
encefalopatia
Qualsiasi malattia caratterizzata da una lesione anatomica, per lo più diffusa, del parenchima encefalico, con caratteristiche evolutive più o meno spiccate, e di natura degenerativa, displasica, tossica, discrasica, dismetabolica o carenziale: rientrano fra le e. le vasculopatie cerebrali legate a un’alterazione del circolo generale o dell’albero arterioso, escluse quelle caratterizzate da lesioni acute a focolaio.
E. epatica
Sindrome di alterato stato di coscienza causata da insufficienza epatica e shunt porto-sistemico. In condizioni normali, nel parenchima epatico vengono metabolizzate e detossificate sostanze provenienti con il sangue portale dall’intestino, originate nel corso della digestione soprattutto proteica: ammoniaca, che si forma per azione della flora batterica sugli amminoacidi e sull’urea endogena; ammine biogene, prodotte dall’azione degli enzimi intestinali sugli amminoacidi aromatici; acidi grassi a catena corta. Nei casi di grave compromissione della funzione epatica il fegato non può più svolgere questa azione detossificante, sia perché è ridotto il numero delle sue cellule efficienti, sia perché, come frequentemente accade nei pazienti affetti da cirrosi, per le anomali comunicazioni che si stabiliscono tra il sistema portale e la vena cava, parte del sangue proveniente dall’intestino ‘salta’ il fegato ed entra direttamente nel sistema venoso generale. A livello cerebrale si realizza allora un’abnorme concentrazione di sostanze tossiche in grado di interferire con il metabolismo energetico del tessuto nervoso (ammoniaca, soprattutto) e di inibire la normale trasmissione degli impulsi neurali (ammine biogene, che agiscono come falsi trasmettitori).
La sindrome che, in considerazione della sua patogenesi, è meglio denominata e. porto-sistemica, è caratterizzata da alterazioni di vario grado dello stato di coscienza, dal torpore allo stupore, al coma (coma epatico). Può presentarsi in forma acuta episodica, che generalmente regredisce a seguito di pronta rimozione delle cause (per es., arresto del sanguinamento di varici esofagee), o in forma cronica, ricorrente o prolungatamente persistente o permanente, suscettibile per lo più di risoluzione, a seguito di adeguato trattamento, senza esiti neurologici stabili. La terapia si fonda sul contenimento della formazione intestinale di ammoniaca e ammine biogene, realizzabile con regime dietetico aproteico e somministrazione parenterale selettiva di amminoacidi (elevata percentuale di amminoacidi ramificati ed estrema riduzione o eliminazione di amminoacidi aromatici).
E. spongiformi trasmissibili
Le e. spongiformi trasmissibili (EST), o malattie da prioni, costituiscono un gruppo di malattie degenerative del sistema nervoso centrale a esito invariabilmente letale. Colpiscono sia l’uomo sia gli animali, ma nella maggior parte dei casi l’uomo non contrae la malattia da animali infetti. I soggetti colpiti presentano gravi lesioni del sistema nervoso centrale caratterizzate da vacuolizzazione del tessuto nervoso (da cui il termine spongiosi), aumento numerico e, a volte, ingrandimento degli astrociti. Spesso è presente anche una più o meno grave perdita di neuroni. A differenza di altre malattie infettive, sono assenti infiltrati di cellule infiammatorie (da cui il termine encefalopatia invece di encefalite).

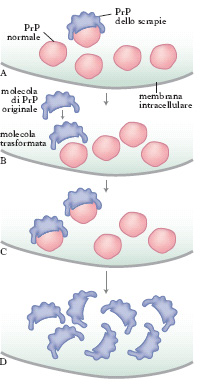
Lesione caratteristica è l’accumulo, soprattutto nel tessuto cerebrale, di una proteina amiloidea chiamata proteina prionica o PrP (➔ prione). L’accumulo di PrP si ritrova esclusivamente nei soggetti colpiti da EST e costituisce pertanto un marker diagnostico importante per distinguere queste e. da altre patologie neurologiche simili, quali, per es., la malattia di Alzheimer. La PrP deriva da un precursore fisiologico situato sulla superficie cellulare la cui funzione è ancora poco conosciuta, anche se si ipotizza un suo ruolo nei meccanismi di interazione tra cellula e cellula. Nelle cellule infette la PrP ‘normale’ va incontro a un’alterazione della sua struttura tridimensionale: due delle quattro catene elicoidali da cui è formata si trasformano, assumendo una configurazione ‘a foglietto’ (fig. 1). In seguito al cambiamento conformazionale, la proteina diviene resistente alla degradazione da parte degli enzimi cellulari, accumulandosi all’interno della cellula fino a provocarne la morte. Secondo alcuni studiosi la prima molecola di PrP patologica si forma spontaneamente. Questa si lega quindi a una molecola di PrP ‘normale’ inducendone la trasformazione e innescando un meccanismo a cascata responsabile della produzione massiva di PrP patologica (fig. 2).
E. spongiforme bovina
La BSE (bovine spongiform encephalopathy; comunemente chiamata malattia della mucca pazza) si è manifestata per la prima volta in Gran Bretagna nel 1986 e sin dalla sua comparsa ha assunto un andamento epidemico. È stato ipotizzato che i primi bovini si siano infettati all’inizio degli anni 1980 in seguito alla contaminazione delle farine di carne con l’agente dello scrapie (➔).
La malattia colpisce animali di età compresa tra i 22 mesi e i 18 anni, con un picco attorno ai 4-5 anni; il tempo medio d’incubazione è compreso tra i 4 e i 6 anni. Dal punto di vista clinico si osservano modificazioni del comportamento, come aggressività, iperestesia a stimoli tattili e uditivi, incoordinazione dei quarti posteriori, tremori muscolari e digrignamento dei denti. L’epidemia della BSE sembra sostenuta da una sola variante, diversa da quelle isolate nello scrapie, e le cui caratteristiche (tempo di incubazione e profilo delle lesioni dopo il passaggio nel topo) rimangono inalterate anche dopo il passaggio in altre specie. Per questo motivo e per la inusuale facilità di trasmissione da una specie a un’altra, la BSE emerge tra le EST come un’entità nosologica a sé stante e, nonostante la sua probabile origine dallo scrapie, distinta da esso.
Malattia di Creutzfeldt-Jakob
La malattia di Creutzfeldt-Jakob (MCJ) può presentarsi in forma sporadica, familiare o come conseguenza di trasmissione accidentale e appartiene alle EST dell’uomo, insieme alla sindrome di Gerstmann-Sträussler-Scheinker (GSS), neurodegenerazione caratterizzata da atassia e anormalità posturali, all’ insonnia fatale familiare (IFF), che causa disturbi del sonno e del sistema nervoso autonomo seguiti da insonnia e demenza, e al kuru (➔). La MCJ è presente in tutto il mondo con un’incidenza di circa un caso per un milione di abitanti, colpisce ugualmente entrambi i sessi e insorge generalmente tra i 50 e i 70 anni con una durata media di circa 5-6 mesi. Le modalità di trasmissione naturale della malattia sono sconosciute. Non sono note evidenze epidemiologiche tali da avvalorare la tesi che le pecore costituiscano il serbatoio animale dell’infezione per l’uomo. Sono noti invece casi di MCJ da trasmissione accidentale iatrogena in seguito a terapia con ormone della crescita estratto da ipofisi umane infette, a trapianto di cornea o dura madre provenienti da donatori affetti da MCJ e a inadeguata sterilizzazione di strumenti chirurgici.
La forma sporadica della MCJ è spesso caratterizzata da una fase iniziale costituita da sintomi aspecifici quali, per es., astenia e perdita di peso. Il quadro clinico progredisce con la comparsa di disturbi della memoria, disturbi psichiatrici quali modificazioni comportamentali, disturbi d’ansia, irritabilità, depressione, insonnia, disturbi della deambulazione, vertigini e disturbi visivi. Nella fase di stato (malattia avanzata), al deterioramento mentale rapidamente progressivo si associano mioclonie (guizzi muscolari), tremori e altri movimenti involontari, segni cerebellari (incoordinazione motoria, disturbi dell’equilibrio), piramidali (deficit dell’attività motoria volontaria, paresi), extrapiramidali (alterazioni dell’attività motoria automatica, quali, per es., i movimenti mimico-emotivi o gli automatismi come l’andare in bicicletta) o visivi (diplopia, allucinazioni visive). Nella fase terminale della malattia si assiste a un peggioramento dei sintomi descritti, alla comparsa in molti casi di crisi epilettiche e coma, fino al decesso che usualmente sopraggiunge per infezioni respiratorie o sistemiche. Molto utile ai fini diagnostici è il riscontro di anomalie elettroencefalografiche.
L’esame del liquor cefalorachidiano rivela una proteina anomala nei pazienti affetti da MCJ. Questa proteina è assente in altre malattie neurodegenerative ed è perciò di grande aiuto per differenziare la MCJ da altre demenze, come, per es., la malattia di Alzheimer. La diagnosi di certezza è tuttavia ancora basata esclusivamente sull’esame istologico del cervello o sull’identificazione della PrP patologica mediante tecniche immunochimiche.
Accanto alla forma sporadica di MCJ, è stata segnalata dapprima in Gran Bretagna una nuova variante ( nvMCJ) per la quale si ipotizza un legame con la BSE. Questa si differenzia dalla forma classica per un esordio precoce, una lunga durata clinica della malattia (superiore a un anno) e caratteristici sintomi di esordio rappresentati da disturbi comportamentali, modificazioni della personalità o depressione.