualmente classificate di conseguenza. Comunque, nonostante i recenti progressi, si conoscono più di un centinaio di condizioni degenerative a eziopatogenesi non ancora definita. Queste possono essere classificate tanto in base alla distribuzione anatomica prevalente delle lesioni (per esempio, processi degenerativi della corteccia cerebrale e della sostanza bianca, dei gangli della base, del talamo e del mesencefalo, ecc.) che alle principali manifestazioni cliniche associate (per esempio, processi dementigeni, disordini del movimento, ecc.). Molte di queste condizioni si manifestano in forme che presentano una relativa uniformità fenotipica a fronte di una notevole variabilità genotipica; in altre parole, forme clinicamente e neuropatologicamente equivalenti possono essere associate a differenti alterazioni geniche o a un genotipo normale. Recentemente, inoltre, nuove tecniche immunoistochimiche e di analisi morfologica quantitativa hanno consentito di individuare forme con manifestazioni cliniche sovrapponibili ma quadri neuropatologici sostanzialmente differenti, che verosimilmente corrispondono a entità distinte. Oggi si ritiene che fattori diversi possano esercitare la loro azione patogena mediante lo stesso o gli stessi meccanismi e/o che i sistemi neuronali siano selettivamente vulnerabili a fattori patogeni di natura differente nei confronti dei quali lo spettro delle possibili reazioni è relativamente limitato. Nella tab. III viene riportato un elenco delle principali malattie degenerative che si rifà ai criteri di classificazione neuropatologica tradizionale, in cui la denominazione di alcune condizioni risulta desueta dal punto di vista clinico. Tuttavia, sebbene i recenti progressi suggeriscano che la classificazione tradizionale sia più che mai da ritenere provvisoria, essa appare ancora vantaggiosa nella pratica, perché la sintomatologia clinica è largamente dipendente dalla sede anatomica piuttosto che dall'origine delle lesioni.
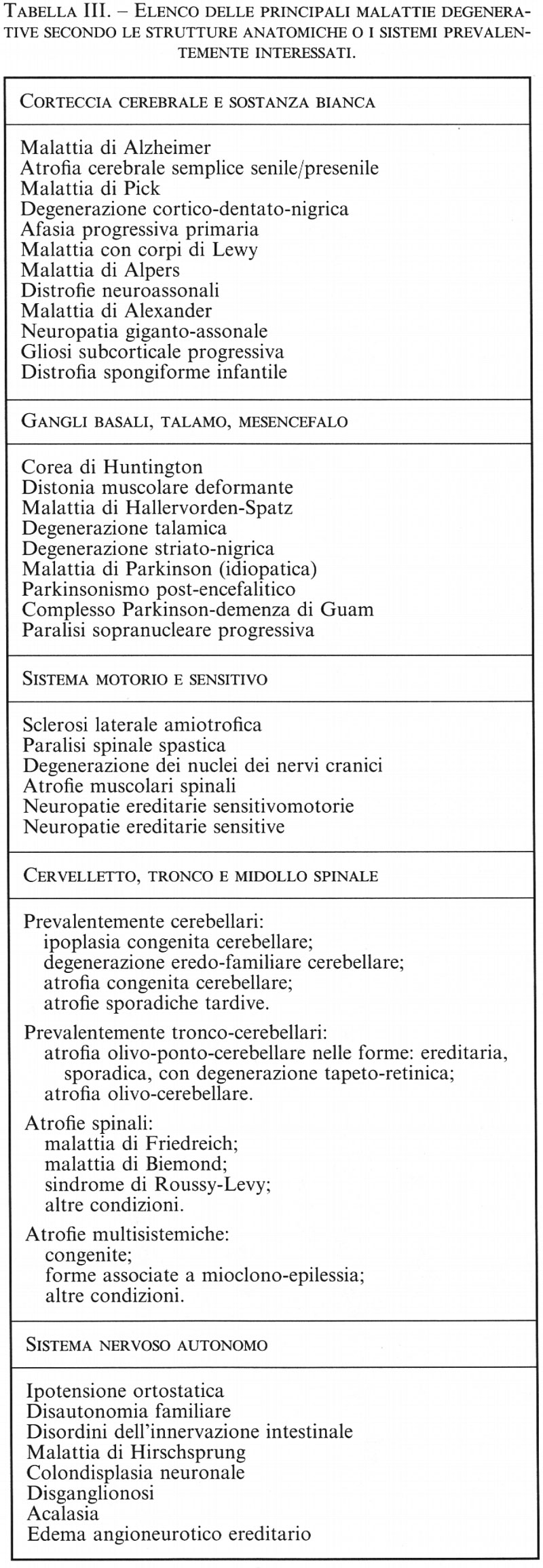
Patogenesi delle malattie degenerative. - Nelle malattie neurodegenerative, la morte neuronale si manifesta secondo un modello le cui caratteristiche (coartazione del corpo cellulare e disintegrazione del DNA nucleare) ricalcano quelle della morte cellulare programmata (apoptosi) che avviene fisiologicamente nel corso dello sviluppo embrionale per eliminare cellule prodotte in eccesso. È stato dimostrato che tanto nelle cellule embrionali che in quelle mature il genoma codifica per un programma di morte cellulare, che nei Mammiferi è soppresso da un gene denominato bcl-2. Non è chiaro il significato della capacità di autoeliminazione della cellula matura, che si ritiene finalizzata alla rimozione di elementi divenuti funzionalmente inaffidabili, senza effetti indesiderati sui tessuti circostanti: infatti l'apoptosi, a differenza della necrosi, non è accompagnata da reazioni infiammatorie. La sopravvivenza e il mantenimento della forma matura dei neuroni dipendono dalla presenza di fattori neurotrofici (NGF, Nerve Growth Factor; BDNF, Brain Derived Neurotrophic Factor; neurotrofine NT3 e NT4/5) liberati dai tessuti circostanti, e da fattori concomitanti, quali l'innervazione da parte di elementi funzionalmente correlati, la presenza di ormoni specifici e, probabilmente, segnali di provenienza gliale. È verosimile che anche le cellule gliali richiedano simili meccanismi trofici, come suggerisce il fatto che la sopravvivenza degli oligodendrociti è promossa dall'assone. Si ritiene che nelle malattie eredodegenerative il programma di morte cellulare possa attivarsi nei neuroni per difetti intrinseci o in risposta a eventi citolesivi che interferiscono con uno o più dei suddetti fattori neurotrofici.
In diverse condizioni neurodegenerative dell'adulto (malattia di Parkinson, malattia di Huntington, atrofie multisistemiche) un ruolo primario o di concausa nella genesi delle lesioni tissutali è svolto, verosimilmente, da un danno ossidativo, progressivo e cumulativo del DNA del sistema nervoso (soprattutto del DNA mitocondriale), correlato all'età dell'individuo. I fattori ossidativi sarebbero rappresentati soprattutto dai radicali liberi dell'ossigeno (O2): anione superossido (O−2), radicale idrossilico (•OH) e perossido d'idrogeno (H2O2). Questi sono prodotti di processi metabolici normali o patologici in cui viene utilizzato ossigeno molecolare (O2) che possono attaccare proteine, acidi nucleici e membrane lipidiche provocando lo sconvolgimento delle funzioni e dell'integrità cellulari. A livello del SN vi sono enzimi coinvolti nel metabolismo neurotrasmettitoriale che generano H2O2 durante la loro normale attività. Per difendersi dagli ossidanti le cellule dispongono di molecole, quali l'acido ascorbico (vitamina C), l'α-tocoferolo (vitamina E) e il glutatione, che hanno spiccate capacità riducenti, e gli enzimi superossidodismutasi, catalasi e glutationeperossidasi (v. radicali liberi: Biologia e patologia).
Sebbene vari fattori possano scatenare processi ossidativi citolesivi, il principale effettore di questi eventi nel SN sembra essere il neurotrasmettitore eccitatorio glutammato, primariamente attraverso l'attivazione di alcuni suoi recettori ionotropi, principalmente NMDA (N-metil-D-aspartato) e KA (acido cainico). Entrambi i recettori, se adeguatamente stimolati, agiscono come canali che attraversano la membrana cellulare e consentono la penetrazione di ioni sodio (Na+), potassio (K+) e calcio (Ca2+) all'interno della cellula. È stato dimostrato che il glutammato, somministrato sperimentalmente, provoca la morte dei neuroni con quadri di tipo degenerativo e che quest'effetto consegue all'attivazione dei recettori NMDA e in molti casi KA ed è mediato dall'innalzamento della concentrazione di Ca2+ intraneuronale. Il processo di degenerazione è dilazionato rispetto al momento in cui si verifica l'incremento della concentrazione del Ca2+ e può essere innescato sia da un'esposizione breve a concentrazioni elevate, sia da un'esposizione lunga a concentrazioni basse. L'attivazione dei recettori del glutammato annovera tra gli effetti Ca2+-dipendenti il possibile coinvolgimento di diverse vie metaboliche capaci di generare fattori ossidanti e l'attivazione di enzimi (proteasi, chinasi, nucleasi) che possono contribuire alla determinazione del danno cellulare. È, ovviamente, possibile che nelle malattie neurodegenerative processi patologici primari compromettano la funzionalità dei neuroni con incremento della loro vulnerabiltà ai fattori ossidanti e/o all'azione eccitocitotossica del glutammato.
Malattia di Alzheimer. - È la causa più comune di demenza: si manifesta nel 5-10% degli individui di età superiore ai 65 anni, e l'incidenza aumenta progressivamente con l'età sino ai 90 anni. In età presenile è più frequente nella quinta decade di vita, ma può comparire anche in giovani adulti. Prevale nelle donne (2:1) ed è uniformemente diffusa nei paesi industrializzati. In maggioranza i casi sono sporadici, ma ben documentati sono anche casi familiari dai quali si deduce una trasmissione ereditaria di tipo autosomico dominante. In alcune famiglie sono state individuate specifiche mutazioni puntiformi del gene APP (Amyloid Precursor Protein) sul cromosoma 21, in altre le analisi di linkage suggeriscono l'esistenza di correlazioni tra la malattia e anomalie dei cromosomi 14 e 19. È una malattia prevalentemente, anche se non esclusivamente, della corteccia cerebrale ed è, quindi, clinicamente caratterizzata dalla perdita delle funzioni corticali superiori: l'esordio, generalmente insidioso, è contrassegnato da disturbi prevalentemente mnesici nelle forme presenili, e depressivo/apatici e/o confusionali in quelle senili; il quadro si deteriora progressivamente per la comparsa di episodi demenziali, turbe del sonno e, nella fase conclamata, di deficit della memoria (per le relazioni spaziali, per i volti, per gli eventi correnti, ecc.), della capacità di giudizio, del linguaggio scritto e orale, del riconoscimento e dell'associazione visiva, e della capacità d'esecuzione di sequenze di movimenti finalizzati alla soluzione di problemi quotidiani. Sono frequenti regressione e disinibizione del comportamento, apparente labilità emotiva, fenomeni di tipo ossessivo e idee deliranti, manifestazioni neurologiche di tipo paretico-spastico e acinetico. Nella fase terminale il paziente è incapace di accudire se stesso, incontinente, non profferisce alcun suono articolato, è amimico, immobile, non manifesta necessità alimentari o d'altro tipo (sindrome acinetico/abulico/apatica), va incontro a cachessia e infine muore, spesso in seguito a broncopolmonite. La durata media della malattia è di circa 5-7 anni. L'elettroencefalogramma mostra un diffuso rallentamento dell'attività elettrica cerebrale. La tomografia computerizzata (TC) e la tomografia a risonanza magnetica nucleare (TRMN) dell'encefalo possono evidenziare - talvolta nelle fasi precoci, costantemente in quelle tardive - l'atrofia cerebrale, prevalentemente corticale, con ampliamento dei solchi e dilatazione ventricolare.
All'esame neuropatologico il cervello appare più piccolo e il suo peso può essere inferiore di un terzo al valore normale per l'età. L'atrofia è generalizzata e simmetrica, ma tendenzialmente accentuata a livello frontale e temporale. Le leptomeningi sono ispessite, i vasi superficiali possono mostrare segni di moderata aterosclerosi. Sulle sezioni coronali del cervello si apprezza l'ampliamento dei solchi e delle scissure della corteccia, il cui spessore è, invece, relativamente conservato. L'atrofia corticale sembra riconducibile, quindi, alla perdita di colonne verticali, piuttosto che di lamine orizzontali di neuroni. I ventricoli cerebrali appaiono arrotondati per la riduzione della quantità di sostanza bianca commisurata alla perdita di prolungamenti assonali. Il grado di atrofia non è, comunque, in relazione con la severità del quadro clinico. I reperti microscopici tipici della malattia di Alzheimer sono le cosiddette placche senili, le degenerazioni neuronali fibrillari e granulovacuolari e i corpi di Hirano. Le placche senili (v. fig. 29) sono formate da aggregati di resti assonali rigonfi di mitocondri, associati o meno, a seconda dell'età della placca, con un core centrale denso di fibrille amiloidi e con processi astrocitari e cellule microgliali. Le placche più antiche sono formate quasi esclusivamente da amiloide. I diversi processi hanno una distribuzione tipica: interessano prevalentemente la corteccia di tutto l'encefalo, ma hanno la concentrazione massima nella parte mediale del lobo temporale (in particolare nell'ippocampo, nel giro paraippocampale, nell'amigdala) e quella minima a livello cerebellare e del bulbo. La morte per degenerazione colpisce soprattutto i neuroni di maggiori dimensioni: la perdita ammonta al 40-45% del totale nei lobi frontali e temporali; relativamente risparmiate sono invece le regioni parietali e occipitali. Nei neuroni superstiti si verifica una riduzione del numero dei dendriti, che risultano inoltre più brevi che nel cervello adulto normale; anche il numero delle sinapsi è marcatamente ridotto. Notevole interesse ha sollevato il riscontro di una cospicua perdita di neuroni e la presenza di degenerazione fibrillare in quelli superstiti nel nucleo basale di Meynert, che fornisce l'innervazione colinergica alla corteccia cerebrale.
Il numero di astrociti fibrillari è significativamente aumentato rispetto a quello riscontrato in controlli di età corrispondente, ma non vi è correlazione tra l'astrocitosi e l'entità delle altre alterazioni. Si ritiene che l'astrocitosi, almeno quella perivascolare, possa essere in rapporto con alterazioni della barriera ematoencefalica. Un'alterazione della parete vasale per deposito di una sostanza proteica (β-amiloide) si riscontra in più dell'80% dei cervelli con malattia di Alzheimer.
I reperti neuropatologici dell'Alzheimer sono presenti anche nel cervello senile: la quantità e la distribuzione delle alterazioni distingue, tuttavia, la condizione patologica dall'invecchiamento fisiologico. Gli studi volti a ricercare una relazione tra severità della demenza e reperti neuropatologici hanno fornito risultati incerti: alcuni dati indicano una significativa correlazione tra grado di demenza (apprezzato con scale di valutazione), deficit colinergico (misurato biochimicamente) e numero di placche neuritiche; altri dati evidenziano una correlazione tra demenza e degenerazione fibrillare. Studi recenti morfometrici e neurochimici suggeriscono che i casi a esordio precoce e quelli a esordio tardivo rappresentino forme distinte della malattia. Esistono casi di malattia di Alzheimer e demenza vascolare combinate e ancora casi atipici in cui non si riscontra degenerazione neurofibrillare: in questi ultimi le manifestazioni cliniche sembrerebbero meno severe.
b) Malattie prioniche
Vengono così definite alcune condizioni, rare nell'uomo ma relativamente frequenti in altri mammiferi, nelle quali vi è un accumulo nel sistema nervoso dell'isoforma abnorme, conosciuta come PrPSC, della proteina prionica o prione, PrPC (SC, scrapie; C, cellulare); si tratta di una sialoglicoproteina della membrana cellulare, espressa in diversi tessuti, soprattutto in quello nervoso, la cui funzione normale è però sconosciuta. Le malattie da prioni erano tradizionalmente classificate come encefalopatie spongiformi trasmissibili o malattie da virus lenti, perché sono contrassegnate da una diffusa spongiosi neuronale, possono essere trasmesse sperimentalmente per inoculazione da un ospite all'altro di specie identica o differente, si sviluppano dopo un lungo periodo d'incubazione, e tali caratteristiche avevano giustificato l'ipotesi di un'eziologia virale.
Patogenesi delle malattie prioniche. - A dispetto della loro rarità, le malattie prioniche hanno suscitato considerevole interesse per le proprietà uniche del loro agente trasmissibile, di natura proteica, per il quale Stanley Prusiner, che lo ha isolato e caratterizzato biochimicamente, ha proposto la denominazione ‛prione' (proteinaceous infectious particle), per distinguerlo dai Virus (v. Prusiner, 1991). Sino alla scoperta dei prioni era universalmente accettato che gli agenti che trasmettono le malattie infettive, anche i più semplici come i Virus, dovessero disporre di acidi nucleici (DNA e RNA) per dirigere la sintesi delle proteine indispensabili alla loro sopravvivenza e duplicazione. Nel caso delle malattie prioniche è stato invece ampiamente dimostrato che frazioni di proteina prionica purificata, estratta da cervelli affetti, trasmettono la malattia ad animali da esperimento; i tentativi di copurificare in queste frazioni acidi nucleici non hanno avuto successo e, d'altra parte, l'infettività di queste frazioni non viene modificata dal trattamento con nucleasi o dall'irradiazione con ultravioletti, che inattivano gli acidi nucleici. L'agente infettivo consiste, quindi, essenzialmente nell'isoforma abnorme della PrPC. Entrambe le forme sono codificate dal medesimo gene (PRNP), che nell'uomo è localizzato sul cromosoma 20, e tra di esse non vi sono differenze amminoacidiche. La modificazione patogena consiste quindi, verosimilmente, nel cambiamento della normale conformazione tridimensionale della proteina da una forma a spirale in una struttura a foglietto ripiegato. Questa trasformazione rende la PrPSC resistente agli enzimi digestivi cellulari e non (proteasi) e ne favorisce il deposito e l'accumulo nei lisosomi intraneuronali, sino a livelli patogeni. L'effetto citolesivo della PrPSC è, infatti, concentrazione-dipendente, sebbene sia ignoto attraverso quali meccanismi si realizzi: verosimilmente, i lisosomi repleti esplodono danneggiando le cellule. La propagazione della PrPSC sembra essere legata alla capacità delle molecole abnormi di indurre per contatto trasformazioni conformazionali di quelle normali. È noto che tale evento avviene a contatto con membrane intracellulari, ma non si sa quali fattori ne favoriscano l'inizio. Si ritiene che le trasformazioni conformazionali patogene siano molteplici e conferiscano alla molecola potenzialità citolesive e tropismo per bersagli cellulari preferenziali differenziati: ciò spiegherebbe la notevole variabilità delle manifestazioni cliniche in relazione con l'accumulo di PrPSC. Esistono ben dimostrati casi familiari ereditari, e anche casi sporadici, nei quali sono state dimostrate diverse mutazioni del gene PRNP: tutte le mutazioni sinora descritte provocano sostituzioni di amminoacidi della PrPC che favoriscono la destabilizzazione della sua normale struttura ad α-elica e innalzano la probabilità di un cambio conformazionale. Fattori genetici (omozigosi per la metionina o la valina al codone 129) predisponenti o favorenti sembrano intervenire, inoltre, anche nelle forme sporadiche e iatrogene. È importante ricordare che non tutte le alterazioni del gene PRNP consentono, di per sé, la diagnosi di malattia prionica: infatti non tutte le mutazioni sono associate alla malattia. Deve essere ancora sottolineato che l'encefalopatia spongiforme, un tempo considerata quale contrassegno morfologico delle malattie da ‛agenti non convenzionali', è presente costantemente solo nei casi tipo Creutzfeldt-Jakob sporadici e in quelli familiari associati a mutazioni del codone 200.
Malattie prioniche umane. - La forma più frequente è la malattia di Creutzfeldt-Jakob, presente in tutto il mondo con un'incidenza di 1 caso per milione di abitanti per anno, per lo più in forma sporadica, ma familiare nel 10-15 % dei casi. L'età media d'insorgenza è di 60 anni e prevale lievemente nelle femmine (1,5:1). La durata media della malattia, che ha sempre epilogo fatale, è 7,6 mesi, ma nel 5-10% dei casi, molti dei quali familiari e a esordio precoce, può superare i 2 anni. Sono noti casi di origine iatrogena, ossia trasmessi accidentalmente nel tentativo di curare altre patologie, attraverso operazioni stereotassiche sul cervello con strumenti inadeguatamente sterilizzati, trapianti di dura o di cornea, trattamento con ormoni di derivazione umana. Anche la malattia di Creutzfeldt-Jakob, come quella di Alzheimer, può essere considerata una malattia prevalentemente, ma non esclusivamente, corticale. Clinicamente, una fase prodromica della durata di alcune settimane, durante le quali compaiono solo manifestazioni aspecifiche (disturbi del sonno, modificazioni del comportamento, depressione, allucinazioni visive o, raramente, acustiche, vertigini, deficit della vista e, talvolta, turbe dell'equilibrio e dell'andatura), precede la comparsa del quadro tipico caratterizzato da demenza a ingravescenza eccezionalmente rapida, con perdita delle funzioni nervose superiori, contrazioni muscolari ripetitive (mioclonie), che possono essere scatenate da rumori o dal semplice contatto, e tipiche alterazioni elettroencefalografiche (complessi trifasici pseudoperiodici) in rapporto o meno con le mioclonie. Nella fase terminale, di durata variabile da settimane a mesi, il paziente, generalmente in coma, presenta manifestazioni cliniche che mutano in rapporto alla progressione del danno strutturale del sistema nervoso, sino alla comparsa di uno stato di decerebrazione o decorticazione. La morte sopravviene, più spesso, in seguito a polmonite ipostatica o a insufficienza cardiovascolare da scompenso autonomico. La TC dimostra, nello stadio tardivo della malattia, segni aspecifici di atrofia cerebrale. La TRMN evidenzia, in parte dei pazienti, modificazioni a livello dei gangli della base che sembrano essere in correlazione con le alterazioni spongiformi dimostrabili istologicamente. I reperti neuropatologici macroscopici, rappresentati essenzialmente da un'atrofia corticale generalizzata e dilatazione ventricolare, di grado variabile in relazione alla durata della malattia, sono aspecifici; caratteristici sono, invece, quelli microscopici, consistenti in una severa, diffusa vacuolizzazione del soma cellulare e dei prolungamenti neuronali (encefalopatia spongiforme), un massivo spopolamento neuronale, astrocitosi e rare placche neuritiche che legano anticorpi anti-PrPC (placche prioniche). La distribuzione di queste lesioni è tipicamente accentuata a livello della corteccia cerebrale, dello striato, del talamo e della sostanza grigia del tronco e del cervelletto, mentre l'ippocampo è generalmente risparmiato. Sono state descritte comunque diverse varianti comprendenti una forma con precoce interessamento della corteccia occipitale (variante di Heidenhain) accompagnata clinicamente da cecità, una forma con prevalente interessamento talamico, una con spopolamento dei neuroni delle corna anteriori spinali che si accompagna a ipostenia e atrofia muscolare (variante amiotrofica), e infine una con esteso coinvolgimento della sostanza bianca.
Un'altra forma, il kuru, è stata osservata solo nella tribù Fore di Papua, nella Nuova Guinea. La malattia, letale, di durata media inferiore a 3 anni, caratterizzata da incoordinazione dei movimenti e spesso demenza, veniva contratta in seguito a cannibalismo rituale: la tribù Fore usava, infatti, onorare i defunti mangiandone il cervello. Tale pratica è stata sospesa da quando, negli anni sessanta, ne è stato scoperto il legame con la malattia, che attualmente è pressoché scomparsa. Neuropatologicamente si riscontrano spopolamento neuronale e astrocitosi, e incostantemente spongiosi e placche prioniche.
Una condizione prevalentemente ereditaria è la sindrome di Gerstmann-Sträussler-Scheinker: la malattia, a esito fatale, esordisce intorno ai 40 anni e ha un decorso clinico di durata superiore ai 2 anni che si manifesta, in genere, con atassia cerebellare, disartria, disfagia e solo tardivamente demenza e mioclono: neuropatologicamente è contraddistinta dalla presenza multifocale di numerose, grosse placche prioniche, da perdita neuronale e da astrocitosi, mentre rara è la spongiosi.
L'insonnia familiare fatale è una malattia prionica, descritta in una famiglia italiana e in una italo-americana; tale malattia, che compare nella mezza età, è caratterizzata da perdita neuronale con gliosi a livello di alcuni nuclei del talamo e dell'ipotalamo e della corteccia cerebrale e cerebellare. Il quadro clinico è dominato inizialmente da insonnia, disautonomia, turbe endocrinologiche, turbe mnesiche con evoluzione verso la demenza e, tardivamente, da atassia e mioclono.
Esistono, infine, forme atipiche, essenzialmente se non esclusivamente dementigene, individuate recentemente grazie a metodi biochimici e genetici. Clinicamente sono difficilmente differenziabili da altre demenze neurodegenerative, in quanto hanno lunga durata, lenta progressione, e mancano il mioclono e le alterazioni elettroencefalografiche tipiche. Anche neuropatologicamente, mancando l'encefalopatia spongiforme, può essere difficile distinguere queste condizioni da un quadro di tipo Alzheimer, a meno di non avere a disposizione tecniche immunoistochimiche per la dimostrazione della PrPC. Appare molto interessante - ma al momento non spiegabile - il fatto che le forme atipiche, a differenza di quanto avviene per le altre, siano trasmissibili solo con estrema difficoltà ai Primati non umani.
Malattie prioniche animali. - Le malattie da prioni degli animali comprendono lo scrapie o virosi neurodegenerativa della pecora, che è la forma più comune, nella quale gli ovini perdono la coordinazione dei movimenti sino a diventare incapaci di reggersi sulle zampe, e inoltre soffrono di un prurito così intenso da indurli a sfregarsi sino a raschiare via (to scrape) parti del mantello. Altre malattie prioniche sono l'encefalopatia trasmissibile del visone, l'atrofia cronica del cervo mulo e dell'alce, l'encefalopatia spongiforme felina e quella bovina. Quest'ultima, nota popolarmente come ‛malattia delle vacche matte', si è manifestata in forma epidemica in Inghilterra negli anni ottanta: l'origine dell'epidemia è stata individuata in un integratore alimentare, attualmente ritirato dal commercio, che conteneva carne e farina di ossa di ovini sottoposte a un metodo di lavorazione che non eliminava l'agente dello scrapie. Sebbene l'epidemia sia ora in fase calante, esiste la preoccupazione che l'infezione possa trasmettersi all'uomo attraverso il consumo di carne contaminata.
c) Malattie da Retrovirus (HIV-1)
La storia della virologia, negli anni ottanta, è stata segnata dalla scoperta di virus a RNA, della famiglia dei Retrovirus, neurotropi, agenti eziologici nell'uomo di sindromi/malattie a evoluzione lenta (Lentivirus): HIV (Human Immunodeficiency Virus) tipo 1, agente eziologico dell'AIDS; HIV tipo 2, agente di una sindrome sovrapponibile all'AIDS; HTLV (Human T-cell Leukemia Virus) tipo I, responsabile di una leucemia a cellule T dell'adulto. Esistono Lentivirus di diverse altre specie animali (scimmia, gatto, bovini, pecora, capra, cavallo) che condividono con quelli umani varie caratteristiche biologiche: spiccato tropismo per gli elementi monocitico-macrofagici, che sembrano costituire il serbatoio naturale e la sede di replicazione del virus; induzione di malattie con un lungo periodo di incubazione ed evoluzione subacuta o cronica; capacità di elusione della sorveglianza immunitaria; diffusione e contagio diretti da ospite a ospite, mediante lo scambio di liquidi biologici, in assenza di vettori/ospiti intermedi; neurotropismo, in quanto attaccano il SNC negli stadi precoci dell'infezione e producono malattie/sindromi neurologiche specifiche.
Poiché le infezioni da HIV-1 e 2 nell'uomo (e quelle da Lentivirus nella scimmia e nel gatto) causano una marcata immunodepressione, le condizioni clinico-neuropatologiche a esse legate sono distinguibili in: infezioni primarie, espressione dell'azione diretta del virus e/o dei meccanismi immunopatologici da esso attivati sui tessuti; infezioni opportunistiche e neoplastiche, espressione, entrambe, della vulnerabilità dell'organismo immunodepresso all'attacco delle noxae patogene più varie (v. tab. IV). Nei pazienti con infezione da HTLV-I, di cui è definitivamente accertato il ruolo eziologico nella malattia neurologica definita ‛mielopatia cronica progressiva', si riscontra, invece, una risposta immunitaria relativamente efficace.
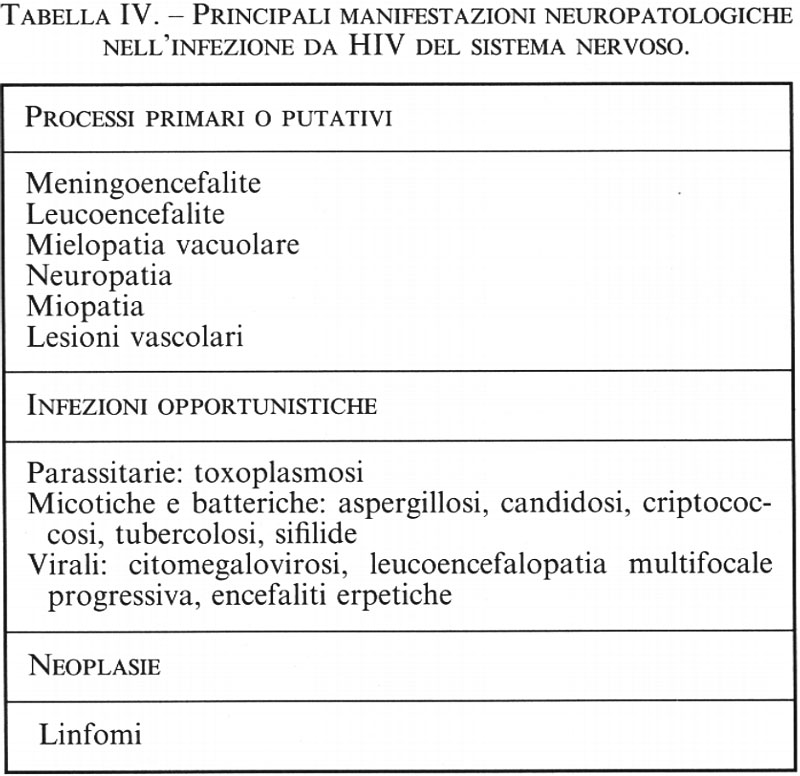
In questo paragrafo verranno trattate esclusivamente le manifestazioni neuropatologiche dell'infezione da HIV-1. Le differenze rispetto all'infezione da HIV-2 sembrano limitate a caratteristiche strutturali minori dei virus e alla loro distribuzione geografica (l'HIV-1 prevale in Europa e in America; l'HIV-2, inizialmente isolato in pazienti della Guinea-Bissau e delle Isole di Capo Verde, è attualmente endemico in Africa occidentale, dov'è il principale agente dell'AIDS); i reperti neuropatologici sono sovrapponibili.
Patogenesi delle lesioni primarie del SNC nell'infezione da HIV-1. - L'HIV-1 raggiunge il SNC attraversando la parete dei vasi sanguigni sia a livello capillare, per infezione dell'endotelio, che a livello delle venule, trasportato da monociti. Infetta, quindi, le cellule che esprimono in superficie recettori CD4 (macrofagi e microglia), a cui si lega attraverso la glicoproteina gp120 dell'involucro virale. L'infezione si distribuisce prevalentemente a livello della sostanza bianca profonda e dei gangli basali piuttosto che nella corteccia. Recentemente, è stato dimostrato che gli astrociti esprimono livelli quantitativamente limitati di alcune proteine e di RNA messaggeri dell'involucro virale. Non esistono prove d'infezione di neuroni e oligodendrociti. La patogenesi del danno tissutale non è ancora definita. I meccanismi ipotizzati comprendono la liberazione di neurotossine (glutammato, citochine, radicali liberi, frammenti della gp120) da parte di monociti e microglia infetti; l'attivazione di meccanismi immunomediati o la liberazione di proteasi da parte di macrofagi attivati; la penetrazione attraverso la BEE alterata di macromolecole tossiche (fattore di necrosi tumorale, gp120). La citotossicità della gp120 è stata dimostrata in vivo e in vitro: nei neuroni riduce l'utilizzazione del glucosio e incrementa il calcio libero intracellulare; negli astrociti altera i meccanismi di regolazione β-adrenergici e deprime la sintesi di proteina gliale fibrillare acida.
Neuropatologia delle lesioni primarie nell'infezione da HIV-1. - In Europa e negli Stati Uniti d'America tra il 40 e il 65% dei pazienti affetti da HIV-1 presenta disordini neurologici e/o psichici, che nel 10% dei casi segnano l'esordio della malattia. Alterazioni neuropatologiche si osservano nell'80% delle autopsie eseguite nei casi di decesso per AIDS. I principali quadri clinico-neuropatologici compaiono ed evolvono in tempi e con frequenza differenti in relazione allo stadio dell'infezione (sieroconversione; sieropositività; complesso AIDS correlato; AIDS conclamato con infezioni opportunistiche; v. immunologia clinica e immunopatologia). La frequenza di specifici disordini varia anche in relazione alle caratteristiche geografiche, razziali, di età e al gruppo a rischio di infezione della popolazione; per esempio, la meningite criptococcica prevale tra i tossicomani consumatori di stupefacenti iniettabili e tra gli Afroamericani; la toxoplasmosi è più comune in Europa che negli Stati Uniti.
Circa i 2/3 dei pazienti con AIDS conclamato presentano demenza e il 90% segni istologici di un'encefalite subacuta (AIDS dementia-complex). I disturbi psichici (apatia, inerzia, amnesia, depressione), suggestivi di un interessamento prevalentemente sottocorticale, evolvono sino a un grave deterioramento psichico con mutismo, mentre i segni neurologici sono relativamente modesti. Eventuali infezioni opportunistiche possono essere escluse con l'esame del liquido cefalorachidiano (neurosifilide, criptococcosi, tubercolosi) o con la biopsia cerebrale (leucoencefalopatia da Papovavirus, toxoplasmosi). La TC e la TRMN mostrano un'atrofia corticale, talora prevalentemente frontale, e alterazioni della sostanza bianca, piccole e maldefinite inizialmente, diffuse nella fase terminale. All'esame neuropatologico si osserva un'atrofia cerebrale generalizzata, marcata a livello fronto-temporale, e un diffuso pallore mielinico. Cellule giganti multinucleate, noduli microgliali (microglia, linfociti, macrofagi) e infiltrati perivenosi cortico-sottocorticali (linfo-monociti e macrofagi) costituiscono il correlato microscopico della encefalite da HIV, o leucoencefalite nel caso di interessamento prevalente della sostanza bianca. Le cellule endoteliali, gli infiltrati perivascolari e le cellule giganti contengono sequenze di acidi nucleici virali, ma solo il numero delle cellule giganti sembra in relazione con il grado di demenza. Nel bambino il quadro microscopico è caratterizzato, oltre che dalle cellule giganti multinucleate e dagli infiltrati perivascolari, da una mineralizzazione dei vasi dei gangli basali e della sostanza bianca.
Nel 50% delle autopsie eseguite su adulti con AIDS conclamato (nel 10% di quelle pediatriche) è riscontrabile una mielopatia vacuolare, che si sviluppa, quasi sempre, in concomitanza con l'encefalopatia e nel 20% circa degli affetti si manifesta con paraparesi spastica progressiva e atassia pseudotabetica. Il quadro neuropatologico è caratterizzato da chiazze di vacuolizzazione della sostanza bianca prevalenti a livello dei cordoni laterali e posteriori nel tratto toracico. Le lesioni, rappresentate in uno stadio iniziale da vacuoli formatisi per slaminamento da edema della mielina e infiltrati macrofagici, possono evolvere, attraverso un quadro di demielinizzazione con astrocitosi e accenni alla rimielinizzazione, sino alla necrosi, associate alla presenza di cellule schiumose e gliofibrosi. La patogenesi è, probabilmente, multifattoriale, verosimilmente tossico-dismetabolica; comunque sono stati esclusi un deficit di vitamina B12 e l'azione di patogeni opportunisti.
L'infezione da HIV-1 può manifestarsi, già durante la prima settimana di sieroconversione, con una sindrome meningoencefalitica, in genere (60% dei portatori) in forma cronica, asintomatica, con pleiocitosi liquorale, e più raramente (2%) in forma acuta accompagnata, occasionalmente, da encefalopatia e/o mielopatia transitorie, con positività liquorale per l'HIV-1 e per anticorpi antivirali. La TC e la TRMN non offrono reperti significativi; i reperti neuropatologici si limitano a infiltrati leptomeningei linfo-monocitari e polimorfonucleari e, talora, a modeste alterazioni della sostanza bianca (pallore e astrocitosi).
Nei pazienti affetti da AIDS sono stati descritti disordini cerebro-vascolari quali infarti embolici ed emorragie cerebrali intraparenchimali, subaracnoidee, epidurali e subdurali. All'origine di queste lesioni vi possono essere una vasculite granulomatosa o una vasculite necrotizzante. Sono stati osservati anche quadri di alterazioni microcircolatorie proliferative a livello leptomeningeo e/o parenchimale.
Sindromi cliniche da compromissione dei nervi cranici e/o del sistema nervoso periferico (polineuropatia infiammatoria demielinizzante acuta o cronica, polineuropatia distale simmetrica, poliradiculopatia, mononeurite multiplex) non sono rare già in fase di sieropositività. Rara una neuropatia sensitiva da ganglioneuronite. L'esame neuropatologico rivela, nella maggioranza dei casi, quadri di demielinizzazione segmentaria, degenerazione assonale e infiltrazione endo- ed epinevrale di elementi mononucleati. La patogenesi delle neuropatie associate all'AIDS è, probabilmente, multifattoriale (autoimmune, tossica, nutrizionale, ecc.), ma per ora rimane oscura. La presenza dell'HIV a livello del nervo periferico è stata dimostrata, solo in alcuni casi di neuropatia associata ad AIDS, su estratti coltivati o mediante dimostrazione diretta dell'RNA virale (ibridazione in situ), mentre le indagini immunocitochimiche e ultrastrutturali sono risultate costantemente negative. In diversi casi la neuropatia periferica è legata a infezioni opportunistiche (infezione da Citomegalovirus, sifilide, ecc.) o a processi neoplastici.
Un quadro di miopatia infiammatoria può comparire in corso di AIDS conclamato. Clinicamente si manifestano ipostenia prossimale a esordio subacuto e, talora, dolori, con innalzamento dei valori sierici della creatinchinasi (CK). L'esame istopatologico del muscolo evidenzia fenomeni di mionecrosi e miofagocitosi, infiltrati mononucleari e macrofagi HIV-positivi a livello endo- e perimisiale e, in taluni casi, corpi citoplasmatici e nemalinici entro le fibre. Un'invasione diretta delle miofibre da parte dell'HIV non è mai stata dimostrata. Si ipotizza che il muscolo diventi il bersaglio di una reazione autoimmune in seguito all'attacco di agenti infettivi opportunisti, oppure, in alternativa, che sia il bersaglio passivo di reazioni infiammatorie provocate dalla presenza del virus entro e attorno la parete dei vasi sanguigni. In pazienti con AIDS, un quadro di mioglobinuria acuta e ipostenia, caratterizzato a livello muscolare dalla presenza di mitocondri anomali contenenti inclusioni paracristalline, è stato recentemente attribuito all'azione tossica della zidovudina (AZT): tali alterazioni sono state osservate, però, anche in malati di AIDS non sottoposti al trattamento con AZT. Una grave atrofia muscolare prevalentemente a carico delle fibre di tipo II, quasi costantemente presente nella fase terminale dell'AIDS, è secondaria allo scadimento delle condizioni generali e non ha significato specifico.
Neuropatologia delle infezioni opportunistiche del SNC in corso d'infezione da HIV-1. - L'AIDS rende i soggetti affetti vulnerabili alle infezioni da parte di vari microrganismi, alcuni dei quali scarsamente virulenti in soggetti immunocompetenti. Tali infezioni, dette opportunistiche, sono la principale causa di decesso in corso d'infezione da HIV-1. Alcune infezioni opportunistiche colpiscono con elevata frequenza il SNC: la loro diagnosi precoce è importante, perché generalmente esse sono sensibili al trattamento farmacologico.
L'infezione opportunistica clinicamente più frequente (10%) è dovuta a Criptococcus neoformans, un lievito ubiquitario responsabile di una meningite cronica delle leptomeningi basali, con proliferazione connettivale reattiva che può incarcerare i nervi cranici e ostruire le vie liquorali. L'esame neuropatologico dimostra un materiale gelatinoso entro gli spazi subaracnoidei e piccole cisti intraparenchimali, talora confluenti, localizzate principalmente a livello dei gangli basali lungo il decorso delle arterie lenticolostriate. Le formazioni gelatinose, denominate ‛criptococcomi', alla TC e alla TRMN simulano un processo occupante spazio, tuttavia istologicamente non mostrano la struttura tipica dei granulomi. Le cisti sono formate da aggregati del microrganismo ammassati nello spazio perivascolare, con scarsa o assente reazione infiammatoria.
La toxoplasmosi è una delle più frequenti cause di disordini neurologici nei pazienti con AIDS: l'incidenza media nelle serie autoptiche è circa del 15%. Si manifesta con ascessi e, raramente, in forma diffusa caratterizzata da sparsi noduli microgliali. Gli ascessi, generalmente multipli, si localizzano al confine tra corteccia e sostanza bianca, nei nuclei grigi della base, meno frequentemente a livello cerebellare e del tronco e raramente a livello spinale. Queste lesioni esercitano un effetto massa e la TC e la TRMN dimostrano immagini tipiche ma non specifiche, per cui per la diagnosi di lesioni isolate è indicata la biopsia. In malati di AIDS adulti, l'infezione può manifestarsi in forma di periependimite e ventricolite, altrimenti tipiche della toxoplasmosi congenita. Istologicamente si riconoscono lesioni acute caratterizzate da un focus necrotico centrale, con emorragie puntiformi, circondato da elementi linfo-monocitari, polimorfonucleati, macrofagi e da vasi in proliferazione. Alla periferia del focus necrotico si trovano tachizoiti liberi, patognomonici di infezione acuta, e bradizoiti incistati (v. fig. 30). I vasi possono essere invasi dai microrganismi e presentare proliferazione intimale o anche una franca vasculite con necrosi fibrinoide e trombosi. Nella fase di organizzazione le lesioni appaiono circoscritte da una ben demarcata area di necrosi coagulativa, circondata da lipofagi e scarsi microrganismi. Le lesioni croniche sono rappresentate da cisti contenenti lipofagi e siderofagi, circondate da gliosi.
Nei paesi industrializzati l'incidenza dell'infezione da Mycobacterium tuberculosis (tubercolosi) si è progressivamente ridotta negli ultimi trent'anni, e tuttavia a partire dagli anni ottanta, in coincidenza con l'incremento dell'AIDS, la velocità di riduzione è bruscamente rallentata (v. malattie emergenti). Altri fattori predisponenti a contrarre la tubercolosi sono l'abuso di alcool e di droghe iniettabili e condizioni di immunodepressione diverse dall'AIDS. È in progressivo aumento l'incidenza di forme della malattia a eziologia atipica (Mycobacterium avium) e a localizzazione primaria extra-polmonare, tra cui la neurotubercolosi. La descrizione classica distingue quattro quadri neuropatologici principali, che possono presentarsi in varia combinazione: 1) tubercoli miliari disseminati; 2) tubercoloma meningeo; 3) meningite acuta; 4) meningite proliferativa. Questi quadri sono divenuti insoliti in seguito alla messa a punto della chemioterapia specifica; in pratica, attualmente le manifestazioni della TBC nel SNC sono rappresentate dalla meningite e dai tubercolomi (v. fig. 31). Nella prima le meningi della convessità sono opache e un denso essudato grigio-verde riempie le cisterne basali, copre la parte anteriore del ponte estendendosi nella cisterna magna e nella scissura silviana. Anche gli spazi subaracnoidei spinali possono essere interessati. I giri sono appiattiti e i solchi ristretti per l'edema della sostanza bianca. Noduli tubercolari possono essere osservati lungo i margini della scissura silviana, in prossimità delle vene della convessità, nella dura della base, nei plessi corioidei e in prossimità dell'ependima. Possono essere presenti idrocefalo e infarti nel territorio dell'arteria cerebrale media e delle lenticolo-striate: gli infarti sono secondari a trombosi da vasculite artero-venosa. I tubercolomi si manifestano invece come lesioni occupanti spazio e sono particolarmente frequenti negli spazi intracranici sottotentoriali (fossa cranica posteriore), soprattutto nei bambini. A questo livello danno origine, quasi sempre, a manifestazioni neurologiche, perché non tendono a organizzarsi e a calcificare come quelli sopratentoriali che spesso cessano di crescere e divengono quiescenti. I tubercolomi si presentano, generalmente, come masse multiple, tondeggianti, di 2-12 mm di diametro, o lobulate. Il centro, necrotico, è circondato da una capsula gelatinosa, piuttosto dura, costituita da collagene, da ammassi di cellule giganti che racchiudono materiale caseoso e da un'area di gliosi reattiva. Sono presenti bacilli tubercolari. La capsula può andare incontro a fibrosi. La parete dei vasi adiacenti è infiltrata di linfociti. I tubercolomi sono meno edemigeni degli ascessi. Talora, nelle forme da M. avium riscontrate soprattutto nei pazienti con AIDS o con infezioni disseminate, le alterazioni neuropatologiche si manifestano sotto forma di meningite cronica o di ascessi multipli, senza la formazione di tubercoli: la risposta infiammatoria tende, infatti, a essere meno pronunciata che nell'infezione da M. tuberculosis.
I pazienti con infezione da HIV-1 sono più a rischio della popolazione normale nei confronti della neurosifilide: in essi la velocità di progressione e la gravità della malattia appaiono più elevate e i test sierologici e liquorali standard (VDRL, Venereal Diseases Research Laboratory, e FTA, Fluorescent Treponemal Antibody) possono essere negativi pur in presenza di Treponema pallidum nel liquor. L'infezione da T. pallidum del SNC non trattata può regredire spontaneamente, rimanere asintomatica, evolvere in meningite acuta o cronica. Questa, a sua volta, può rimanere asintomatica o rappresentare la base per lo sviluppo della neurosifilide meningovascolare (cerebrale e spinale) e parenchimale (paralisi generale e tabe dorsale), o della sifilide gommosa. Un'encefalite necrotizzante gravissima, con massiva invasione treponemica del parenchima cerebrale, è stata osservata in ammalati di AIDS che erano stati considerati guariti da una pregressa infezione treponemica: per questa forma è stata proposta la definizione di sifilide ‛quaternaria'. La meningite acuta, a prevalente distribuzione basale, è caratterizzata da leucocitosi, ipergammaglobulinemia, positività del test VDRL liquorale, e può accompagnarsi a idrocefalo. La neurosifilide meningovascolare coinvolge le meningi, i vasi e il parenchima; soprattutto le arterie di piccolo e medio calibro vanno incontro a fibrosi, stenosi e trombosi (endoarterite obliterante), con conseguenti infarti.
La neurosifilide parenchimale, caratterizzata da invasione del tessuto nervoso da parte delle spirochete con alterazioni infiammatorie perivascolari diffuse e degenerazione e perdita neuronale con astrogliosi e proliferazione microgliale, non si realizza mai indipendentemente da una componente meningovascolare. Raramente si associano gomme miliari o nodulari. Nella forma cerebrale (paralisi generale o demenza paralitica) il cervello è atrofico, le lesioni interessano principalmente la corteccia prefrontale e il corpo striato. È presente una ependimite granulare; frequenti sono anche i depositi di ferro intraparenchimali. La tabe dorsale è una forma tardiva di neurosifilide spinale parenchimale caratterizzata da infiammazione e degenerazione dei gangli e delle radici spinali dorsali (in queste ultime compaiono processi di demielinizzazione) a cui consegue la degenerazione dei cordoni posteriori; si associa spesso ad atrofia ottica primaria. Le gomme sono rare lesioni tondeggianti, con effetto massa, simil-granulomatose con un centro di necrosi coagulativa circondata da cellule epitelioidi, cellule giganti multinucleate, linfociti e plasmacellule formanti una parete cellulare a sua volta incapsulata in tessuto fibrotico con marcata proliferazione vasale. Possono trovarsi sparse nel nevrasse, ma sono in genere localizzate sulla superficie spinale e cerebrale perché la loro capsula fibrosa origina dal connettivo meningeo e vascolare.
Il Citomegalovirus (CMV) è il più frequente patogeno virale opportunista nei malati di AIDS: segni di infezione da CMV si riscontrano nel 15-20% delle autopsie. Il quadro più comune è quello di una encefalite subacuta diffusa con formazione di noduli microgliali nella sostanza grigia, associati o meno a cellule con inclusioni virali. Il quadro istologico è sovrapponibile a quello dell'encefalite da HIV-1 ma, a differenza di quest'ultimo, il CMV può infettare tutte le cellule del SNC e tende a localizzarsi preferenzialmente nell'ependima e nella regione subependimale: ne risulta una ventricoloencefalite con necrosi tissutale massiva, emorragie, ventricolite e infiammazione dei plessi corioidei. Cellule citomegaliche con prominenti inclusioni intranucleari e intracitoplasmatiche vengono facilmente identificate tanto nelle lesioni che alla loro periferia.
Nei pazienti con AIDS il virus Herpes simplex (HSV) può causare un'encefalite acuta con un quadro clinico-patologico del tutto sovrapponibile a quello osservato negli individui non immunocompromessi. Macroscopicamente il cervello mostra segni di infiammazione, congestione e rammollimento accentuati a livello dei lobi temporali; sono presenti anche emorragie puntiformi o estese. Nel giro di 2 settimane dall'esordio dell'infezione si hanno franca necrosi e colliquazione tissutali. Istologicamente le lesioni interessano un'area notevolmente più estesa di quella riconoscibile all'esame macroscopico. Allo stadio iniziale si osserva congestione del microcircolo a livello cortico-subcorticale con emorragie puntiformi. Le inclusioni intranucleari eosinofile (corpi di Cowdry tipo A) sono suggestive della diagnosi. Il quadro lesionale evolve verso la necrosi tissutale con infiltrati mononucleari perivascolari e subaracnoidei, gliosi, satellitosi e neuronofagia. Nel neonato l'infezione, generalmente, interessa diffusamente il cervello che va incontro a necrosi estesa con idroanencefalia, poroencefalia, lesioni multicistiche. Il mezzo diagnostico più sensibile e specifico dell'infezione da HSV è la dimostrazione immunoistochimica o con ibridazione in situ del microrganismo in una biopsia cerebrale.
L'infezione da virus varicella zoster (VZV) dà conto di circa il 12% delle infezioni erpetiche nei pazienti con AIDS: i quadri neuropatologici associati comprendono un'encefalomielite, una leucoencefalite, una vasculopatia cerebrale, l'Herpes zoster oftalmico e l'encefalite trigeminale.
La leucoencefalopatia multifocale progressiva (PML), o malattia di Richardson, è stata descritta inizialmente come disordine della sostanza bianca in pazienti immunocompromessi (malattie mieloproliferative, linfoproliferative o granulomatose). L'agente patogeno responsabile è il Papovavirus JC. All'esame macroscopico il cervello rivela chiazze irregolari, mal definite, di distruzione granulare della sostanza bianca, con dimensioni variabili da pochi millimetri a un intero lobo cerebrale o emisfero cerebellare. Microscopicamente le lesioni multifocali appaiono, nelle colorazioni per la mielina, come ammassi stellari che ricordano l'aspetto di una galassia. Le lesioni consistono in aree di demielinizzazione, nel centro delle quali sono presenti lipofagi e pochi assoni risparmiati. Alla periferia delle lesioni vi sono oligodendrociti con i nuclei ipertrofici e cromatina rimpiazzata da materiale virale opalescente, la cui natura è riconoscibile con tecniche immunoistochimiche o con ibridazione in situ. Alla periferia delle lesioni si trovano, ancora, astrociti giganti e rari infiltrati linfoplasmacellulari perivascolari e subaracnoidei. Nei pazienti con AIDS l'incidenza di PML varia dall'1 al 6% e l'estensione e la gravità delle lesioni tendono a essere maggiori che nei pazienti non infetti da HIV, con tendenza alla necrosi tissutale. In fase iniziale le lesioni possono rimanere misconosciute alla TC, mentre la TRMN evidenzia lesioni sia nella sostanza bianca che in quella grigia.
Neoplasie del SNC in corso d'infezione da HIV-1. - Con il prolungarsi del tempo di sopravvivenza, in seguito alla terapia con zidovudina e al trattamento delle infezioni opportunistiche, nei pazienti affetti da AIDS vengono descritti con sempre maggiore frequenza linfomi B primari del SNC che, dopo la toxoplasmosi, rappresentano la seconda causa di lesioni cerebrali occupanti spazio. Nelle lesioni singole la diagnosi differenziale è spesso possibile solo mediante biopsia. A livello cerebrale sono frequenti anche metastasi di linfomi sistemici che nei pazienti con AIDS presentano un'alta prevalenza di elevata malignità e scarsa risposta alla terapia.
BIBLIOGRAFIA
Adams, J. H., Duchen, L. W. (a cura di), Greenfield's neuropathology, London 19925.
Artigas, J., Grosse, G., Niedobitek, F. (a cura di), The central nervous system in AIDS, Berlin-New York 1993.
Cervâos-Navarro, J., Ferszt, R., Klinische Neuropathologie, Stuttgart 1991 (tr. it.: Neuropatologia clinica, Verona 1992).
Cervâos-Navarro, J., Urich, H., Metabolic and degenerative diseases of the central nervous system: pathology, biochemistry, and genetics, San Diego, Cal., 1995.
Lambert, H. P., Infections of the central nervous system, in Handbook of infectious diseases (a cura di E. H. Kass, T. H. Weller, S. M. Wolff e D. A. Tyrrel), Philadelphia-London 1991.
Prusiner, S. B., Molecular biology of prion diseases, in ‟Science", CCLII, 1991, pp. 1515-1522.
Rose, F. C., Bynum, W. F. (a cura di), Historical aspects of the neurosciences, New York 1982.