
gomma
Sostanza dotata di proprietà elastiche, di varia origine e natura.
Botanica
Essudato incolore o bruno, derivante dall’alterazione di cellule dei tessuti corticali o dell’alburno, che si forma a causa di un processo patologico delle piante dovuto in genere a traumi o a parassiti (gommosi o mal della g.); la g. si riunisce in grandi cavità prodotte dalla distruzione delle cellule e molto spesso esce alla superficie del corpo della pianta. Sono soggette a gommosi ciliegio, pesco, mandorlo, gli agrumi, l’olivo, il fico ecc. Si distinguono gommosi non parassitarie, traumatiche e parassitarie. Nelle gommosi non parassitarie la pianta è predisposta alla malattia in quanto gruppi di cellule non raggiungono il loro completo sviluppo e vanno soggette a gommificazione; le gommosi traumatiche (da ferite, da freddo) si verificano in ogni tessuto anche non predisposto e rappresentano spesso una protezione della pianta, mentre altre volte si ha una vera degenerazione gommosa; le gommosi parassitarie sono quelle determinate da batteri e funghi diversi (Clasterosporium, Phytophthora, Coryneum ecc.). In alcuni casi la gommosi non è una vera malattia, in altri rovina i rametti, a volte può determinare anche la morte di tutta la pianta.
Chimica
Prodotto, naturale o sintetico, solido, con proprietà elastiche, facilmente deformabile. Le g. naturali sono prodotti derivanti dalla metamorfosi di alcuni costituenti delle cellule vegetali (pareti cellulari, amido) di diverse piante che all’aria induriscono rapidamente e che hanno proprietà adesive. La denominazione g. sintetiche comprende diversi polimeri che hanno, più o meno spiccatamente, alcune proprietà della g. naturale; dei vari composti finora prodotti come sostituenti della g. naturale nessuno (salvo la g. poliisoprene) ne possiede tutte le proprietà chimiche e fisiche (per questo spesso si preferisce parlare, anziché di g. sintetica, di elastomeri, sostituenti della g. ecc.), anche se alcuni di questi prodotti sono adatti per impieghi nei quali la g. naturale non si può usare.
Provenienza della g. naturale
Le piante che possono fornire g. naturale sono oltre 300, appartenenti a famiglie diverse, tuttavia sono poche quelle importanti dal punto di vista industriale, agrario e commerciale. Alla famiglia Euforbiacee appartengono: Hevea brasiliensis del Pará; Hevea guyanensis, della Guiana; Hevea pauciflora, del Rio Negro e Rio Uaupés; diverse specie di Manihot e in particolare Manihot glaziovii, del Brasile. Questa specie resiste alla siccità e cresce rapidamente, tanto che si può cominciare a incidere al sesto anno, ma poiché non resiste ai venti, è stata sostituita da Manihot dichotoma.
Alla famiglia Moracee appartengono: Castilloa elastica del Messico, molto coltivata in America; Ficus elastica, dell’India, penisola malese, e molti altri Ficus coltivati in queste regioni. Ficus elastica può essere inciso ogni tre anni.
Alla famiglia Apocinacee appartengono: Hancornia speciosa del Brasile meridionale; Landolphia tomentosa, owariensis e altre congeneri dell’Africa e Madagascar; Kickxia (o Funtumia) elastica, dell’Africa occidentale; Mascarenhasia elastica, dell’Africa orientale e altre specie del Madagascar. Hancornia speciosa vive nei terreni rocciosi, aridi: è un arbusto di 3 m, con scarso fogliame, ha il frutto a bacca, edule, detta mangaba, donde il nome di mangabeira dato alla pianta. Kickxia elastica, albero alto 20-30 m, produce la maggior parte del caucciù esportato dall’Africa occidentale. Le piante appartenenti al genere Landolphia sono liane e alcune specie abbondano nell’Africa tropicale, dove forniscono il cosiddetto caucciù d’erba.
Alla famiglia Asteracee appartengono: Parthenium argentatum, detto guaiule, alcune specie di Solidago, Scorzonera e Taraxacum. Il primo è un arbusto degli altipiani del Messico, nel quale la g. non è contenuta in tubi laticiferi, bensì nelle cellule del parenchima fondamentale dei rami, nei raggi midollari ecc., e perciò la g. si ottiene dopo macinazione e levigazione di tutta la pianta: tale g., molto resinosa, serve soltanto a determinati usi; la coltura è estesa negli Stati Uniti meridionali. Nell’ex URSS vi sono estese coltivazioni di alcune scorzonere e di Taraxacum megalorrhizon, detto dai Russi krim-saghyz.
Primi sviluppi dell’uso della gomma
Nota in Europa subito dopo la scoperta dell’America, la g. naturale fu studiata scientificamente molto più tardi. Nel 1736, da Quito, C.-M. de La Condamine inviò all’Accademia delle scienze di Francia alcuni campioni di g., avvertendo che questa era ottenuta dal liquido, bianco come latte, che colava dalle incisioni fatte dai nativi della provincia di Esmeralda su una pianta chiamata Hevea. La stessa pianta cresceva lungo le rive del Rio delle Amazzoni, e il prodotto ottenibile per indurimento del suo latice era chiamato cahuchu(c), da cui il francese caoutchouc. Lo studioso francese informava altresì che gli Indiani si servivano della g. per vari usi, per farne torce, bottiglie ecc. Lo stesso Condamine si servì del latice per impermeabilizzare le tele con cui ricopriva i suoi strumenti astronomici. Poco dopo fu posto il problema del solvente più adatto. L.-A.-P. Hérissant e P.-J. Macquer (1763) proposero l’olio di Dippel, l’olio chiaro di trementina, l’etere ben rettificato; l’italiano G. Fabroni indicò il solvente economico della g.: il petrolio. La prima applicazione con vero carattere industriale (G. Priestley, 1770) fu la fabbricazione della g. da cancellare. In seguito i tecnici si orientarono verso la fabbricazione di impermeabili: in Inghilterra furono prodotti abiti formati da una tela raddoppiata con interposta una foglia di g.; T. Hancock costruì il primo masticatore e il primo mescolatore (1820-21) e più tardi fabbricò il primo filo elastico (1838). Nel 1839 C. Goodyear ottenne un prodotto elastico mescolando alla g. greggia zolfo e riscaldandola sopra il punto di fusione di questo. Il processo, chiamato poi vulcanizzazione, fu brevettato. A. Parkes scoprì e brevettò (1843) un nuovo solvente, il solfuro di carbonio, e in seguito (1846) realizzò tre procedimenti di vulcanizzazione a freddo con cloruro di zolfo. Nel 1921, l’inglese S.J. Peachey propose la vulcanizzazione a freddo, sfruttando la reazione fra l’idrogeno solforato e l’anidride solforosa per ottenere una deposizione di zolfo colloidale nella gomma.
Aspetti chimici
La g. naturale è costituita da un polimero dell’isoprene, e precisamente dal polimero cis- (➔ isoprene). A contatto di solventi quali idrocarburi aromatici (benzene, xileni) o alifatici (gasolina) o clorurati (tricloroetilene, tetracloruro di carbonio ecc.), la g. rigonfia, dando una massa di consistenza gelatinosa, che poi si disperde formando una soluzione. Per la presenza dei doppi legami nel poliisoprene, la g. reagisce addizionando diversi elementi (idrogeno, cloro, zolfo, ossigeno ecc.); alcune di queste reazioni hanno particolare importanza dal punto di vista pratico: così l’ossigeno, l’ozono, i perossidi ecc. causano ossidazioni che modificano le caratteristiche e le proprietà del prodotto (dando origine al cosiddetto ‘invecchiamento’). Il cloro dà origine a un prodotto resistente agli alcali e agli acidi (clorocaucciù), utilizzato nella preparazione di vernici, di adesivi ecc. La reazione fra g. e zolfo viene sfruttata nella operazione di vulcanizzazione; si ammette di solito che lo zolfo entri in combinazione legando le varie catene, reagendo sia con i doppi legami sia con gli atomi di idrogeno dei gruppi metilenici adiacenti al doppio legame. Si ottengono prodotti (g. vulcanizzata) a struttura reticolata, nei quali il movimento delle singole catene è limitato e che quindi risultano elastici ma poco plastici, poco solubili, poco rigonfiabili nei solventi ecc.; se la quantità di zolfo che reagisce con la g., anziché essere dell’ordine dell’1-3%, è maggiore, si ottengono prodotti duri, rigidi, come, per es., l’ebanite.
Produzione e commercio della g. greggia
La g. greggia è ottenuta da incisioni praticate in alberi di selva e in alberi di piantagione. La g. di selva, che ha perduto quasi interamente la sua importanza, è estratta da Hevea brasiliensis allo stato naturale nelle foreste. Quella di piantagione, che rappresenta la produzione di gran lunga più importante, è ottenuta dalla stessa Hevea diffusamente coltivata in territori della Malaysia, dell’Indonesia, del Vietnam, della Thailandia, di Sri Lanka, dell’India.
Nel 1900 la produzione mondiale di g. era data esclusivamente da alberi di selva e ammontava a circa 54.000 t, di cui metà provenienti dal Brasile e metà dalle altre regioni, principalmente dall’Africa. Lo sviluppo dell’automobilismo con la relativa richiesta di g. per pneumatici fece salire enormemente il prezzo della materia prima, che nel 1909 arrivò a una punta di 12 scellini per libbra. La situazione però era sul punto di mutare per lo svilupparsi delle piantagioni di Hevea in Oriente. Il governo brasiliano aveva posto il veto all’esportazione di semi di Hevea, ma già nel 1876 l’inglese H. Wickam era riuscito a esportarne circa 70.000 sotto il nome di ‘campioni botanici’ e dalla piccola parte di essi che rimase in vita ebbero origine le attuali piantagioni. La prima produzione sensibile (145 t) giunse sul mercato nel 1905 e nel 1913 la produzione delle piantagioni malesi superava già quella di selva del Brasile, nel 1920 era il decuplo e nel 1938 rappresentava la quasi totalità. Attualmente, la grande maggioranza della materia prima impiegata nel mondo proviene da Malaysia, Indonesia e Thailandia e in misura minore da altri paesi asiatici. Gli stessi paesi in via di sviluppo hanno nel tempo cominciato a essere anche consumatori.
Per quanto riguarda il commercio della g. greggia, esso è influenzato, come avviene per altre materie prime della zona tropicale, e molto più spiccatamente, dalla concentrazione dell’industria derivata in paesi lontani da quelli di origine del greggio: USA, Gran Bretagna, Francia, Germania, Italia, anche se tale scenario si è modificato in virtù del rapido sviluppo industriale di paesi quali la Cina, la Corea e il Brasile.
Lavorazione industriale della g. naturale
Si può operare a partire dalla g. greggia coagulata oppure direttamente dal latice. Il secondo metodo è entrato nella pratica industriale solo in epoca recente. Il primo, di gran lunga più importante, si divide in due branche, una delle quali comprende la preparazione delle mescolanze e la loro successiva lavorazione, mentre l’altra comprende la preparazione di soluzioni della g. greggia in solventi organici, le quali possono essere impiegate tal quali oppure per gommare tessuti o per ricavarne oggetti.
Lavorazione da mescole
La preparazione e la lavorazione delle mescolanze si svolgono in fasi successive. La prima è la plastificazione della g. greggia, che ha lo scopo di conferire notevole plasticità alla g. per facilitare la diffusione degli ingredienti. La g. viene fatta passare ripetutamente in un mescolatore a cilindri: questi ultimi, eventualmente riscaldati o raffreddati per mezzo di vapore o di acqua, ruotano in modo da provocare lo stiramento della g. in ‘foglie’ sempre più sottili; oppure, la g. passa attraverso un estrusore, dal quale viene espulsa in forma di un grosso tubo, tagliato poi a pezzi che si lasciano raffreddare all’aria; oppure ancora, tagliata a pezzi, viene riscaldata per un determinato tempo in stufa. Contribuisce alla plastificazione l’ossidazione in contatto con l’aria ambiente.
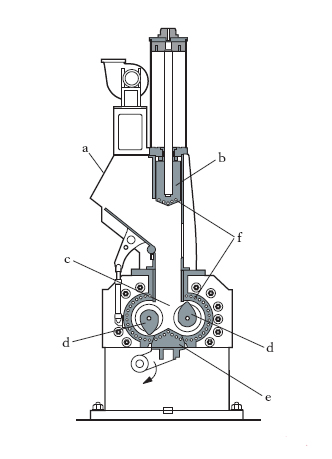
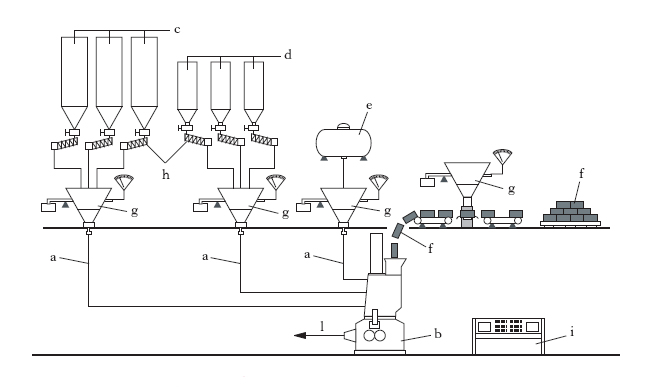
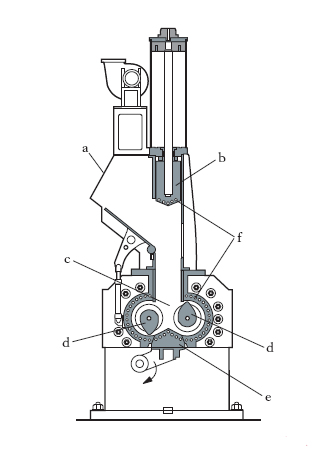
L’aggiunta e dispersione di ingredienti ha lo scopo di formare la mescolanza adatta ai vari impieghi; gli ingredienti, alcuni liquidi, altri in forma di polveri, sono: vulcanizzanti, acceleranti, plastificanti rinforzanti, antinvecchianti, cariche inerti, coloranti. In genere, acceleranti e antiossidanti sono aggiunti prima, a parte, a formare la cosiddetta ‘mescola madre’ che si aggiunge poi alla ‘mescola’ definitiva. Con passaggi successivi attraverso i cilindri si produce la necessaria dispersione degli ingredienti in modo da ottenere una mescola sufficientemente omogenea. L’operazione può essere eseguita anche in un mescolatore a camera chiusa (fig. 1) in cui la g. e gli ingredienti, caricati nella tramoggia a, vengono spinti dal pistone b nella camera c dove i rotori d effettuano la mescolazione. Il prodotto viene poi estratto dal basso attraverso la porta e. Un sistema di serpentine f provvede al raffreddamento delle varie parti della macchina. In fig. 2 è mostrato lo schema di preparazione della mescola: attraverso i condotti a vengono immessi nel mescolatore a camera chiusa b i diversi componenti: carbone, proveniente dai silos c; cariche bianche (caolino, carbonato di calcio), per appesantire il prodotto, provenienti dai silos d; oli plastificanti, provenienti dal serbatoio e; dall’alto vengono aggiunte le balle di gomma f. La dosatura precisa di ogni componente è eseguita da bilance automatiche g e coclee dosatrici h, tutte collegate a un elaboratore elettronico i che gestisce l’intero processo, alla fine del quale la mescola, uscente da l, viene avviata alle successive lavorazioni. Quando nelle mescole si raggiungono alte temperature, i vulcanizzanti vengono aggiunti a parte, per evitare una prematura vulcanizzazione.
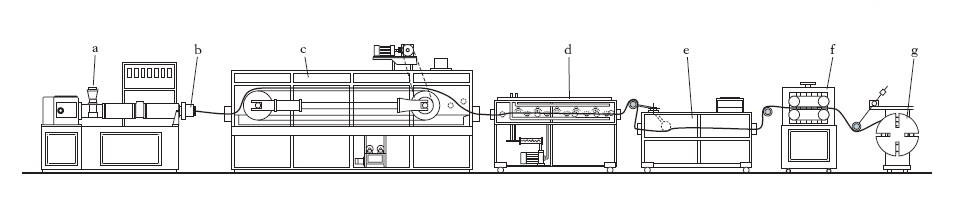
La preparazione dei semilavorati ha lo scopo di ridurre la mescola in foglie, tubi, sbarre e profilati vari, oppure tessuti gommati. Le foglie sono ottenute a caldo per calandratura. Per mezzo della calandra si esegue pure la gommatura dei tessuti, che consiste nel forzare la mescola tra i fili del tessuto (frizionatura) ed eventualmente nell’applicare sopra il tessuto una foglia di mescola (fogliettatura). Per la produzione di profilati si utilizza il processo di lavorazione per estrusione (fig. 3). Il semilavorato proveniente dal processo di mescolazione viene introdotto nell’estremità a del cilindro di uno speciale estrusore. All’interno del cilindro ruota una grossa vite, che miscela e comprime il semilavorato, spingendolo verso l’altra estremità del cilindro, dove è fissata una testa di estrusione b. La g., costretta a passare in una feritoia della testa di estrusione (detta matrice o filiera), ne assume in sezione la forma: se circolare ne deriverà un cilindro, se rettangolare una piattina ecc. La linea di estrusione è completata da un forno di vulcanizzazione in continuo c, in bagno di sali fusi, da una unità di lavaggio in acqua calda d e da una unità di refrigerazione e. Alla fine del processo il prodotto, per mezzo del trascinatore a cingoli f, è raccolto su tamburi rotanti g, oppure su un nastro trasportatore, oppure direttamente a mano. Accoppiando due estrusori si possono ottenere profilati da due mescole diverse, in particolare con effetti vari di colori. Durante l’operazione la g. può essere direttamente applicata su un’anima o un tubo centrale, facendo passare questo attraverso un’apposita testa di estrusione; tale operazione ha un interesse pratico notevolissimo per l’applicazione delle coperture di g. su tubi con interposizione di tessuto oppure su cavi elettrici. I prodotti ottenuti con la calandratura e l’estrusione possono avere già la forma definitiva e non richiedono allora che di essere vulcanizzati; in altri casi però essi devono essere ulteriormente confezionati.
La formatura ha lo scopo di dare al prodotto la forma definitiva o una forma adatta a successive lavorazioni, per es., di stampaggio. Può essere eseguita a mano, oppure per mezzo di adatte macchine. L’eventuale stampaggio è effettuato a caldo, disponendo l’oggetto, preconfezionato, in uno stampo metallico entro cui viene forzato mediante presse idrauliche.
La vulcanizzazione ha lo scopo di migliorare le caratteristiche meccaniche ed elastiche della g.; la lavorazione più importante per l’industria è quella a caldo, consistente nel portare e mantenere a una data temperatura, mediante impiego di vapor d’acqua, per un determinato tempo, l’oggetto da vulcanizzare; di solito si opera a temperature dell’ordine di 140-170 °C e per tempi variabili da pochi minuti a più di un’ora, in relazione alle dimensioni e al tipo dell’oggetto. Durante l’operazione l’oggetto crudo deve essere sostenuto, per non perdere la forma, con stampi e involucri metallici o con altri mezzi, e di solito pressato, sia per mantenerne la forma definitiva completandone la stampatura, sia per evitare la formazione di bolle d’aria, di gas o di vapore. Il calore può essere fornito dai piani delle presse, a tale scopo riscaldati, o per mezzo di un fluido riscaldato disponendo i pezzi da vulcanizzare entro autoclavi o in presse autoclavi. La pressione necessaria viene esercitata, oltreché meccanicamente da presse, anche da fluidi (aria, acqua, anidride carbonica, azoto) immessi dall’esterno nell’autoclave di vulcanizzazione oppure immessi o addirittura formati nell’interno dell’oggetto da vulcanizzare quando questo sia cavo (per es., palloni da gioco). Altri sistemi di vulcanizzazione, industrialmente meno importanti, consistono nell’esporre l’oggetto a vapori di cloruro di zolfo o nel trattarlo con una soluzione di cloruro di zolfo. Gli oggetti vulcanizzati vengono poi sottoposti a operazioni di finitura. Comune alla gran massa degli oggetti stampati è la cosiddetta sbavatura, che consiste nell’asportare la pellicola di g. (bava) che si insinua tra le varie parti che compongono lo stampo.
Lavorazione da soluzioni
La lavorazione della g. attraverso le sue soluzioni in solventi (benzina o benzene o loro miscele) è eseguita in impastatrici chiuse e segue la preparazione delle mescole. La gommatura dei tessuti, che costituisce la principale applicazione di tale lavorazione, viene eseguita con macchine spalmatrici, nelle quali il tessuto è fatto passare in tensione, rinviato più volte, per mezzo di rulli e cilindri sotto un coltello contro il quale viene deposta la soluzione da spalmare; dopo ogni passaggio, lambisce tavole riscaldate che fanno evaporare il solvente (che di solito viene recuperato). Mediante immersioni successive di adatte forme nelle soluzioni contenute in vaschette, e facendo poi sgocciolare l’eccesso di soluzione ed evaporare il solvente, si possono ottenere oggetti aventi le forme più svariate.
Lavorazione da latice
Utilizza o latice normale da piantagione o latice concentrato o latice vulcanizzato. La concentrazione ha lo scopo di aumentare il tenore in g. del latice e può essere eseguita per evaporazione, per via meccanica (per centrifugazione e filtrazione) o per via chimica. La prima fase di lavorazione consiste nella preparazione degli impasti, allo scopo di aggiungere al latice i vari ingredienti necessari per gli oggetti desiderati. Segue la preparazione degli oggetti, che può avvenire per immersione negli impasti o per estrusione di essi; per la gommatura di tessuti si procede per immersione o per spalmatura. La preparazione di oggetti per immersione delle forme negli impasti avviene: per semplici immersioni ed evaporazioni successive fino a raggiungere lo spessore desiderato; per mezzo di deposizione per coagulazione e immersione (immersione della forma dapprima in un bagno coagulante e poi nel latice); per deposizione su stampo caldo di latice contenente speciali addensanti-coagulanti; per deposizione elettroforetica su stampo (le particelle di g. cariche negativamente portate in un campo elettrico migrano all’anodo, dove si scaricano e coagulano). L’estrusione può essere eseguita in due modi: estrusione del latice fluido da una opportuna filiera e successivo passaggio in un bagno coagulante oppure estrusione con contemporanea coagulazione dell’estruso. Dopo la deposizione degli oggetti deve essere evaporato il solvente
Fabbricazione della g. sintetica
I primi tentativi di fabbricazione della g. sintetica furono orientati verso la sintesi dell’isoprene e la sua polimerizzazione, con risultati però non soddisfacenti. Durante la Prima guerra mondiale si produssero invece alcune migliaia di tonnellate di ‘gomma metile’ (detta anche metilcaucciù) utilizzando il metilderivato dell’isoprene, più facile da sintetizzare. Dopo diversi anni (1930) si giunse, in Germania, a migliori risultati, polimerizzando in emulsione il butadiene addizionato di stirene o di nitrile acrilico (i prodotti furono detti rispettivamente Buna S e Buna N); nello stesso periodo in Russia e in Italia venivano preparate varietà di g. polimerizzando il butadiene mediante sodio metallico. Successivamente, negli USA si riuscì a ottenere prodotti aventi proprietà analoghe alla g. polimerizzando soluzioni di polisolfuro di sodio e cloruro di etilene o polimerizzando il cloroprene (il polimero fu detto g. al neoprene) o polimerizzando l’isobutilene da solo (il prodotto fu detto g. butile) o addizionato di piccole percentuali di isoprene o altri dieni. In tempi più recenti si è riusciti, mediante polimerizzazione stereospecifica, a preparare un prodotto con la struttura della g. naturale (g. poliisoprene); sempre per polimerizzazione stereospecifica è stato possibile produrre un polimero dell’isoprene con la struttura trans, caratteristica della guttaperca.
Con i diversi tipi di g. sintetica prodotti per polimerizzazione in emulsione è possibile disporre anche di prodotti da impiegare sotto forma di latice (specie per preparare masse porose e simili); però, rispetto al latice di g. naturale, i latici sintetici hanno in genere l’inconveniente di avere particelle troppo grosse e di richiedere aggiunte troppo forti di composti stabilizzanti.
La lavorazione industriale della g. sintetica si svolge sostanzialmente nelle stesse fasi che caratterizzano l’industria della g. naturale: mescola, formatura, vulcanizzazione. Si hanno anche lavorazioni da soluzioni e da latice. Questi latici risultano da emulsioni dei vari elastomeri sintetici, da soli o addizionati ad altri componenti capaci di aumentare le proprietà adesive.
La produzione annuale di elastomeri sintetici rispetto a quella totale di g. è superiore al 60%.
Classificazione delle g. sintetiche
Seguendo l’uso introdotto inizialmente negli USA, le g. sintetiche vengono correntemente classificate in g. di tipo R, non resistenti agli oli minerali (g. butile, g. etilene-propilene, al polibutadiene); g. di tipo S, resistenti ai prodotti petroliferi, a loro volta divise in g. a elevatissima resistenza (g. al polisolfuro), a elevata resistenza (g. nitrile, g. poliuretaniche) e a media resistenza (al neoprene, polietilene-clorosolfonato); g. di tipo T, resistenti alla temperatura, divise in due sottogruppi comprendenti g. resistenti ad alte e basse temperature (g. siliconiche) e g. resistenti all’aria calda e agli oli minerali (g. acriliche e g. fluorurate).
G. acriliche
Prodotti di polimerizzazione di esteri dell’acido acrilico. Tali g. sono caratterizzate da una elevata resistenza all’invecchiamento, agli agenti ossidanti, al calore, da una bassa permeabilità ai gas, ma anche da una scarsa resistenza all’acqua e ai solventi contenenti atomi di ossigeno. Nonostante il loro carattere saturo sono suscettibili di autovulcanizzazione perché, in presenza di agenti alcalini (metasilicato di sodio, idrato potassico, ossido di piombo), il radicale −COOR dell’unità monomerica
(dove R è un radicale idrocarburico) elimina con l’atomo di carbonio terziario di un’altra catena una molecola di alcol e si forma un legame covalente tra due atomi di carbonio di due catene. Di solito, però, si preferisce ricorrere alla formazione di copolimeri con monomeri capaci di introdurre nella catena un doppio legame o un atomo di cloro, utilizzabili per una successiva reazione di reticolazione.
G. butadiene-stirene
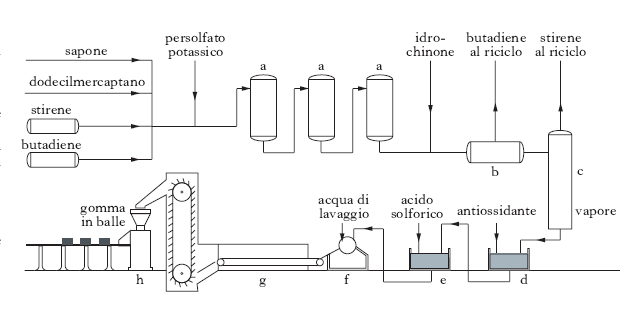
Nota anche con i nomi di Buna S, GRS (da Government Rubber Styrene type, perché la produzione negli USA avvenne, durante la Seconda guerra mondiale, in base a un programma governativo), e SBR, è costituita da un copolimero del butadiene e dello stirene (di solito nel rapporto all’incirca di 3:1). Il processo di copolimerizzazione (fig. 4) consiste nell’emulsionare con sapone i due prodotti monomeri, accuratamente depurati; nell’aggiungere una sostanza regolatrice della grandezza molecolare del polimero (dodecilmercaptano) e un iniziatore della polimerizzazione (persolfato potassico); nel riscaldare la miscela a circa 50 °C in apparecchi metallici internamente vetrificati, per 12-15 ore, dopo di che la polimerizzazione si arresta aggiungendo piccole percentuali di idrochinone. A questo momento la polimerizzazione è avvenuta per il 70-75%; non conviene spingerla oltre, perché con gradi di conversione maggiori si otterrebbero polimeri con caratteristiche tecnologiche inferiori. Eliminati i monomeri inalterati ancora presenti, il latice, addizionato di un antiossidante, viene coagulato con acido solforico; dopo il passaggio attraverso un filtro e un essiccatore, le particelle solide ottenute si comprimono in balle. Si ottiene un prodotto vulcanizzabile con zolfo, come la g. naturale, e che si usa, addizionato con nerofumo, specie per pneumatici di veicoli, per tubi, cinghie, suole ecc. Rispetto alla g. naturale presenta un migliore comportamento all’invecchiamento, ma una minore elasticità e una resistenza minore alla lacerazione.
Una g. butadiene-stirene, detta g. fredda, è stata ottenuta operando la polimerizzazione a bassa temperatura: tale prodotto ha caratteristiche meccaniche migliori, struttura più regolare, maggiore resistenza all’abrasione (i pneumatici fabbricati con g. fredda hanno una durata maggiore del 30% rispetto a quella degli pneumatici ordinari). La g. fredda si ottiene mediante polimerizzazione effettuata intorno a 5 °C; l’operazione viene arrestata quando si sono raggiunti gradi di conversione dell’ordine del 60% (ciò che richiede 12-15 ore). Si può far avvenire la polimerizzazione anche a −40 °C, però in questo caso bisogna introdurre sostanze capaci di iniziare la reazione anche a queste basse temperature; adatti si sono dimostrati alcuni diazocomposti, miscele di perossido di benzoile con un sale ferroso, o di una poliammina con un perossido organico e un sale ferroso ecc. Le g. stirene-butadiene vengono classificate in base a una numerazione standardizzata, stabilita dall’Office of rubber reserve, che è indicativa delle caratteristiche del prodotto. Grazie al largo impiego che esse trovano in vari settori, sono al primo posto nei consumi, assorbendo il 35% circa del mercato globale di elastomeri sintetici.
Medicina
In patologia, nodulo profondo della pelle, o di altri tessuti, che evolve verso il rammollimento e l’ulcerazione, dando esito a una sostanza di aspetto gommoso. La forma più nota e caratteristica è rappresentata dalla g. sifilitica (o luetica), manifestazione della sifilide terziaria, il cui contenuto è costituito da un liquido denso e filante, di aspetto gommoso. Le g. sifilitiche della pelle danno luogo a ulcerazioni a stampo, spesso d’aspetto reniforme, seguite da una cicatrice piana e regolare; quelle del palato o del setto nasale danno luogo a perforazioni più o meno ampie e persistenti; quelle degli organi interni sono tanto più gravi quanto più importante è l’organo che viene colpito. Sono da rammentare inoltre le g. tubercolari, espressione della tubercolosi cutanea o scrofuloderma (➔), e quelle secondarie a sporotricosi o ad altre micosi profonde.