
petrolio
petrolio Liquido oleoso, più o meno denso e viscoso, di colore da giallo a bruno scuro e a nero, dotato di fluorescenza da verde ad azzurra, di odore caratteristico, costituito prevalentemente da idrocarburi liquidi che contengono disciolti idrocarburi naturali solidi o gassosi, accompagnati da relativamente piccole percentuali di composti ossigenati, solforati, azotati. È detto anche olio minerale. Si ritrova in sacche o falde permeanti rocce porose a profondità variabili da poche decine di metri a qualche kilometro.
Composizione chimica
La composizione elementare dei p. è compresa entro i seguenti limiti: carbonio 79-89%; idrogeno 9,5-15%; azoto 0,02-2%; ossigeno 0,1-7%; zolfo 0,01-6%. Gli idrocarburi costituenti il p. appartengono alla serie paraffinica, naftenica e aromatica.
Gli idrocarburi paraffinici presenti nei p. sono tutti quelli della serie CnH2n+2 a partire da n=1 fino a n=30-40, del tipo a struttura sia lineare sia ramificata; a temperatura ordinaria quelli contenenti fino a 4 atomi di C sono gassosi, quelli fino a 16 sono liquidi e quelli superiori sono solidi. Gli idrocarburi olefinici sono presenti in minime quantità (mentre olefine e diolefine si formano, in quantità anche rilevanti, durante i trattamenti termici del petrolio).
I composti naftenici dei p. sono costituiti da idrocarburi ciclici, del tipo (CH2)n, con lunghe catene laterali; predominano i derivati del ciclopentano e del cicloesano, che sono le strutture cicliche più stabili.
Gli idrocarburi aromatici sono rappresentati dal benzene e dai suoi derivati (toluene, xileni ecc.), nonché da idrocarburi a più anelli e loro derivati. Sono presenti, infine, idrocarburi che partecipano alle varie serie, risultando costituiti da anelli aromatici e naftenici su cui sono ancorate catene, lineari o ramificate, di idrocarburi della serie paraffinica.
La maggior parte dei composti ossigenati nei p. è di natura acida (acidi grassi alifatici, acidi naftenici). I composti solforati, talvolta presenti in percentuali notevoli, sono costituiti da idrogeno solforato, da mercaptani, da solfuri, da disolfuri. I composti azotati, rappresentati da derivati della chinolina, della piridina, del pirrolo, da basi azotate aromatiche e non aromatiche, sono contenuti nei p. generalmente in quantità minori rispetto a quelli solforati ma, in ogni caso, i p. che contengono più zolfo contengono anche più azoto. Nei p. sono presenti inoltre composti resinosi e asfaltici, sostanze di natura complessa, costituite dalla condensazione di nuclei aromatici e naftenici, di elevato peso molecolare ed esistenti nel p. in forma colloidale. Nel p. sono contenute anche piccolissime percentuali di composti metallorganici a base di vanadio, ferro, nichel ecc.; spesso, inoltre, il p. contiene la cosiddetta salamoia, cioè una soluzione salina acquosa intimamente miscelata con la fase idrocarburica; la salamoia viene separata prima del frazionamento primario (➔ dissalazione).
Formazione
Naftogenesi
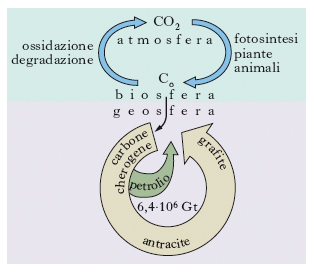
Benché sia teoricamente possibile che gli idrocarburi si formino anche attraverso processi inorganici, è ormai largamente accettata la loro natura organica (naftogenesi). Gli sviluppi conseguiti dalla geochimica organica hanno infatti evidenziato come il carbonio, che entra a far parte della sostanza organica attraverso il processo della fotosintesi, giochi un ruolo fondamentale. Con la morte degli organismi, la sostanza organica è degradata a opera dei batteri e il carbonio organico viene in parte ossidato in CO2 e in parte riutilizzato dagli organismi per sintetizzare nuova materia vivente (ciclo del carbonio organico; fig. 1). Il carbonio che è sfuggito nel corso del tempo geologico a questi due processi si è accumulato in grandi quantità nei bacini sedimentari e in determinate rocce ha raggiunto concentrazioni maggiori dello 0,3-0,5%, rendendole materiali potenzialmente in grado di generare petrolio (rocce madri).
Tra i diversi litotipi che costituiscono le rocce madri, in ordine di importanza si hanno: le rocce madri argillose, quelle calcaree e dolomitiche, le silicee e quelle carboniose. La sostanza organica contenuta nei sedimenti è costituita da molecole piuttosto semplici (biomonomeri: glicerolo, acidi grassi, amminoacidi, zuccheri, fenoli), le quali derivano dalle molecole più complesse (biopolimeri: lipidi, proteine, carboidrati, lignine, tannini), che costituiscono in origine la sostanza organica prima della scomposizione operata dai batteri. Parallelamente alle trasformazioni che subiscono i sedimenti per diventare rocce sedimentarie (costipamento, cementazione, litificazione), anche la materia organica in essi contenuta si modifica e in particolare i biomonomeri si ricondensano in molecole più complesse chiamate geopolimeri.
Cherogene (o kerogene)
Il prodotto più importante dei geopolimeri è rappresentato dal cherogene, che costituisce la sostanza dalla quale, attraverso successive modificazioni, si genererà p. attraverso un processo di trasformazione detto maturazione. Altri composti organici che si affiancano al cherogene sono i cosiddetti markers biologici o fossili geochimici, i quali, inglobati nei sedimenti, hanno subito lievi modifiche e sono ancora in grado di dare indicazioni sulla natura originaria della sostanza organica presente nel sedimento stesso.
Il cherogene ha una struttura molto complessa alla quale non è possibile attribuire una formula chimica generale. Esistono invece diversi tipi di cherogene in relazione ai tipi di sostanza organica da cui deriva; per differenziare questi differenti cherogeni vengono utilizzati i rapporti atomici dell’idrogeno e dell’ossigeno rispetto al carbonio; si distingue così un cherogene di tipo I con alto rapporto H/C e basso rapporto O/C, tipico degli ambienti di sedimentazione lacustre; un cherogene di tipo II, con alto rapporto H/C e basso rapporto O/C, caratteristico degli ambienti marini; un cherogene di tipo III con basso rapporto H/C e alto rapporto O/C, che si forma in ambienti di sedimentazione continentale, in prossimità della costa. Questi 3 tipi di cherogene determinano anche la densità degli idrocarburi che da essi derivano, così che il primo tipo tende a produrre generalmente p., il secondo p. e gas e il terzo prevalentemente gas.
Esistono diversi metodi che consentono di valutare il grado di maturazione del cherogene presente nelle rocce, tra cui quello più semplice è la valutazione del colore; all’inizio infatti il cherogene immaturo è generalmente giallo-verdastro e diventa sempre più scuro man mano che aumenta la sua maturazione in quanto aumenta il suo contenuto di carbonio. Un altro metodo è rappresentato dalla valutazione della riflettanza della vitrinite, cioè dalla misura della quantità di luce riflessa da questa sostanza carboniosa di origine vegetale che costituisce uno tra i componenti più diffusi del cherogene.
Stadi di trasformazione del cherogene
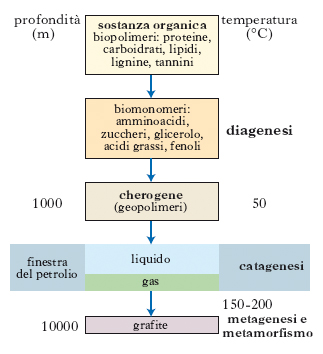
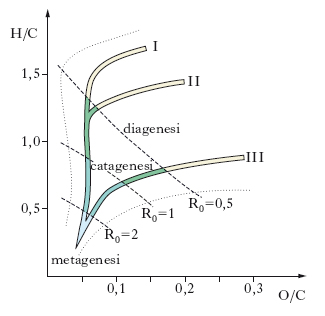
Il processo di trasformazione della materia organica in cherogene e da questo in p. avviene attraverso 3 stadi successivi chiamati: diagenesi, catagenesi e metagenesi; un quarto stadio è rappresentato dal metamorfismo, che porta i sedimenti alla perdita quasi completa dei propri caratteri originari attraverso una più o meno completa ricristallizzazione delle rocce e del loro contenuto organico (fig. 2 e 3).
Nel primo stadio (diagenesi) i processi fondamentali che si verificano sono la trasformazione del sedimento in roccia e della sostanza organica in cherogene. Durante questa fase i sedimenti raggiungono profondità sui 1000 m e temperature di 50 °C; l’acqua presente nel sedimento viene espulsa per compattazione e nella materia organica, a seguito dell’attività batterica, si ha la distruzione dei biopolimeri e la formazione dei geopolimeri. Sempre durante la diagenesi, in presenza di una materia organica quasi esclusivamente composta di resti vegetali, si formano inoltre ligniti e carboni bruni.
Nel secondo stadio (catagenesi) il processo più importante è la trasformazione del cherogene in p., a cui si accompagna anche la formazione di metano. Durante questa fase, per effetto della subsidenza, lo spessore dei sedimenti può raggiungere e superare anche i 2000 m e la temperatura aumentare fino a 150 °C. In queste condizioni la pressione sui sedimenti e sulla materia organica è molto elevata (1000-1500 bar), così che, a seguito della ulteriore compattazione, le rocce diminuiscono la loro porosità e permeabilità, viene espulsa altra acqua, e il cherogene, completando la sua maturazione, produce tutto il p. possibile.
Questo processo di maturazione, oltre che dalla pressione e dalla temperatura, è fortemente dipendente anche da un altro fattore: il tempo di cottura. In sostanza, la maturazione può essere raggiunta in tempi diversi in relazione alle differenti temperature a cui la materia organica è sottoposta: in aree dove il gradiente geotermico è elevato il cherogene può maturare e quindi produrre p. in tempi più brevi, mentre in aree dove il gradiente geotermico è più basso i tempi di maturazione saranno necessariamente più lunghi. In funzione di questi fattori esisteranno dunque una profondità e una temperatura critica che determineranno il livello necessario affinché inizi il processo di maturazione del cherogene, e una profondità e una temperatura critica alle quali il processo di maturazione si completa e il cherogene avrà così generato tutto il p. possibile. Il range entro il quale si verifica questo processo è chiamato finestra del p. (oil window; fig. 2).
Durante la metagenesi il cherogene, continuando nella sua maturazione, diventa stracotto (overcooked), perde ulteriormente idrogeno e ossigeno e si arricchisce sempre più in carbonio; durante questa fase cessa in ogni caso la formazione di idrocarburi liquidi e si genera solo metano che in parte deriva anche dal cracking termico del p. che si era generato durante la catagenesi. I carboni in questa fase si trasformano in antracite. Con l’aumento ulteriore della temperatura (150-200 °C) e a profondità superiori ai 5-6 km si entra nel campo del metamorfismo; in questa fase gli idrocarburi sono assenti e quel che resta del cherogene si trasforma in grafite.
Manifestazioni petrolifere
Quando il p. fuoriesce a giorno oppure nel sottosuolo in conseguenza di scavi o dell’esecuzione di pozzi, si hanno quelle che vengono dette manifestazioni petrolifere. Vengono distinti 2 tipi di manifestazioni: quelle superficiali e quelle profonde. Le manifestazioni superficiali (fig. 4) si evidenziano quando le rocce entro le quali il p. ha migrato intersecano la superficie terrestre, e vengono classificate in solide, liquide e gassose: tra quelle solide particolare importanza rivestono gli scisti bituminosi, mentre quelle liquide consistono generalmente in un gocciolamento libero di olio, il quale risale in superficie attraverso le fratture della roccia o attraverso strati porosi e permeabili; infine, le manifestazioni gassose sono emissioni superficiali di gas e sono generalmente associate a quelle liquide. Il gas può essere solo metano o anche anidride carbonica e idrogeno solforato. Alle manifestazioni gassose è spesso legato il fenomeno del vulcanismo sedimentario: il gas che fuoriesce in superficie può trascinare con sé sedimenti argillosi e acqua o anche olio e sabbia; questo materiale viene generalmente deposto con la venuta a giorno e nel tempo può dar luogo a un cono di fango con una morfologia simile a quella di un apparato vulcanico. Le manifestazioni profonde si verificano a seguito di scavi di pozzi e trincee di varia natura; come quelle superficiali vengono distinte in solide, liquide e gassose.
Le manifestazioni petrolifere rivestono un ruolo importante nella ricerca di idrocarburi e vanno tenute in debito conto.
Lavorazione del grezzo
Il complesso delle lavorazioni eseguite sul p. grezzo per ottenere la gamma di prodotti desiderati viene definito genericamente raffinazione del grezzo. Per ottenere i diversi prodotti finiti, le raffinerie di p. eseguono 2 successivi gruppi di operazioni: dapprima un frazionamento primario sul grezzo per ottenere correnti di prodotti intermedi, consistenti in frazioni aventi diverso intervallo di ebollizione e in un residuo; quindi processi specifici per ogni frazione, seguiti da miscelazioni, aggiunte di additivi ecc., per ottenere da tali intermedi (o semilavorati) i prodotti finiti da immettere sul mercato.
Le raffinerie
Le raffinerie sono costituite fondamentalmente da: impianti di lavorazione; serbatoi per la materia prima (il grezzo), gli intermedi e i prodotti finiti; sistemi per movimentare il grezzo, gli intermedi e i prodotti finiti; servizi ausiliari per generare e distribuire energia elettrica, vapore, acqua depurata, aria compressa, azoto, combustibili per uso interno. Le raffinerie vengono classificate in base alla potenzialità e al ciclo di lavorazione. Di fatto non esistono due raffinerie che lavorino il grezzo in modo del tutto identico; le differenze sono determinate dalle condizioni di mercato dei prodotti e dalle caratteristiche dei grezzi. Si può operare una distinzione di massima tra raffinerie a combustibili che destinano il residuo a olio combustibile e/o a bitume e raffinerie a lubrificanti; queste ultime, assai meno numerose, dispongono di impianti di distillazione sotto vuoto più complessi e di unità per trattare distillati e residuo da vuoto ottenendo frazioni lubrificanti (deasfaltazione, estrazione con solventi, deparaffinazione).

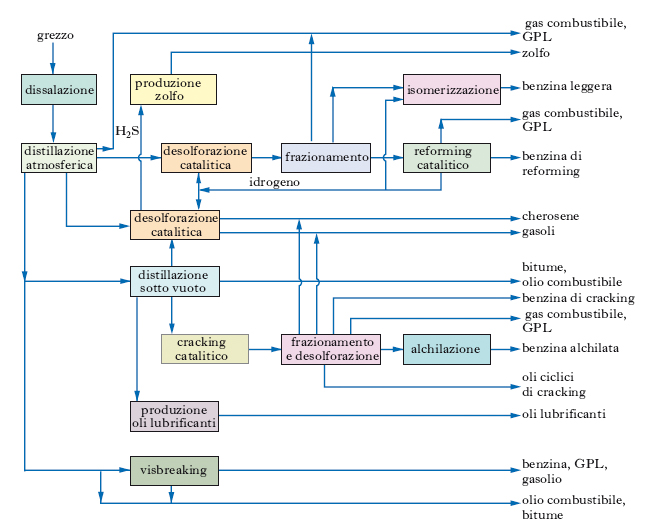
Le raffinerie si classificano per lo più in 2 grandi gruppi: a) raffinerie a ciclo semplice (hydroskimming), che non alterano sostanzialmente le rese dal grezzo; i principali impianti sono distillazione, desolforazione, reforming; b) raffinerie di conversione, che modificano le rese dal grezzo a favore di determinati prodotti, in genere verso la massima resa in benzina; oltre agli impianti citati dispongono di unità di cracking ed eventualmente di alchilazione. Le fig. 5 e 6 riportano gli schemi tipici dei due gruppi di raffinerie. Le raffinerie a ciclo semplice sono dotate anche di impianti di isomerizzazione, per produrre benzine ad alto numero di ottano senza aggiunta di antidetonanti al piombo, ma composte soltanto di idrocarburi, eventualmente con aggiunta di composti ossigenati quali componenti ad alto potere indetonante. Queste raffinerie possiedono spesso anche impianti di conversione termica, soprattutto visbreaking (viscosity-breaking), per aumentare la resa in gasolio a scapito dell’olio combustibile; ciò rende meno netto il confine tra i 2 gruppi principali di raffinerie. Un’altra evoluzione della struttura delle raffinerie di entrambi i tipi riguarda l’adozione di impianti per produrre idrogeno mediante conversione con vapor d’acqua di gas di raffineria ed eventualmente anche di frazioni liquide. Tale processo è considerato tipicamente petrolchimico, ma dato il crescente fabbisogno di idrogeno in raffineria, soprattutto per processi di idrodesolforazione, il reforming catalitico (tradizionale fornitore netto di idrogeno in raffineria) può essere insufficiente a fornire tutto l’idrogeno necessario. Alcune compagnie petrolifere hanno introdotto in raffineria anche processi di gassificazione di residui, concettualmente analoghi a quelli da tempo usati per trasformare i carboni fossili in CO e H2; ciò consente di convertire completamente determinati residui in gas, poi impiegato per la produzione di energia elettrica.
Frazionamento primario
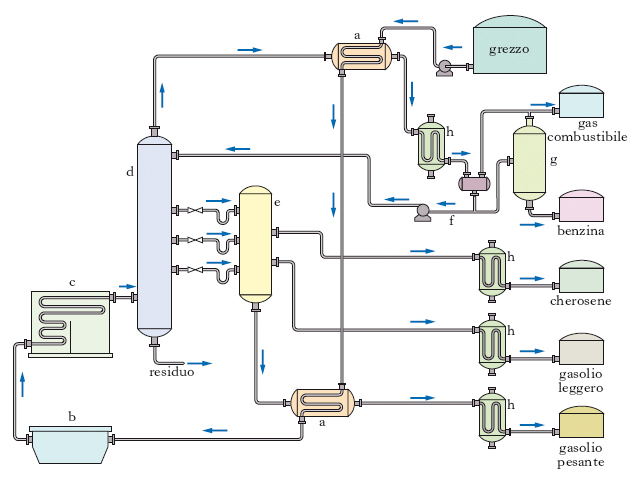
Un primo trattamento del p. mira a suddividerlo non in singoli composti ma in frazioni, cioè in miscele, ancora molto complesse, di numerosi composti. Questo frazionamento primario si effettua basandosi sulla diversa volatilità, a sua volta legata al peso e alla complessità molecolare, presentata dai singoli componenti; esso consiste in una distillazione (detta anche topping), che separa le cosiddette frazioni primarie (semilavorati): gas e benzine fino a 180-200 °C, petrolio (cherosene) fino a circa 250 °C, gasoli fino a 350-380 °C, residuo che non distilla. Lo schema di processo di un tipico impianto di distillazione atmosferica del grezzo è riportato nella fig. 7. Il grezzo da alimentare al topping deve essere per quanto possibile privo di acqua e di sali. Allo scopo la distillazione è preceduta da un’operazione di dissalazione. L’alimentazione passa attraverso una serie di scambiatori a per ricuperare al massimo il calore delle diverse frazioni distillate e del residuo; lo scambio avviene ovviamente con le correnti in uscita via via più calde. L’alimentazione preriscaldata negli scambiatori di calore attraversa il dissalatore b e quindi passa nel forno c alla temperatura richiesta (350-380 °C) ed entra quindi nella colonna di distillazione d. I prelievi laterali (cherosene, gasolio leggero e gasolio pesante) vengono inviati a colonnine di stripping e alimentate con vapore surriscaldato. La benzina di testa costituisce in parte il riflusso f e in parte viene inviata alla colonna di stabilizzazione g per privarla delle frazioni gassose che può contenere. Tutti i prodotti in uscita passano attraverso gli scambiatori h, mentre il residuo viene generalmente inviato all’unità di distillazione sotto vuoto, che opera a pressione ridotta (30-100 mbar); l’abbassamento di pressione comporta un abbassamento della temperatura di ebollizione dei vari composti, che possono così distillare senza decomporsi. Si ottengono in tal modo frazioni destinate alla produzione di lubrificanti e agli impianti di cracking catalitico e un ulteriore residuo, che può essere destinato alla produzione di bitumi, di oli combustibili, o agli impianti di cracking termico.
Le frazioni ottenute con la distillazione subiscono ulteriori trattamenti, primo fra tutti quello di idrodesolforazione, prima di diventare prodotti finiti. Quelle più pesanti e i residui possono venir sottoposti a processi di conversione.
Idrodesolforazione
I processi di raffinazione con idrogeno, dei quali fa parte l’idrodesolforazione, hanno lo scopo di allontanare alcuni elementi nocivi presenti nei grezzi e nelle loro frazioni, come lo zolfo, l’azoto, l’ossigeno e i metalli legati nelle molecole organometalliche. Essi comprendono un ampio spettro di processi catalitici, dal pretrattamento della benzina con cui alimentare il reforming fino alla desolforazione dei residui. Anziché dalla rottura di legami C−C, le reazioni di idroraffinazione sono caratterizzate essenzialmente dalla rottura dei legami C−S, C−O, C−N, che porta ad allontanare zolfo, azoto e ossigeno sotto forma di H2S, H2O e NH3. Durante il processo si ha anche la saturazione delle eventuali olefine presenti nell’alimentazione. I processi di idrodesolforazione, realizzati in tutte le raffinerie, utilizzano l’idrogeno generato dal processo di reforming eventualmente integrato con idrogeno prodotto in appositi impianti. Si trattano sia frazioni leggere (GPL e benzine) sia intermedie (cherosene, gasolio) per ottenere prodotti finiti conformi alle specifiche riguardanti il contenuto di zolfo. Si trattano anche alimentazioni da inviare a processi che impiegano catalizzatori sensibili allo zolfo e/o all’azoto. A seconda del tipo di processo e di carica, le condizioni operative variano da ca. 300 °C fino a ca. 360 °C, sotto pressioni di idrogeno da 20 a 40 bar. I catalizzatori sono fondamentalmente ossidi e solfuri semiconduttori di metalli dei gruppi VI A e VIII A, soprattutto Co−Mo.
Processi di conversione
Lo scopo principale dei processi di conversione, termici o catalitici, è quello di modificare le rese dal grezzo a favore di determinati prodotti, come la benzina o il gasolio, più leggeri di quelli di partenza. Ciò si realizza provocando la rottura (cracking ) delle molecole più complesse per formare molecole più piccole e quindi prodotti più leggeri. Dei trattamenti esclusivamente termici conservano importanza il visbreaking, il cracking termico e il coking. Il visbreaking è impiegato soprattutto per aumentare la resa in distillati medi. Il cracking termico è il più antico processo di conversione dei distillati medi in benzina; opera a temperature più alte del visbreaking per fornire essenzialmente gas, benzina e residuo. Il coking è stato sviluppato per trasformare i residui in gas, distillati e coke facendo permanere per lungo tempo l’alimentazione ad alta temperatura. Nonostante la superiorità dei processi catalitici, quelli termici hanno tuttavia una qualche importanza, soprattutto perché consentono di trattare anche i residui, cosa non facile con i processi catalitici.
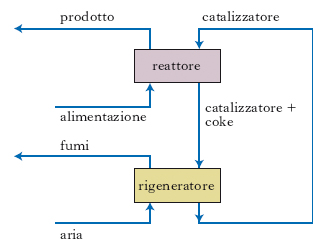
I processi catalitici di conversione più importanti sono il cracking, che trasforma alimentazioni costituite da distillati medio-pesanti, come gasoli da vuoto o da processi termici, in benzina con rese elevate, e l’idrocracking. Quest’ultimo è un processo molto versatile, che necessita però di alte pressioni e consuma quantità rilevanti di idrogeno, ed è quindi assai costoso. Il primo processo di cracking catalitico (processo Houdry) a letto fisso fu presto abbandonato a favore dei processi a letto mobile (houdryflow, thermophor). In questi il catalizzatore, sotto forma di sferette con diametro di 2-3 mm, circola tra il reattore e il rigeneratore (fig. 8). La rigenerazione continua del catalizzatore è necessaria perché su di esso si depositano i residui carboniosi (coke) sottoprodotti delle reazioni di cracking, che vengono quindi bruciati nel rigeneratore. In seguito sono stati unicamente impiegati processi a letto fluidizzato: il principio è lo stesso, con la differenza che il catalizzatore è in polvere e viene mantenuto in sospensione nella fase vapore idrocarburica. Il reattore può essere sistemato a fianco o sopra il rigeneratore, secondo la tecnologia di progetto. Il catalizzatore fluidizzato passa dall’uno all’altro per gravità o per differenza di pressione; il ritorno nel recipiente a livello o pressione superiore è assicurato variando la densità apparente del catalizzatore mediante immissione di vapor d’acqua (o di aria nel caso del rigeneratore). Il catalizzatore svolge più funzioni: oltre ad accelerare le reazioni di cracking funziona anche da supporto per il coke che si forma e da trasportatore di calore dal rigeneratore al reattore. I processi moderni di cracking operano a temperature intorno ai 500 °C e a pressioni di poco superiori a quella atmosferica, con rese di conversione in benzina superiori al 50%, rispetto alla carica pesante alimentata; producono anche GPL e piccole quantità di olefine C3 e C4.
I primi catalizzatori di cracking erano composti da silice e allumina sostanzialmente amorfe. A partire dal 1940 si impiegarono catalizzatori sintetici, sempre costituiti da allumino-silicati. Un netto progresso si è avuto all’inizio degli anni 1960 con l’introduzione dei catalizzatori cristallini zeolitici, molto più selettivi, che ha consentito di eliminare il reattore catalitico di cracking e di portare a termine le reazioni direttamente nella linea di trasferimento del catalizzatore dal rigeneratore; tale linea viene definita in tal caso riser reactor. I processi più recenti sono in effetti processi di riser cracking in cui il condotto di sollevamento può essere interno o esterno al rigeneratore, per meglio sfruttare il calore di combustione del coke.
Industria e geopolitica del petrolio
{kind=link}
(v. fig. 9)
Cenni storici
La storia dell’industria petrolifera si fa incominciare dal giorno (27 agosto 1859) in cui a Titusville, un villaggio di legnaioli della Pennsylvania, fu perforato il primo pozzo di petrolio. Prima di allora, il p. utilizzato era soltanto quello che risultava dal trasudamento in superficie o che poteva essere scremato da venature e da pozzi d’acqua. La trivellazione del pozzo di Titusville, permettendo di utilizzare in quantità commerciali il p. per ricavarne il cherosene, aprì improvvisamente ai produttori ampie prospettive di guadagno in un mercato che era stato già preparato convenientemente dai commercianti del cherosene, ottenuto per distillazione del carbone. Il salto produttivo del grezzo, nella sola Pennsylvania nord-occidentale, negli anni immediatamente successivi alla perforazione del pozzo di Titusville, è già di per sé sufficiente a indicare il passaggio dalla fase primitiva della estrazione del p. a quella industriale e commerciale moderna: dai 2000 barili del 1859 si passò, 10 anni dopo, a 4.800.000 barili e, nel 1871, a 5.205.000 barili.
Nel periodo, che possiamo definire ‘pionieristico’, precedente la formazione delle grandi società petrolifere, le condizioni del mercato del p. erano disordinate, tumultuose. Piccole società, con capitali assai modesti, si inserivano con facilità in uno dei rami del processo produttivo: o in quello della perforazione o in quello della vendita o in quello della fabbricazione dei barili per la conservazione e il trasporto, quasi sempre senza nessun vincolo o rapporto con gli altri settori della produzione. Nei primi tempi, alla nuova attività industriale si dedicarono i reduci della guerra civile americana, che vi trovarono un mezzo di rapidi guadagni. Rendeva allettante l’impresa petrolifera anche il principio comune alla legislazione anglosassone che il proprietario del suolo era al tempo stesso proprietario del sottosuolo, quindi dei prodotti minerari. Come reazione all’incertezza e all’instabilità determinate dalla concorrenza sfrenata e dall’irresponsabilità dei pionieri, alcune società cominciarono a collegarsi per ridurre i rischi e controllare meglio il mercato. La Standard oil co. sorse così come associazione di azionisti di varie compagnie, allo scopo di mettere ordine nelle varie fasi dell’attività petrolifera. Con la creazione della Standard oil co. un nuovo periodo si apre nella storia del p., periodo dominato dalla figura di J.D. Rockefeller. L’organizzazione di Rockefeller s’interessava di tutte le fasi successive alla produzione del p.: dalla raffinazione al commercio. La sua potenza crebbe con l’aumento dell’importanza, nello sviluppo dell’industria meccanica, dei lubrificanti, sempre più richiesti dalla diffusione e dal perfezionamento del motore a scoppio. L’industria automobilistica concorse in maniera decisiva ad allargare le possibilità d’impiego della produzione petrolifera.
Il monopolio che si trovò a esercitare la Standard oil co. nel mercato petrolifero statunitense finì con il suscitare l’intervento del governo di Washington, culminato nel 1911 con una sentenza di scioglimento dell’organizzazione; delle 33 compagnie che ne risultarono, 3 cominciarono a ingrandirsi, soprattutto sviluppando le loro attività all’estero: la Standard oil of New Jersey (poi Exxon o Esso), la Mobil oil e la Standard oil of California. Tuttavia il monopolio di Rockefeller aveva cominciato a essere intaccato già nel 1901, quando, in seguito alla scoperta dei ricchi giacimenti texani, furono costituite la Texas oil co. (Texaco) e la Gulf oil. Nel 1907 nacque la Royal dutch shell, dalla fusione di una compagnia olandese che si dedicava ad attività di estrazione nelle Antille Olandesi e di una società britannica convertitasi al trasporto dei prodotti petroliferi. Nel 1908, infine, un’importante scoperta fatta in Iran in circostanze avventurose determinò la nascita della Anglo-persian oil co., la futura British Petroleum. Nacquero così le ‘sette sorelle’: 3 come risultato della frantumazione del gruppo Rockefeller (Exxon, Mobil oil e Socal), altre 2 statunitensi (Texaco e Gulf oil), una anglo-olandese (Royal dutch shell) e una inglese (British Petroleum).
Con la Seconda guerra mondiale l’industria petrolifera compì altri passi in avanti, soprattutto con lo sfruttamento delle risorse dell’America Meridionale, in particolare di quelle importantissime del Venezuela, e con l’intensificazione delle ricerche e della coltivazione dei giacimenti nel Vicino e Medio Oriente, dimostratisi sempre più necessari all’industria europea in un’epoca di vasti consumi di prodotti energetici sostitutivi del carbone. Il forte aumento della domanda di idrocarburi, in concomitanza con la scoperta di giacimenti sempre più ricchi, agevolò l’ingresso di nuove compagnie nell’industria petrolifera. La nuova ripartizione delle concessioni, in particolare nei Caribi e nel Vicino e Medio Oriente, favorì i nuovi arrivati, le compagnie nazionali dei paesi consumatori e le società indipendenti (società che provvedono al trasporto, alla raffinazione e alla distribuzione dei prodotti ma che, di norma, non dispongono in proprio di grezzo e devono acquistarlo da terzi).
Malgrado l’accresciuta concorrenza, la supremazia delle sette sorelle rimase intatta fino agli inizi degli anni 1970, allorché cominciarono a manifestarsi due importanti novità: da una parte, l’ingresso dell’industria petrolifera in una fase di costi crescenti, che seguiva un lungo periodo di costi decrescenti; dall’altra parte, un capovolgimento dei rapporti di forza tra le compagnie e gli Stati petroliferi, a favore di questi ultimi. Fino al 1950 il dominio delle compagnie sui paesi petroliferi era stato incontrastato, e il capitale petrolifero aveva potuto impossessarsi della maggior parte della rendita mineraria.
La storia delle relazioni tra paesi petroliferi e compagnie subì una svolta decisiva nel 1960, con la creazione dell’Organization of the Petroleum Exporting Countries (➔ OPEC). Fin dal momento della sua nascita l’OPEC riuscì non solo a impedire ribassi del prezzo di riferimento, ma anche a modificare, a vantaggio dei paesi membri, i termini dei contratti di concessione. L’essere diventati proprietari, a tutti gli effetti, della maggior parte dei giacimenti situati sui loro territori ha conferito ai governi dei paesi esportatori, soprattutto a quelli del Golfo Persico, un ruolo geopolitico considerevole.
Le crisi petrolifere
Il conflitto arabo-israeliano del 1967 è stato uno degli avvenimenti che hanno avuto conseguenze determinanti per un radicale mutamento del mercato mondiale del p.; processo, questo, che sarebbe comunque avvenuto, certo più lentamente, in conseguenza di una naturale evoluzione delle rivendicazioni dei paesi produttori nei confronti dei paesi consumatori. In tal senso va interpretato l’allineamento su di una politica di prezzi crescenti, come strumento di pressione indiretta sullo Stato di Israele e come elemento di ritorsione nei riguardi dei paesi consumatori che appoggiavano le sue posizioni o che tendevano a mantenere un atteggiamento neutrale nei confronti dei contendenti. Da un punto di vista strettamente economico, questo orientamento era condiviso, di fatto, anche da altri paesi produttori estranei al conflitto, come per es. il Venezuela, al fine di bilanciare il continuo aumento dei prezzi dei beni strumentali necessari alle rispettive politiche di sviluppo nazionale e dei beni di consumo importati per elevare il livello di vita dei propri abitanti. Un ulteriore inasprimento delle condizioni poste dai paesi produttori si ebbe con la riapertura del conflitto arabo-israeliano nell’ottobre 1973, quando i paesi arabi seguirono una linea politica tendente a fare del p. (si parlò di una vera e propria guerra del p.) un’arma di ritorsione che in qualche modo riequilibrasse le sorti del conflitto con Israele, risoltosi per loro in maniera disastrosa. Agli aumenti del prezzo (primo shock petrolifero) seguirono infatti una riduzione dei ritmi di produzione, una riduzione delle esportazioni verso alcuni paesi e, per un breve periodo, un embargo completo nei confronti degli USA e dei Paesi Bassi. Ciò nonostante, la domanda di grezzo sui mercati mondiali riprese a crescere fino alla fine degli anni 1970, allorquando si verificò il secondo shock petrolifero: la rivoluzione islamica iraniana, disorganizzando l’apparato produttivo di questo paese, ridusse del 13% il quantitativo di grezzo complessivamente fornito dall’OPEC. Il timore di penuria, da parte del mondo industrializzato, fece subire una nuova impennata ai prezzi, che raggiunsero per un breve periodo punte elevatissime. Ma quest’evento si verificò in un momento in cui la domanda già dava segni di riflusso e già si stava intensificando l’estrazione del p. in nuove zone di produzione, in paesi non appartenenti all’OPEC (specialmente nel Mare del Nord e in Alaska). Pertanto i prezzi si adeguarono alla nuova situazione di abbondanza e, dal 1982, presero a diminuire, dapprima lentamente, poi con ritmo accelerato. Anche l’invasione del Kuwait da parte dell’Iraq, nel 1990, e la crisi mondiale che ne seguì, non riuscirono a far ripartire i prezzi al rialzo se non per poche settimane, malgrado la scomparsa dalla scena petrolifera di due grandi produttori, appunto l’Iraq e il Kuwait.
Per i paesi consumatori, le crisi degli anni 1970-90 hanno avuto l’effetto positivo di suscitare un processo di riflessione sul modo più razionale di utilizzare le risorse energetiche disponibili, nel quale il p., dopo gli eccessivi entusiasmi del precedente periodo, tornava a configurarsi come risorsa energetica di primaria grandezza, ma relativamente costosa e limitata in quantità perché non rinnovabile. Le politiche di contenimento e di razionalizzazione dei consumi hanno fatto leva su vari provvedimenti, quali l’adozione di misure di risparmio energetico, la sostituzione del p. con altre fonti, l’intensificazione della ricerca di fonti rinnovabili.
Tra i fattori determinanti per un radicale mutamento del mercato mondiale del p., e per la conferma dell’improrogabilità della transizione dallo sfruttamento di fonti energetiche non rinnovabili alla definizione di modelli economici sostenibili, si colloca il drammatico conflitto esploso nel febbraio 2022 con l’invasione russa dell’Ucraina: esso ha ribadito sul piano geopolitico la stringente necessità di un affrancamento dalla dipendenza dal flusso di idrocarburi proveniente dall’Est - essendo esso il principale strumento a sostegno delle mire imperialistiche russe -, confermando sul piano ambientale l'urgenza di una riconversione verso fonti rinnovabili per la generazione di energia a emissioni zero in grado di rallentare il riscaldamento globale.
Riserve e nuove scoperte di giacimenti
Valutazione delle riserve
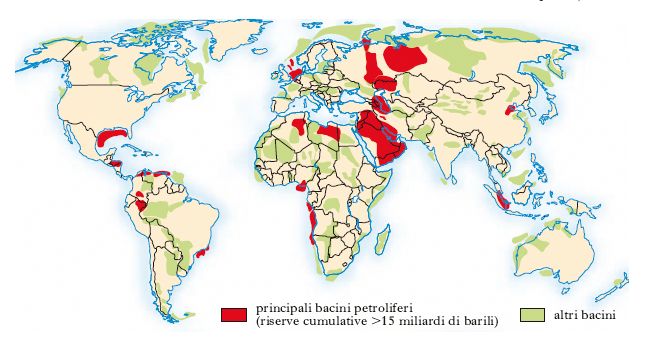
Negli anni 1990, il rapporto tra le riserve mondiali accertate e la produzione annuale è costantemente diminuito: secondo le stime ufficiali che prendono in considerazione dati medi globali (2000), la durata delle riserve sarebbe limitata a una quarantina d’anni (la produzione di gas naturale continuerebbe invece per un periodo più lungo, oltre 60 anni); molto lontani da questo valore intermedio sono i dati relativi alla durata delle riserve dei paesi OPEC (80 anni) e di quelli non-OPEC (15 anni). Si sta peraltro imponendo sempre più la necessità di acquisire maggiore certezza nell’approvvigionamento e, dunque, di trovare nuovi giacimenti in paesi politicamente meno instabili rispetto a quelli mediorientali, dove è concentrato il 65% delle riserve accertate. Un valido contributo in questo senso deriva dalla progressiva diminuzione dei costi d’esplorazione, favorita in larga misura dai progressi tecnologici. È perciò divenuto redditizio lo sfruttamento di pozzi, anche offshore, situati a elevate profondità: sono numerosi i paesi che hanno implementato questo tipo di esplorazione, soprattutto nei tratti di mare a NO dell’Australia e a E delle Filippine, nel Mare Cinese Meridionale, nel Mare di Barents e nel Mare del Nord; le prospezioni compiute nel Golfo di Guinea hanno portato al ritrovamento di giacimenti di potenzialità superiore a 95 milioni di t. Fra le aree di maggiore interesse emerge quella caspica, soprattutto nelle tre repubbliche di Azerbaigian, Kazakistan e Turkmenistan; fondamentale è stato il contributo dato dalle compagnie petrolifere internazionali che, a differenza delle repubbliche ex sovietiche, dispongono delle tecnologie avanzate necessarie per raggiungere i giacimenti meno accessibili. Le nuove cospicue scoperte hanno reso la regione del Mar Caspio la terza in ordine di importanza, dopo quelle del Golfo Persico e della Siberia: il suo ruolo sarà sicuramente cruciale per definire le dinamiche economiche del 21° sec. e garantire ai paesi occidentali un minor grado di dipendenza dal p. mediorientale; per di più, le royalties e le tasse di transito che ne derivano per i paesi della regione rappresentano per essi un importante strumento di affrancamento dall’egemonia russa. Il quadro geopolitico regionale particolarmente complesso rappresenta, però, un serio elemento di disturbo per i progetti di esportazione. A tutto ciò si aggiungono altri problemi: le dispute sui diritti di proprietà e di sviluppo dei pozzi; le scelte relative alla realizzazione e al potenziamento dei corridoi per l’export; il congestionamento del Bosforo. Importanti sono, infine, le questioni di ordine tecnico (infrastrutture per l’esplorazione e il commercio; equipaggiamenti per l’esplorazione dei pozzi; manutenzione della rete degli oleodotti). Pur in questo difficile contesto, è stata resa possibile l’apertura degli oleodotti Baku-Novorossijsk (1997), Baku-Tbilisi-Supsa (1999), Baku-Tbilisi-Ceyhan (2006) e dell'oleodotto Odessa-Brody (2002), che ha aperto una via alternativa agli approvvigionamenti petroliferi dell'Occidente.
Coltivazione e sviluppo dei giacimenti
Indipendentemente dalle dibattute considerazioni sull’entità delle risorse petrolifere mondiali, importanti innovazioni tecnologiche hanno consentito di incrementare sensibilmente il fattore di recupero dei giacimenti. Per i campi situati in rocce relativamente porose e permeabili sono stati perfezionati i metodi di monitoraggio sismico tridimensionale (3-D): introducendo tecniche in grado di seguire il movimento di p., gas e acqua durante il drenaggio produttivo degli strati a opera dei pozzi, è possibile prevedere il comportamento del campo in diverse condizioni di estrazione per individuare quella dal rendimento maggiore. Anche le tecniche di recupero migliorato o assistito attraverso iniezioni di fluidi consentono incrementi dei fattori di recupero e si avvalgono sempre più frequentemente dei sistemi di movimentazione di miscele multifase (multiphase flow). Per quanto riguarda la coltivazione di campi petroliferi rimasti isolati, si preferisce l’utilizzazione delle perforazioni direzionali, che fra l’altro comportano cospicue riduzioni dei costi rispetto al metodo dell’iniezione. Grazie allo sviluppo di motori volumetrici da profondità per l’azionamento dello scalpello, collocabili poco sopra lo scalpello stesso, e soprattutto di sensori per il rilevamento di p., gas e acqua, tipicamente a raggi gamma, posizionati anch’essi in prossimità dello scalpello e in grado di operare a temperature e pressioni elevate (rispettivamente ca. 200 °C e ca. 1800 bar), è possibile seguire accuratamente la direzione di avanzamento del pozzo, anche orizzontale, con la facoltà di eseguire variazioni di 90° nello spazio di 100 m. Un’innovazione riguarda i cosiddetti pozzi intelligenti: con l’ausilio di valvole e separatori a centrifuga, regolati da stazioni di controllo in grado di elaborare in tempo reale i dati forniti dai sensori di profondità, essi permettono di utilizzare simultaneamente differenti diramazioni, da alcune delle quali si può recuperare p. e da altre acqua, separata dai fluidi estratti, da iniettare in zone sterili. Notevoli progressi sono stati effettuati anche nella coltivazione dei giacimenti di offshore profondo; oltre a piattaforme petrolifere ancorate a fondali profondi più di 1000 m, sono operative navi di perforazione dotate di sistemi di posizionamento dinamico riferiti alla rete GPS che consentono, riducendo al minimo gli spostamenti della nave rispetto alla collocazione della testa di produzione sul fondale, lo sfruttamento di campi fino a 3000 m di profondità.
Giacimenti di p. non convenzionale
Circa la coltivazione e lo sviluppo dei giacimenti di p. non convenzionale, costituiti da depositi di sabbie bituminose e scisti bituminosi di grande potenzialità (si stima, per es., che le formazioni sabbiose del Canada contengano più p. dei giacimenti dell’Arabia Saudita), interessanti prospettive sembra offrire un metodo di estrazione fondato sull’iniezione di vapore ad alta temperatura: quando riscaldato, il p. proveniente dal bitume si raccoglie in forma liquida al di sotto del livello di iniezione, da dove può essere condotto in superficie secondo le ordinarie procedure dell’industria petrolifera. Tale metodo consentirebbe di ovviare alle rilevanti implicazioni ambientali di quei sistemi di produzione di p. non convenzionale che prevedono l’estrazione in stabilimento con formazione di residui minerali potenzialmente inquinanti, che devono essere smaltiti attraverso onerosi trattamenti.
Aree di produzione e consumo
Nel corso della prima metà del 20° sec. l’area di maggior produzione e di maggior consumo è stata l’America Settentrionale, con gli USA al primo posto. Ma, a partire dal 1948, gli USA da esportatori sono diventati importatori netti, e da allora hanno visto costantemente ridursi il loro ruolo di produttori nel contesto mondiale. La produzione di p. in USA nel 2007 è stata pari a 254.492.000 t. Il Vicino e Medio Oriente occupano complessivamente il posto di maggior rilievo tra le grandi aree produttrici, anche se la Russia detiene il primo posto nella graduatoria mondiale dei produttori (470.676.000 t di p. nel 2007). L’Europa occidentale, i cui fabbisogni ammontano a circa 1/5 della produzione mondiale, deve importare un quantitativo di p. pari a 3/4 dei suoi consumi, dato che non può fare assegnamento su consistenti risorse interne, se si eccettuano i giacimenti del Mare del Nord. I paesi costieri del Mediterraneo costituiscono, invece, un’area di importazione e trasformazione del grezzo di provenienza mediorientale.
Le altre grandi aree geografiche producono proporzionalmente alle proprie riserve. Nel 2006, il continente africano – le cui riserve sono concentrate in Nigeria, in Algeria e in misura più limitata in Libia – contribuiva per circa l’8% alla produzione mondiale; l’America Latina (in particolare Messico e Venezuela, mentre Brasile, Argentina, Colombia, Ecuador, Trinidad e Tobago sono produttori minori) forniva circa il 13% del totale. La Cina produce circa il 4,5% (185.976.000 t) del totale, consumando però circa il 9%.
A livello globale, a partire dal 2008 la crisi finanziaria ed economica ha inciso sulla produzione e sulla domanda di p., complicando ulteriormente il quadro già reso incerto e fluttuante dal progressivo diminuire della disponibilità e dagli irrigidimenti dei paesi produttori.
L’Italia
A partire dagli anni 1960 l’Italia si è decisamente orientata verso una larga partecipazione del p. al suo bilancio energetico, fino a raggiungere un’aliquota massima nel 1971, quando il grezzo copriva il 77,5% dei consumi di energia. Nel corso dei 20 anni successivi il contributo del p. al soddisfacimento del fabbisogno nazionale è sceso al 55%, anche per effetto della politica energetica perseguita, che si è prefissa l’obiettivo di attenuare i rischi dell’eccessiva dipendenza dalle importazioni. A partire dagli ultimi anni del 20° sec. numerosi giacimenti sono stati individuati come quelli presso l’Aquila e nel canale d’Otranto, mentre si stanno sfruttando anche i campi petroliferi in Val d’Agri (Basilicata). Anche in Sicilia (oltre a quelli di Gela e Ragusa) sono stati individuati dei giacimenti presso Masseria Vecchia e Pizzo Tamburino. Il 75% della produzione nazionale – che nel 2008 è stata di circa 5.200.000 t con un piccolo decremento rispetto al 2007 (5.839.000 t) – viene dai campi della Basilicata e della Sicilia.
Nel corso degli anni 1950 e 1960 la domanda petrolifera italiana, sulla scia degli altri paesi industrializzati, si è indirizzata verso l’area mediorientale. L’Arabia Saudita e l’Iran sono sempre stati i primi fornitori di quest’area, ma negli ultimi decenni del 20° sec. il baricentro delle importazioni si è spostato in Africa, in particolare in Libia, che da sola ha assicurato il 27,3% delle importazioni italiane nel 2006, divenendo il primo fornitore in assoluto. Come riflesso positivo della favorevole posizione rispetto ai traffici petroliferi, l’Italia, fin dai primi anni 1950, ha sviluppato un’industria di raffinazione tra le più consistenti e tecnologicamente avanzate di tutto il mondo.
Aspetti legislativi
Le tipologie di accordi di investimento petrolifero più diffuse, anche se soggette a profonde differenziazioni all’interno degli ordinamenti dei singoli paesi, sono in linea generale: concessione, production sharing, contratto di servizio, prestazione di servizi.
La concessione è il tipo di contratto più antico e tuttora ampiamente utilizzato, benché stia cedendo il passo ad altre forme contrattuali. Sotto il profilo giuridico, si configura come attribuzione, da parte dello Stato, dell’esclusività della ricerca e della produzione di idrocarburi su un’area e per una durata determinate (entrambe tendono a ridursi in misura sensibile in rapporto alle concessioni più antiche) a una compagnia petrolifera (a volte è imposta anche la partecipazione dell’ente petrolifero statale), che acquisisce la proprietà degli idrocarburi portati in superficie, mentre quella delle risorse del sottosuolo resta allo Stato. Dal canto suo, la compagnia è tenuta a corrispondere allo Stato una royalty (sotto forma di un’aliquota della produzione o del suo controvalore in denaro) e una imposta sul reddito.
Il production sharing è un contratto stipulato tra l’ente petrolifero statale, titolare dei diritti minerari, e la compagnia petrolifera privata, la quale si assume però tutti i rischi derivanti dall’investimento. Infatti, solo a produzione avviata la compagnia è autorizzata a detrarre, dalla quantità di p. prodotta, quella che viene chiamata cost oil, destinata a recuperare i costi sostenuti; la parte residua di produzione, detta profit oil, viene ripartita, in proporzioni variabili da paese a paese, tra l’ente statale e la compagnia. Questa è poi tenuta al pagamento di una tassa sul reddito calcolata in base alla propria quota di profit oil. Non è raro peraltro il caso che l’ente nazionale partecipi fin dall’inizio ai rischi dell’esplorazione: in tal caso, anche la quota di cost oil viene ripartita in base alle spese sostenute.
Il contratto di servizio è un contratto analogo a quello di production sharing, dal quale si differenzia perché sia il recupero dei costi sostenuti dalla compagnia sia la sua remunerazione avvengono in valuta e non in p., per il quale la compagnia gode del diritto di acquistare quote di produzione a un prezzo sensibilmente inferiore a quello di mercato.
La prestazione di servizi è un contratto con il quale la compagnia si limita a effettuare le operazioni per conto dello Stato, fornendo la tecnologia e il know-how necessari, dietro pagamento di un corrispettivo per il servizio reso e con il diritto preferenziale di acquistare il p. a prezzo agevolato. Tale tipo di contratto consente allo Stato di mantenere il massimo controllo sulle attività minerarie.